Abstract
Heart failure is associated with profound alterations in cardiac intermediary metabolism. One of the prevailing hypotheses is that metabolic remodeling leads to a mismatch between cardiac energy (ATP) production and demand, thereby impairing cardiac function. However, even after decades of research, the relevance of metabolic remodeling in the pathogenesis of heart failure has remained elusive. Here we propose that cardiac metabolic remodeling should be looked upon from more perspectives than the mere production of ATP needed for cardiac contraction and relaxation. Recently, advances in cancer research have revealed that the metabolic rewiring of cancer cells, often coined as oncometabolism, directly impacts cellular phenotype and function. Accordingly, it is well feasible that the rewiring of cardiac cellular metabolism during the development of heart failure serves similar functions. In this review, we reflect on the influence of principal metabolic pathways on cellular phenotype as originally described in cancer cells and discuss their potential relevance for cardiac pathogenesis. We discuss current knowledge of metabolism-driven phenotypical alterations in the different cell types of the heart and evaluate their impact on cardiac pathogenesis and therapy.
1. Introduction
Cardiovascular disease remains a major cause of morbidity and mortality in both developing and developed countries worldwide. Heart failure (HF) afflicts 27 million patients globally and despite advances in treatment, it still constitutes a rising socioeconomic burden, primarily due to an expanding, aging population [1]. Current HF treatment is primarily targeted at the reduction of cardiac workload via the inhibition of neuroendocrine pathways. However, derangements in cardiac energy metabolism have also been implicated in the pathogenesis of HF, and there is an increasing interest in the modulation of cardiac metabolism as an alternative or complementary therapeutic approach [2].
The healthy heart predominantly relies on the mitochondrial oxidation of fatty acids (60–90%) and of glucose and lactate (10–40%) for the generation of adenosine 5′-triphosphate (ATP), which is necessary to sustain the contraction–relaxation cycle of the cardiac muscle [3]. The contribution of each of these substrates to cardiac ATP production varies and to a large extent depends on extra-cardiac factors (exercise, diet, fasting, etc.). This flexibility in substrate utilization allows the healthy heart to dynamically alter fatty acid and carbohydrate metabolism depending on substrate availability. Increases in circulating fatty acid levels decrease the uptake of glucose and vice versa [4]. During HF this metabolic flexibility becomes compromised. In the failing heart, the ability to oxidize fatty acids is reduced, while the glycolytic conversion of glucose to lactate increases [5]. Mechanistically, this shift away from mitochondrial oxidative metabolism, and consequently the reduction in ATP production, is commonly considered to compromise cardiac function. The failing heart is believed to become “energy-starved” [6,7], as reflected, among other things, by the decline in the cardiac creatine phosphate (CrP) and ATP levels, and in the CrP/ATP ratio in HF patients as assessed by NMR [8,9,10]. However, it is still debated whether the observed reduction in cardiac high-energy phosphate levels in HF patients is sufficient to cause serious cardiac dysfunction [11].
In this review, we investigate substrate and energy metabolism from an entirely different perspective and consider whether the rewiring of cardiac substrate metabolism may have consequences far beyond the mere synthesis of ATP needed to maintain adequate cardiac muscle contraction. Recent developments in cancer research have convincingly demonstrated that the rewiring of cellular substrate metabolism is a crucial event in driving cellular phenotype [12] and parallels have been drawn recently between oncometabolism and substrate metabolism of the failing heart [13]. Here, we discuss the implications of these novel insights regarding the rewiring of metabolic fluxes for cardiac disease. Thereto, we will discuss the principles of influential metabolic changes in the main metabolic pathways first described in cancer, such as the Warburg effect. Next, we review the evidence for metabolic control of cellular phenotype in the main cell types of the cardiovascular system, namely fibroblasts, endothelial cells, vascular smooth muscle cells, immune cells and cardiomyocytes. Finally, the potential implications of the metabolic rewiring for the pathogenesis and treatment of cardiac disease are discussed.
2. Metabolic Rewiring in Cancer: Possible Implications for the Heart
Most of our current knowledge regarding the influence of metabolic changes on cell phenotype and function stems from research on cancer cells. These groundbreaking studies revealed that reprogramming metabolic pathways ensures the survival and proliferation of malignant cells. Non-dividing differentiated cells largely rely on mitochondrial oxidative phosphorylation to maintain cellular homeostasis. On the other hand, rapidly proliferating, undifferentiated cells, such as cancer cells, are characterized by high rates of glycolysis and increased metabolism of glutamine (glutaminolysis) [14]. The reprogramming of these metabolic pathways, often referred to as oncometabolism, therefore constitutes a hallmark of cancer and is essential for the growth of a tumor [15]. During neoplasia, cancer cells display high rates of metabolic turnover and switch from mainly catabolic to anabolic pathways in order to facilitate the generation of precursor molecules for rapid cell growth and proliferation. The most acknowledged change in cancer cell metabolism is metabolic switching to high rates of glycolysis and increased lactate production. In the cardiovascular field, high rates of glycolysis are traditionally associated with hypoxia or ischemia (anaerobic glycolysis), but in tumors, glycolysis can be upregulated when oxygen is abundantly present. This is commonly referred to as aerobic glycolysis. Aerobic glycolysis was first documented in tumor cells by Otto Warburg almost a century ago and is therefore referred to as the Warburg effect [16]. The question that emerges is if the heart also has the intrinsic capacity to increase glycolysis under conditions when oxygen availability is not limiting. In other words, is the Warburg effect operational in the cardiac muscle?
The shift from mitochondrial oxidative to glycolytic metabolism observed in tumors is reminiscent of the shift from fatty acid to glucose metabolism observed in the hypertrophic and failing heart [11,17,18]. In cancer cells, enhanced glycolysis supports an anabolic, proliferative state of the cells, even if it is at the expense of total ATP synthesis. It has been estimated that the normal beating heart invests 20–30% of its ATP in basal metabolism and in maintaining cellular integrity [19]. It is important to realize that cardiac remodeling also depends on growth (cardiomyocyte hypertrophy), proliferation of fibroblasts (fibrosis) and endothelial cells (angiogenesis), as well as infiltration of immune cells (migration). Furthermore, it has also been demonstrated that cardiomyocyte hypertrophy is associated with the partial re-activation of the cell cycle and this is often considered an attempt to initiate proliferation [20].
This raises a number of intriguing questions. Is the metabolic remodeling observed in the hypertrophic and failing heart meant to optimize energy metabolism (and thus pump function), or does it rather serve to support anabolic processes and to allow cardiac cells to adapt their phenotype? Which cell types constituting the heart are subject to metabolic rewiring? What implications does this all have in the setting of insulin resistance and diabetes, when the cardiac muscle becomes even more dependent on the oxidation of fatty acids? To address these questions, we first discuss the various metabolic pathways that contribute to the metabolic rewiring in cancer cells in more detail.
2.1. The Warburg Effect
In terms of energy yield, the conversion of glucose to lactate is far less efficient than the complete oxidation of glucose, yielding only 2 ATP instead of up to 36 ATP [21]. Despite the lower ATP yield, the switch toward aerobic glycolysis is considered highly advantageous for tumor cells. An increased glucose uptake promotes increased flux through pathways that use glycolytic intermediates as a starting point. This includes the pentose phosphate pathway (PPP) and the hexosamine biosynthetic pathway (HBP) that use glucose-6-phosphate and fructose-6-phosphate as substrates (Figure 1). The PPP leads to the formation of precursor molecules of nucleic acids, needed by proliferating cells. A substantial part of the glycolytic intermediates is also used for the synthesis of amino acids, thereby stimulating protein synthesis [22].
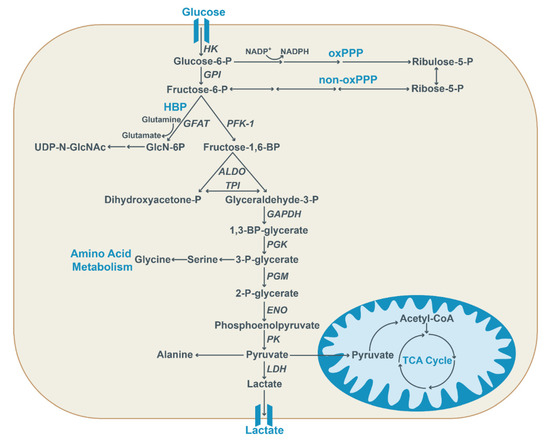
Figure 1.
Glycolysis provides important intermediates for anabolic processes. Glucose is transported into the cell via glucose transporters and first converted into glucose-6-phosphate by hexokinase (HK) and subsequently converted into fructose-6-phosphate. Alternatively, it acts as a substrate for the oxidative pentose phosphate pathway (oxPPP) to generate ribulose-5-phosphate, thereby also generating reducing equivalents in the form of NADPH. Fructose-6-phosphate is converted into fructose-1,6-phosphate by phosphofructokinase 1 (PFK-1) or it can be utilized in the non-oxidative pentose phosphate pathway (non-oxPPP), ultimately resulting in ribose-5-phosphate generation. Moreover, the hexosamine biosynthetic pathway (HBP) utilizes fructose-6-phosphate, together with glutamine, to generate glucosamine-6-phosphate (GlcN-6P) via glutamine:fructose-6-phosphate aminotransferase (GFAT). Subsequently, the end product UDP-N-acetylglucosamine (UDP-N-GlcNAc) is generated in a multi-step conversion. Glyceraldehyde-3-phosphate generated by aldolase (ALDO) and triose-phosphate isomerase (TPI) further undergoes glycolysis. After conversion into 1,3-BP-glycerate via glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), the glycolytic intermediate 3-P-glycerate is generated via phosphoglycerate kinase (PGK) and converted into pyruvate by pyruvate kinase (PK). It can also be used for the synthesis of amino acids. Pyruvate is either converted into lactate by lactate dehydrogenase (LDH), used to generate amino acids, or enters the mitochondria to fuel the TCA cycle.
Originally, the Warburg effect was attributed to mitochondrial dysfunction. Later studies showed that mitochondrial oxidative capacity in cancer cells was marginally affected in many cases [16], suggesting that direct activation of the glycolytic pathway was the primary cause of the Warburg effect. Although various signaling pathways (PI3K-Akt-mTOR) and transcription factors such as Forkhead, the tumor suppressor p53 and the oncogene Myc have been implicated in the stimulation of aerobic glycolysis [23,24], the transcription factor hypoxia-inducible factor-1a (HIF-1a) is considered the major driver of the Warburg effect.
The activity of HIF-1a is primarily regulated at the level of protein stability and, as the name implies, is dependent on oxygen levels. HIF-1a stability is controlled by the prolyl hydroxylase (PHD)-dependent hydroxylation of proline residues in HIF-1a. This reaction requires oxygen and alpha-ketoglutarate as substrates and yields succinate as an end product (Figure 2). Under normoxic conditions, the PHD-dependent hydroxylation of HIF-1a marks it for ubiquitin-mediated degradation. However, when oxygen is limiting, PHD activity is suppressed, resulting in HIF-1a stabilization and subsequent induction of HIF-1a-dependent genes [25]. Notably, the end product succinate, which also is an intermediate of the tricarboxylic acid (TCA) cycle, acts as an inhibitor of PDH activity and thereby causes stabilization of HIF-1a. This implies that independent of oxygen levels, a rise in cellular succinate levels—for instance, due to changes in TCA cycle activity—also prevents HIF-1a degradation. The latter provides a mechanistic explanation for the Warburg effect. Hypoxia-independent stabilization of HIF-1a by changes in cellular succinate levels was first documented in cancer cells [26,27,28] and later shown to be operative in macrophages as well [29]. These studies reveal the close interconnection between TCA cycle activity and HIF stability.
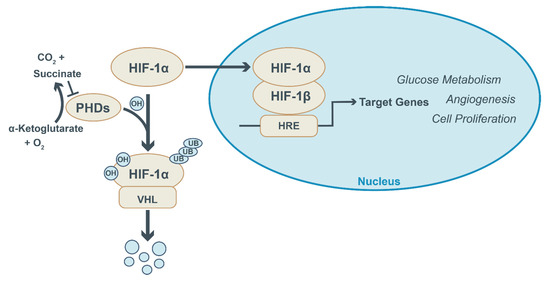
Figure 2.
HIF-1a signaling and regulation. Under normoxic conditions, prolyl hydroxylase enzymes (PHDs) hydroxylate HIF-1a and mark it for degradation by the von Hippel–Lindau E3 ubiquitin ligase (VHL). HIF-1a hydroxylation by PHDs requires alpha-ketoglutarate and oxygen as substrates and yields succinate and CO2. PHD activity is suppressed under hypoxic conditions and following the accumulation of succinate. This prevents HIF-1a degradation and allows its translocation into the nucleus, where it dimerizes with HIF-1b and binds to HIF-responsive elements (HRE) in the promotor region of genes. This activates the expression of target genes involved in glucose metabolism, angiogenesis and cellular proliferation.
HIF-1a stabilization results in the concerted induction of the glucose transporter (GLUT1), nearly all glycolytic enzymes (Table 1) and the monocarboxylate transporter-1 (MCT-1) involved in the export of lactate. It also induces the expression of the kinase pyruvate dehydrogenase kinase-1 (PDK1). PDK1 phosphorylates the pyruvate dehydrogenase (PDH) complex, thereby inhibiting PDH and diverting pyruvate away from mitochondrial oxidation [30]. Finally, it induces the expression of angiogenic factors like angiopoietin-like-4 (ANGPTL4) and vascular endothelial growth factor (VEGF) [31,32]. ANGPTL4 also inhibits the activity of lipoprotein lipase on the endothelial luminal surface and, in this way, limits the delivery of fatty acids as a substrate for mitochondrial oxidation [33].
Accordingly, HIF-1a promotes the use of glucose at all levels, from the import of glucose down to the export of lactate. Moreover, at the same time, it inhibits the mitochondrial oxidation of glucose (via PDK1) and indirectly that of fatty acids (via ANGPTL4), and promotes oxygen supply to cells (via VEFG-stimulated angiogenesis).
It is well feasible that similar mechanisms are operative in the myocardium. Perhaps the commonly observed increase in glycolysis and its partial uncoupling from glucose oxidation during cardiac hypertrophy and failure [17,34,35] are indications of the Warburg effect being operative in the diseased heart.
2.2. Non-Canonical Functions of Glycolytic Enzymes
When discussing the glycolytic pathway from a metabolic perspective, one also has to take into account that many of the glycolytic enzymes have been shown to have pleiotropic functions and are directly involved in cellular signaling. These reported actions are listed in Table 1. For instance, the expression of hexokinase-2 (HK2), the isozyme predominantly found in insulin-sensitive tissues like the heart, markedly increases during tumorigenesis [36,37]. Activation of the Akt-mTOR pathway evokes a HIF-1a-dependent increase in the expression of HK2 [38,39]. The isozyme is found both in the cytoplasm and bound to mitochondria, depending on cellular conditions.

Table 1.
Non-canonical functions of glycolytic enzymes.
Table 1.
Non-canonical functions of glycolytic enzymes.
| Enzyme Isoforms | TF Regulation | Non-Canonical Functions | Ref. |
|---|---|---|---|
| HK-1/2 | HIF/Myc | Apoptosis | [40,41,42] |
| GPI | HIF | Secreted growth factor (cell migration), Apoptosis | [43,44,45] |
| PFK-M/L/P | HIF/Myc | YAP/TAZ Signaling | [45,46,47,48] |
| ALDO-A/C | HIF | Activates AMPK | [49,50,51] |
| TPI | HIF | n.d. | [52] |
| GAPDH | HIF | RNA-binding, Apoptosis | [53,54] |
| PGK-1 | HIF | Protein kinase activity (autophagy) | [55,56] |
| PGAM-1 | HIF | Actin binding (cell migration) | [57] |
| ENO-1/2 | HIF/Myc | Apoptosis, cell migration, tRNA transport | [58,59,60] |
| PKM-1/2 | HIF/Myc | Protein kinase activity | [61,62] |
Importance of the transcription factors (TFs) HIF-1a and Myc in the transcriptional regulation of isoforms of glycolytic enzymes and reported non-canonical functions of these enzymes. For TPI, non-canonical functions have not been described (n.d.). HK—hexokinase, GPI—phosphoglucose isomerase, PFK—phosphofructokinase, ALDO—aldolase, TPI—triose-phosphate isomerase, GAPDH—glyceraldehyde-3-phosphate dehydrogenase, PGK—phosphoglycerate kinase, PGAM -phosphoglycerate mutase, ENO—enolase, PKM—pyruvate kinase (muscle isoform). Numbers and capitals refer to the different isoforms of these enzymes.
Following its phosphorylation by Akt, HK2 translocates to the outer mitochondrial membrane [63]. This inhibits apoptosis by interfering with the mitochondrial calcium- and reactive oxygen species (ROS)-mediated opening of the mitochondrial permeability transition pore [40,41]. Indeed, overexpression of HK2 promoted glycolysis and suppressed cancer cell death, an effect that depended on its N-terminal mitochondrial binding domain [64,65]. The enolase-1 isozyme (ENO1) is another glycolytic enzyme that is increased in metastatic cancer cells. Remarkably, ENO1 is also found on the cell surface of these cells where it interacts with plasminogen, thereby promoting ECM degradation and cell migration [58]. It would be interesting to explore if ENO1 has comparable non-canonical functions in cardiac fibrosis.
The pyruvate kinase muscle isozyme (PKM) catalyzes the last step of the glycolytic pathway. Alternative mRNA splicing of PKM gives rise to two mutually exclusive isoforms, containing either exon 9 (PKM1) or exon 10 (PKM2). PKM1 is normally found in more differentiated cells, whereas PKM2 is considered the embryonic form and is present at higher levels in proliferating cells, including cancer cells [66,67]. Both PKM1 and PKM2 form tetramers with high enzymatic activity. However, PKM2 is also present in the enzymatically less active dimeric and monomeric form. Not only does it divert glucose metabolism toward anabolic pathways, but it also exerts many other cellular functions as well. PKM2 is a target of tyrosine kinases [68] and interacts with phosphorylated tyrosine residues of proteins [69]. Both events interfere with the binding of the allosteric activator fructose-1,6-bisphosphate to PKM and thereby prevent the formation of the active tetrameric form. Through this, PKM activity and function are directly linked to growth factor-dependent tyrosine kinase signaling. Monomeric PKM2 has been shown to translocate toward the nucleus upon ERK1/2 phosphorylation, where it modulates gene transcription by acting as a co-activator of HIF-1a and of the beta-catenin/TCF4 complex [70,71]. Beta-catenin induces transcription of oncogenic Myc, a transcriptional regulator of several glycolytic enzymes (Table 1), and of Cyclin-D1, required for cell cycle progression. Via this mechanism, the formation of PKM2 by alternative splicing can be considered a hallmark of the Warburg effect as it promotes glycolysis and the use of glycolytic intermediates for biosynthetic purposes [72].
Notably, the development of cardiac hypertrophy is associated with the reactivation of the fetal gene program. This includes a general increase in the expression of various glycolytic enzymes, along with an isoform shift of several glycolytic enzymes to their fetal isoforms, including enolase (ENO3→ENO1) [73,74] and PKM (PKM1→PKM2) [75,76]. The functional significance of these isoform changes for the heart remains to be established. As far as HK2 is concerned, the non-canonical function of this enzyme in protecting the heart against apoptosis has been firmly established [77,78]. Strikingly, selective pharmacological activation of the PKM2 isoform protected against diabetes-induced kidney dysfunction [79]. It remains to be tested if PKM2 activation has similar effects in heart disease. Collectively, these studies suggest that the switch of glycolytic enzymes from one isoform to another not only has effects on glycolytic rate control but also serves discrete roles in cellular signaling.
2.3. Pentose Phosphate Pathway
As discussed, increased uptake and conversion of glucose to glucose-6-phosphate also fuels the PPP. The PPP promotes cancer cell proliferation by using glucose-6-phosphate and fructose-6-phosphate to generate ribose-5-phosphate via the oxidative branch (oxPPP) and the nonoxidative branch (non-oxPPP) of the PPP, respectively (Figure 1) [80]. Ribose-5-phosphate provides the substrate for the de novo synthesis of nucleotides that become incorporated in DNA, RNA, NAD(P) and ATP. The PPP gets activated in case of an increased requirement for nucleotide synthesis in highly proliferative cancerous cells. In addition, in the oxidative branch of the PPP, NADP is reduced to NADPH. NADPH is required for the de novo synthesis of fatty acids. The fatty acids, in turn, act as building blocks for phospholipids, needed for the formation of cellular membranes by proliferating cells. Furthermore, NADPH provides protection against oxidative stress by converting the antioxidant glutathione (GSH) to its reduced form [81].
In the heart, modifications in oxPPP flux have been implicated in the maintenance of the redox balance: Following pressure overload as well as myocardial infarction, mice exhibited increased activity of glucose-6-phosphate-dehydrogenase (G6PDH), the first and rate-limiting enzyme of the oxPPP, indicative of an increased oxPPP flux. G6PDH-deficient mice, in which oxPPP flux is reduced, exhibited exacerbated cardiac remodeling in response to myocardial infarction and pressure overload and experienced increased redox stress [82]. Rats that developed left ventricular hypertrophy and failure exhibited enhanced PPP flux as well, as evidenced by the accumulation of PPP intermediates and increased G6PDH activity [83]. Additionally, overexpression of hexokinase in mice amplified PPP flux and attenuated cardiac hypertrophy in response to isoproterenol [84]. On the other hand, in a dog model of pacing-induced heart failure, it was observed that the increase in G6PDH supplies the NADPH needed by ROS-generating enzymes and thereby fuels oxidative stress [85].
2.4. Hexosamine Biosynthetic Pathway
The HBP utilizes the glycolytic intermediate fructose-6-phosphate as substrate and accounts for 2–5% of total glucose metabolism. Enhanced flux through the HBP has been documented in cancer cells [86,87,88]. The HBP is highly dependent on glutamine availability, as glutamine is converted together with fructose-6-phosphate into glucosamine-6-phosphate(GlcN-6P) and glutamate by the rate-limiting enzyme glutamine:fructose-6-phosphate aminotransferase (GFAT) (Figure 1) [89]. As discussed in more detail later, in this way the glycolytic pathway and HBP are linked to glutamine metabolism. The end product of the HBP is UDP-N-acetylglucosamine (UDP-GlcNAc). Importantly, UDP-GlcNAc is a substrate for post-translational modifications of proteins by N-glycosylation, O-glycosylation and O-GlcNAcylation, which are essential for the regulation of cellular functions. For example, modification and subsequent activity modulation of glycolytic and lipid biosynthesis-associated enzymes by O-GlcNAcs have been documented in various cancers as prominent modulators of cellular metabolic pathways [90,91]. Specifically, O-GlcNAcylation of the glycolytic enzyme phosphofructokinase (PFK-1), a key regulatory enzyme in the glycolytic pathway, inhibits PFK-1 activity and diverts glycolytic flux toward the PPP, proving advantageous for cancer growth under conditions of oxidative stress [92]. In addition, the O-GlcNAcylation of proteins at Ser/Thr residues may compete with phosphorylation at the same site and may have divergent effects. For instance, O-GlcNAcylation of the oncogene Myc at Thr58 prevents phosphorylation of Thr58 and blocks its ubiquination and degradation [93,94]. Taken together, via the O-GlcNAcylation of proteins the HBP connects cellular metabolism with signaling pathways that are critical for cell growth and proliferation.
As far as the heart is concerned, it has been shown that various proteins involved in cardiac contraction, including Troponin I and T, are subject to O-GlcNAcylation [95]. In addition, it has been documented that challenging the cardiomyocytes or the heart in situ with pro-hypertrophic stimuli increases global protein O-GlcNAcylation [96]. Global O-GlcNAcylation is increased in cardiac tissue of advanced heart failure patients, patients with aortic stenosis as well as in murine models of cardiac hypertrophy and failure [97,98]. This increase in O-GlcNAcylation is, at least in murine hearts, accompanied by elevated levels of the cardiac O-GlcNAc transferase [99]. Pharmacological inhibition or cardiac deletion of O-GlcNAc transferase exacerbated cardiac dysfunction in association with decreased global O-GlcNAcylation [97,98]. Conversely, pharmacologically preserved O-GlcNAc levels in isolated adult cardiomyocytes provided protection against H2O2-induced cell death [100]. Evidence is accumulating that the modulatory effect of important regulatory proteins such as AMPK and HDAC4 on cardiac hypertrophy may be mediated by their effect on the HBP [96,101]. Enhanced flux through the HBP and consequent chronic GFAT activation has been suggested to contribute to the development of cardiac hypertrophy, as GFAT-associated O-GlcNAcylation appears to induce mTOR-mediated pro-hypertrophic signaling [102].
2.5. Glutaminolysis
Next to glucose, glutamine is an important carbon source for the biosynthesis of macromolecules required for the proliferation and maintenance of redox capacities within cancer cells [103]. Glutamine is the most abundant non-essential amino acid in circulation and is the main source of nitrogen needed for nucleotide synthesis. Glutamine fuels glutaminolysis, which is one of the major anaplerotic pathways. During glutaminolysis, glutamine is first converted into glutamate and ammonia via the enzyme glutaminase (Figure 3). Glutamate is then further converted into the TCA intermediate alpha-ketoglutarate by glutamate dehydrogenase, thereby fueling the TCA cycle and producing carbon sources for the generation of macromolecules (Figure 3) [104]. Reductive carboxylation of alpha-ketoglutarate yields citrate that acts as a source of cytosolic acetyl-CoA needed for the biogenesis of fatty acids and phospholipids [105]. Reductive carboxylation in tumor cells is believed to become important under conditions when HIF-1a is active or when mitochondrial function is impaired [106].
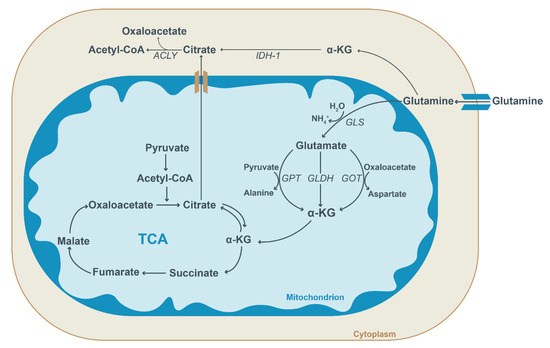
Figure 3.
Schematic overview of glutamine metabolism. In the cytosol, glutamine is converted into alpha-ketoglutarate (α-KG) or transported into mitochondria. Within the mitochondria, glutamine is first converted into glutamate by glutaminase (GLS) and then to α-KG. Glutamate conversion into α-KG is facilitated by three enzymes: glutamate pyruvate transaminase (GPT), glutamate dehydrogenase (GLDH) and glutamate oxaloacetate transaminase (GOT). Subsequently, α-KG enters the TCA cycle and aids the generation of succinate, thereby replenishing the TCA cycle (anaplerosis). Alternatively, it can be used to generate citrate (reductive carboxylation) which can be transported to the cytosol. In the cytoplasm, isocitrate dehydrogenase (IDH-1) facilitates the conversion of α-KG into citrate. Citrate is utilized for acetyl-CoA and oxaloacetate generation by ATP-citrate lyase (ACLY). Cytosolic acetyl-CoA forms the substrate for lipogenesis and the acetylation of proteins.
The role of glutaminolysis in the heart has not been clearly defined. Under normal conditions, the role of glutamine in the anaplerosis of the TCA cycle intermediates seems limited [107]. However, cardiac glutaminolysis was shown to be activated in a rat model of right ventricular hypertrophy [108]. Notably, the induction of transporters and enzymes involved in glutamine handling is Myc-dependent, again pointing to an important role of this oncogene in metabolic rewiring [109].
2.6. Metabolism and Post-Translational Modifications
In addition to UDP-GlcNAc, an end-product of the HBP pathway discussed above, acetyl-CoA also serves as an important metabolic intermediate for the post-translational modification of proteins. Protein acetylation occurs in the mitochondrial, cytoplasmic as well as nuclear compartments. In the cytosol, mitochondria-derived citrate is converted into acetyl-CoA and oxaloacetate by the enzyme ATP-citrate lyase (ACLY) (Figure 3) [110]. The acetyl unit can be used for lipogenesis or for the acetylation of lysine-residues in proteins via lysine acetyltransferases [110]. Acetylation affects not only the activity and stability of a large number of metabolic enzymes but also that of important regulatory proteins, such as the tumor suppressor p53 [111] and the peroxisome proliferator-activated receptor gamma, coactivator PGC-1a [112], the latter acting as a master regulator of mitochondrial biogenesis and respiration. In addition, the acetylation/deacetylation of histones provides epigenetic control of gene transcription [113].
Interestingly, the spectrum of posttranslational mechanisms associated with metabolic intermediates was recently extended by the discovery of another and comparable posttranslational modification process, which is based on the addition of succinate to lysine residues [114]. The substrate for this process appears to be the TCA cycle intermediate succinyl-CoA, but this still remains to be proven definitively. Whereas acetylation neutralizes the positive charge of lysine residues, succinylation converts it into a negatively charged residue. Interestingly, increasing the degree of protein succinylation, via genetic deletion of the desuccinylating enzyme Sirtuin-5 in mice, has been associated with the development of hypertrophic cardiomyopathy [115], suggesting that succinylation might also be of importance for the heart.
The combined data demonstrate that in cancer cells, alterations in substrate and energy metabolism directly influence the behavior of these cells. On the one hand, these alterations affect energy supply and balance, on the other, they modulate cellular phenotype and thereby cellular function. Changes in the fluxes through metabolic pathways provide substrates for the synthesis of biomolecules and for the post-translational modification of proteins. Metabolic enzymes themselves can also exert non-canonical functions and thereby regulate non-metabolic cellular processes.
3. Metabolic Rewiring in Cells of the Diseased Heart
Could it be that mechanisms similar to the metabolic rewiring in cancer cells are in place in cells of the cardiovascular system and play a role in cardiac disease? In the following section, we will reflect on the regulation of cellular phenotypes by metabolic rewiring in the principal cell types of the cardiac tissue, namely endothelial cells, fibroblasts, vascular smooth muscle cells and the cardiomyocytes themselves. We will first discuss the role of infiltrating immune cells, macrophages in particular, not only because these cells play an important role in cardiac disease, but equally relevant, because the concept of metabolic rewiring as a determinant of cell function has been developed further for immune cells. In fact, it led to the development of a new niche in immunology research: immunometabolism [116].
3.1. Immune Cell Metabolism
It is commonly accepted that the rewiring of immune cell metabolism is an integral and crucial element of the immune response [117]. Indeed, the fate and function of various types of immune cells, including macrophages, dendritic cells and T-cells, is determined by changes in cellular metabolism.
The connection between metabolic cues and macrophage phenotype and function has been a focal point of interest in recent years [118,119]. Macrophages adjust their phenotype in response to environmental stimuli, such as tissue injury, microbial infection, or T-cell signaling [120]. Historically, based on in vitro experiments, a distinction is made between two polarized states of the macrophages. Classically activated M1 macrophages exert pro-inflammatory functions to protect against microbial infections, whereas alternatively activated M2 macrophages serve a more anti-inflammatory role and coordinate tissue repair [121]. Recently, it has been recognized that macrophage polarization in vivo does not confer a strict M1/M2 phenotype, but rather presents a continuum along the two extremes. M1 macrophages are activated by IFNγ secreted by type 1 T-helper (Th1) cells, and by Toll-like receptor (TLR) ligands, such as LPS. They exhibit a pro-inflammatory profile and produce TNFα, IL-6 and IL-12 as well as reactive oxygen and nitrogen species, providing them with the tools needed for microbicidal actions [122]. In contrast, cytokines such as IL-4 and IL-13 that are secreted by type 2 T-helper (Th2) cells provoke M2 polarization, leading to the expression of anti-inflammatory mediators, such as TGFβ, IL-10 and IL-1 receptor antagonists, and resulting in high phagocytotic activity [122,123]. For the heart, it has been shown that in response to cardiac ischemia, the first wave of infiltrating macrophages mainly comprises M1-like macrophages, involved in initiating an inflammatory response, whereas the second wave consists of M2-like macrophages involved in tissue repair [124].
Polarization toward an M1-activated state leads to a metabolic switch, marked by an increased glycolytic flux along with an increased expression of the glucose transporter GLUT1 and various glycolytic enzymes [125,126,127,128]. Most of the glucose is converted into lactate rather than oxidized, indicative of aerobic glycolysis [129]. Notably, overexpression of GLUT1 in mouse macrophages has been shown sufficient to promote an M1-like phenotype, demonstrating the dependence of macrophage phenotype on metabolism [127].
The increased aerobic glycolysis in activated M1 macrophages also fuels the oxidative branch of the PPP, thereby promoting the formation of NADPH needed to facilitate microbicidal respiratory bursts via the NAPDH oxidase [127,130]. The mitochondrial oxidation of fatty acids is reduced in LPS-stimulated M1 macrophages [129,131]. The decline in mitochondrial oxidative phosphorylation has been linked to altered TCA cycle activity, more specifically to breaks at the level of isocitrate dehydrogenase and of succinate dehydrogenase (SDH), along with a rise in citrate and succinate levels [132,133]. Succinate stabilizes HIF-1a and thereby stimulates glycolysis and induces secretion of pro-inflammatory cytokines such as IL-1b [29,132]. Part of the citrate is transported from the mitochondria to the cytosol where ACLY can convert it into oxaloacetate and acetyl-CoA required for fatty acid synthesis. The switch to glycolytic ATP production results in mitochondrial hyperpolarization, which together with succinate-driven reverse electron transport involving respiratory complex I and II greatly stimulates mitochondrial ROS production in respiratory complex I [132].
By contrast, in IL-4-activated M2 macrophages, uptake and oxidation of fatty acids are increased due to transcriptional activation via STAT6 and PGC-1b leading to increased expression of enzymes involved in fatty acid uptake, mitochondrial transport and beta-oxidation [134]. Moreover, glutamine uptake and anaplerotic glutaminolysis are enhanced in M2 macrophages, thereby stimulating the HBP and TCA cycle activity. The glutamine-dependent generation of alpha-ketoglutarate strongly promotes the anti-inflammatory M2 phenotype [135]. The detailed metabolic studies of macrophages and other immune cells provide unequivocal evidence of the biological significance of metabolic rewiring in cells, other than tumor cells.
3.2. Endothelial Cell Metabolism
Healthy endothelial cells inhibit the proliferation of smooth muscle cells, adhesion of leukocytes, aggregation of platelets, and maintain appropriate vasodilation via the release of vasoactive substances [136]. Within the myocardium, cardiomyocytes are in direct interaction with endothelial cells via their adjacent position to capillaries, which ensures an adequate supply of nutrients and oxygen [137]. Although endothelial cells have immediate access to oxygen provided by the circulating blood, they do not rely on oxidative metabolism and are instead highly glycolytic [138]. Approximately 85% of ATP production in endothelial cells is achieved via aerobic glycolysis, a dependence similar to highly glycolytic cancer cells [139]. These high glycolytic rates subsequently provide glycolytic intermediates to fuel side pathways, such as the PPP and HBP, which play a role in maintaining cell viability and migration [140]. Notably, under conditions of hyperglycemia, overactivation of the HBP promotes O-linked glycosylation of signaling proteins, eventually impairing endothelial nitric oxide synthase (eNOS) activity and angiogenic capacity [141,142].
The low oxygen need of endothelial cells is advantageous, as it allows endothelial cells to sprout into low-oxygenated tissue and to spare the available oxygen for surrounding perivascular cells [143]. Although endothelial cells are capable of increasing oxidative phosphorylation when needed, they primarily produce ATP via glycolysis to generate new blood vessels [139]. The role of other metabolic pathways in endothelial cells has not been completely elucidated yet. However, endothelial cells utilize carbons derived from fatty acids for the production of nucleotide precursors as well as glutamate and aspartate for angiogenic proliferation [144,145]. Pharmacological inhibition of carnitine palmitoyl transferase-1 (CPT1), a rate-limiting enzyme of mitochondrial fatty acid oxidation, did not disturb energy balance, but inhibited nucleotide synthesis required for DNA replication and in this way reduced endothelial cell proliferation [145].
Following exposure to pro-angiogenic growth factors, endothelial cells switch to an active state and become migratory tip cells or proliferating stalk cells. Under VEGF stimulation, GLUT1 expression is increased, leading to higher glucose transport in proliferating endothelial cells [146]. Nearly all glucose is converted into lactate and excreted, and thus not used for the synthesis of nucleic acids or amino acids. Accordingly, in contrast to cancer cells, the high glycolytic activity of endothelial cells is not meant to sustain anabolic processes in the proliferating cells. However, glutamine remains within endothelial cells, indicating a significant contribution of this amino acid in the generation of the biomass needed for cell proliferation. Pharmacological or genetic inhibition of glutamine utilization leads to a marked reduction in TCA cycle intermediates and induces proliferative arrest [147,148]. Due to their dependence on glycolysis, endothelial cells contain fewer mitochondria than cells driven by oxidative phosphorylation, and experimental inhibition of mitochondrial ATP production does not affect vessel sprouting [139]. By contrast, suppression of glycolysis in endothelial cells by inhibiting phosphofructokinase-2/fructose-2,6-biphosphatase 3 (PFKFB3) or PKM2, potently inhibits angiogenesis by inhibiting endothelial cell migration and proliferation [149,150,151]. The latter observations support the concept that therapeutic interventions at the level of metabolism can be used to modulate angiogenesis [152].
3.3. Vascular Smooth Muscle Cell Metabolism
An important function of vascular smooth muscle cells (VSMCs) is to regulate vessel tone, and thereby blood flow and blood pressure. Due to their specialized role, VSMCs express a distinctive set of signaling molecules and contractile proteins, including smooth muscle alpha-actin, smooth muscle myosin heavy chain and calponin [153]. Within mature, healthy blood vessels, they maintain a low proliferative rate and produce small amounts of ECM proteins [153]. However, in response to mechanical or neurohumoral stimulation secondary to hypertension or vascular injury, VSMCs undergo phenotypic switching toward a more synthetic phenotype. Synthetic VSMCs are characterized by increased cellular proliferation and migration in addition to the upregulated synthesis of ECM components and decreased expression of contractile proteins [153].
VSMCs exhibit a Warburg-like metabolism, which is further increased after acquiring a synthetic phenotype. VSMCs utilize aerobic glycolysis to source a third of the total ATP, and 90% of this glycolytic flux results in the generation of lactate [154]. Increased levels of lactate in the extracellular environment further reinforce the VSMC synthetic phenotype [155]. Oxidative phosphorylation is also used to produce ATP. If fatty acid oxidation is specifically stimulated, aerobic glycolysis is partially substituted, indicating that VSMCs exhibit metabolic flexibility and that aerobic glycolysis is not essential in all metabolic conditions [156]. Exposure of VSMCs to 2-deoxyglucose, a glucose analog inhibiting glycolysis at the level of HK, reduces proliferation in a dose-dependent fashion, showing that aerobic glycolysis, although not essential for cell survival and maintenance of the cytoskeleton, nevertheless plays an important role in VSMCs proliferation [157]. High glucose environments as well as induced upregulation of cellular glucose metabolism promote anti-apoptotic pathways in VSMCs via protein kinase C (PKC) activation [158] and inactivation of glycogen synthase kinase 3-beta (GSK3b) [159]. The increase in glycolytic activity in synthetic VSMCs partially depends on elevated HK2 expression secondary to the activation of STAT3 or the PI3K-AKT pathway [160,161]. Pharmacological inhibition of HK2 prevents the migration as well as the proliferation of pulmonary VSMCs. Likewise, activation of ENO1 stimulated the proliferation and de-differentiation of pulmonary VSMCs [162], once more illustrating that metabolic cues drive the transition from a contractile to synthetic VSMC phenotype.
3.4. Fibroblast Metabolism
Fibroblasts provide structural support to the myocardium via the synthesis of extracellular matrix (ECM) components [163]. Under physiological conditions, ECM homeostasis is maintained via cell–matrix interactions and biochemical signaling in so-called quiescent fibroblasts. However, pathological stimuli cause a switch from a quiescent to an activated state, eventually resulting in their transition into myofibroblasts. Myofibroblasts are characterized by elevated levels of alpha-smooth muscle actin (aSMA) containing stress fibers and display increased collagen production, mobility and contraction [164]. TGF-beta, a cytokine commonly produced by M2 macrophages in the process of wound repair, is a potent inducer of fibroblast to myofibroblast differentiation [165].
Fibroblast activation is both induced and accompanied by metabolic reprogramming toward a more glycolytic state: High glucose environments activate cardiac and kidney fibroblasts and promote their proliferation and transition into myofibroblasts in vitro [166,167]. Likewise, cardiac fibroblasts isolated from diabetic rat hearts exhibit a myofibroblast phenotype characterized by increased aSMA expression and contractility [168]. Fibroblasts from the lung tissue of pulmonary hypertension patients or fibrotic kidneys exhibit increased glycolytic gene expression as well, despite deriving from a normoglycemic environment [166,169]. Induction of myofibroblasts via TGF-beta leads to an increased expression of glycolytic enzymes such as HK2, PFK and LDHA via HIF-1a [166,170,171]. The inhibition of glycolysis with PFKFB3 inhibitors in turn reduced TGF-beta-induced effects in vitro and attenuated the development of fibrosis in a murine model of lung fibrosis [172]. The increased aerobic glycolysis leads to an accumulation of succinate, stabilizing HIF-1a and further stimulating fibroblast activation [172,173]. Likewise, hypoxia enhanced myofibroblast formation via HIF-1a in a mouse model of lung fibrosis [174]. Enhanced mitochondrial fragmentation through the overexpression of mitochondrial fission factor (MFF) limits mitochondrial respiration and enforces a glycolytic shift, thereby also inducing myofibroblast-like characteristics such as increased aSMA and calponin expression [175]. On the other hand, supplementation with 2-deoxyglucose, which effectively inhibits aerobic glycolysis, leads to decreased aSMA expression and a less pronounced myofibroblast phenotype [171]. The metabolic shift toward increased glycolysis in cardiac fibroblasts is accompanied by transcriptional activation and secretion of ECM proteins as well as increased O-GlcNAcylation via increased glucose flux through the HBP [176,177]. Apart from glycolytic reprogramming, myofibroblasts also exhibit increased glutaminolysis, providing the cells with citric acid cycle intermediates necessary for their increased energetic demands [173]. Inhibition of glutaminolysis via reduced glutaminase activity in mice with liver fibrosis suppressed myofibroblast accumulation and the progression of fibrosis [173]. The myofibroblast phenotype can also be reversed, as has been documented for hepatic stellate cell-derived myofibroblasts: These cells revert to a quiescent state when HIF-1a, glycolytic enzymes, glutaminolysis or lactate accumulation are inhibited [178]. Zhao and colleagues reported that the inhibition of glycolysis as well as the stimulation of fatty acid oxidation reduced the production of ECM proteins by dermal fibroblasts [179]. Collectively, these observations lend support to the concept that cardiac fibrosis may also be reversed via metabolic interventions.
3.5. Cardiomyocyte Metabolism
As reviewed in the previous paragraphs, phenotypical switches in the various cell types that constitute the cardiac muscle are associated with changes in cellular metabolism and, conversely, forced changes in cellular metabolism influence cellular phenotype and function. In view of the interest in the significance of “energy starvation” as a causative factor in the development of cardiac disease, it is quite surprising that little is known about the significance of metabolic rewiring in cardiomyocyte remodeling. To some extent, this can be explained by the fact that adult cardiomyocytes are non-proliferating, terminally differentiated cells that are notoriously difficult to study under in vitro conditions. Some information can be extracted from in situ studies that made use of cardiomyocyte-restricted knockout or overexpression of genes involved in fatty acid or glucose metabolism. Table 2 provides a summary of such studies, focusing on the effect of specific genetic interventions on intermediary metabolism and on the development of cardiac hypertrophy. It is worth noting that every intervention which limits fatty acid metabolism, irrespective of whether it is at the level of cardiomyocyte fatty acid uptake or mitochondrial beta-oxidation, led to an increase in glucose metabolism and was associated with increased hypertrophy. Conversely, stimulation of fatty acid metabolism by the cardiac-restricted knockout of acetyl-CoA carboxylase (ACC) had the opposite effect. These observations fit with the idea that increased glycolysis is linked to hypertrophic growth. However, the outcomes of studies with interventions at the level of glucose metabolism are more divergent (Table 2), being influenced by the level at which glycolysis is blocked. In some cases, glycolytic flux was affected in its entirety. In other cases, such as the cardiac-restricted overexpression of a kinase-deficient PFK2, the PPP and HBP were stimulated, while at the same time glycolytic flux was reduced [180,181].
Table 2.
Cardiac-restricted deletion or overexpression of metabolic genes affects hypertrophic growth.
Alternatively, compensation by other isoforms of the inhibited enzyme, or differences in the measurement of glycolytic flux might be involved. To illustrate, cardiac-restricted knockout of GLUT4 led to a counterintuitive increase rather than a decrease in glycolysis, due to a compensatory increase in cardiac GLUT1 expression [191].
Although adult cardiomyocytes are considered to be terminally differentiated, it has also been demonstrated that hypertrophy leads to the partial re-activation of the cell cycle and the re-expression of fetal genes [20,195]. The neonatal heart is highly dependent on glucose as substrate and neonatal cardiomyocytes still have the ability to proliferate (reviewed in [196]). During cardiac postnatal development, the loss of the ability to proliferate coincides with the switch from glycolysis to mitochondrial fatty acid oxidation. Along this line of thinking, attempts are currently being made to intervene in the metabolism of induced pluripotent stem cell (iPSC)-derived cardiomyocytes, with the aim of promoting a more differentiated, oxidative, adult-like phenotype of these cells [197].
In hypertrophying cardiomyocytes, the re-entry of the cell cycle leads to polyploidy and binucleation instead of cell division and allows cardiomyocytes to increase in size. It is tempting to speculate that, in analogy to proliferating cancer cells, this incomplete cytokinesis represents the attempt of hypertrophic cardiomyocytes to initiate proliferation without the need to give up their structural integrity and function. Following this line of reasoning, the hypertrophic switch to a more glycolytic metabolism and the re-expression of fetal genes should be considered part of the proliferative growth program.
4. Metabolic Cross-Talk between Cells
It is common knowledge that at the whole-body level there is metabolic interdependency between tissues. Emerging evidence indicates that there is also extensive metabolic cross-talk between the different cell types within an organ. For instance, within skeletal muscle, lactate is exchanged between white and red muscle fibers [198]. Equally intriguing is the observation that lactate produced by tumor cells via the process of aerobic glycolysis induces the polarization of tumor-associated macrophages toward an M2-like phenotype [199]. It has also been proposed that the secretion of lactate by tumor cells has a damaging effect on non-cancerous neighboring cells through the acidification of the microenvironment [200,201].
Research on metabolite signaling has been boosted by the recent discovery of G-protein coupled receptors that act as selective metabolite sensors, for instance for long-chain fatty acids (GPR40, GPR120) [202], succinate (GPR91) [203] and lactate (GPR81) [204]. In fact, lactate binding to GPR81 interferes with Toll-like receptor-mediated activation of the inflammasome (NLPR3), thereby providing a mechanistic explanation for the earlier noted anti-inflammatory effect of lactate [205]. Lactate has also been found to inhibit monocyte migration and the secretion of proinflammatory cytokines [206]. However, it is not always easy to discern if observed effects are related to lactate–GPR81 signaling, to metabolic effects (substrate competition), or to lactate-mediated acidification of the microenvironment, all of which elicit cellular responses. Notably, pH lowering appears to promote exosome secretion and, in this way, cross-talk between cells [207].
Succinate accumulates in the myocardium under certain pathological conditions, particularly low oxygen states, and is subsequently released into the circulation [208,209]. Succinate-mediated activation of GPR91 has been found to induce renovascular hypertension [203] and cardiomyocyte hypertrophy in vitro and in vivo [210]. These observations suggest that the myocardium also makes use of metabolic signals to communicate between (neighboring) cell types. It will be interesting to explore in what manner the activation (or inhibition) of these recently discovered metabolic receptors modulates cardiac phenotype and function.
5. Metabolic Rewiring in the Failing Heart
It is commonly acknowledged that cardiac hypertrophy and failure are associated with alterations in cardiac substrate and energy metabolism [5,7,11]. However, even after decades of research, it is still debated if this type of metabolic remodeling should be considered an adaptive or maladaptive response [211].
The picture even becomes more complicated when the distinct forms of heart failure are taken into consideration, namely heart failure with reduced ejection fraction (HFrEF) and with preserved ejection fraction (HFpEF). HFrEF is characterized by systolic dysfunction and is associated with reduced fatty acid and increased cardiac glucose uptake. As such, the switch in cardiac metabolism in HFrEF fits the traditional picture. Generally speaking, the switch from fatty acids to glucose has usually been considered an energy-efficient, adaptive response, given that the oxidation of glucose yields more ATP per oxygen atom used, than that of fatty acids (reflected in a higher ATP/O ratio for glucose). However, it is well-established that in the hypertrophic and failing heart a substantial part of the glucose is not oxidized, but converted into lactate instead [212], reducing the total ATP yield [34,35]. Whether the lactate is produced via anaerobic or aerobic glycolysis remains to be determined. If aerobic glycolysis (Warburg effect) is involved, this would imply that a mismatch between oxygen supply and demand is not necessarily the driving force for the increase in cardiac lactate production. As discussed in this review, it is well conceivable that the increase in glycolytic flux primarily serves to support cardiac anabolic processes instead of sustaining energy metabolism.
By contrast, HFpEF is characterized by diastolic dysfunction. In Western societies, the prevalence of HFpEF is rapidly increasing and is more frequently observed in individuals with obesity and type 2 diabetes [213]. These individuals usually suffer from insulin resistance, which profoundly affects systemic metabolism. Under these conditions, the heart is forced to rely even more on the use of fatty acids as substrate. If we assume that glycolytic metabolism is required to support hypertrophic growth, then the inability to increase glycolysis should have consequences for the structural remodeling of the heart. In line with this is the observation that in a pig model of cardiometabolic disease, cardiomyocyte size was reduced compared to the controls, despite the presence of marked hypertension [214]. Furthermore, earlier studies by Stanley et al. (reviewed in [215]) showed that a high-fat diet attenuated the increase in cardiac mass in rats subjected to pressure overload. These findings support the idea that the hypertrophic growth of cardiomyocytes depends on an increased glycolytic flux.
This line of thinking will also have consequences for the design of metabolic therapies for the diseased heart. There is an increasing interest in the use of metabolic interventions, as an adjunct to current pharmacological therapies that target the various neuro-hormonal pathways, to treat heart failure [2]. Generally speaking, the goal of metabolic therapies has been to improve cardiomyocyte energy metabolism by stimulating ATP synthesis. Primarily because of the higher ATP/O ratio of glucose, so far nearly all metabolic interventions in clinical studies with HF patients have been aimed at promoting cardiomyocyte glucose utilization through pharmacological inhibition of enzymes involved in mitochondrial fatty acid uptake and beta-oxidation [2]. However, what effect will this have on other cell types? For instance, is it possible that by inhibiting fatty acid oxidation and by stimulating glycolysis one promotes cardiac inflammation by favoring the formation of the more pro-inflammatory M1 macrophages over the anti-inflammatory M2 macrophages? The limited success of metabolic interventions so far may well be related to the fact that these interventions not only increase glycolytic flux and alter cardiomyocyte energy metabolism, but they also modulate the phenotype and function of other cardiac cell types.
Surprisingly, one of the most promising classes of drugs now appear to be the sodium-glucose co-transporter-2 inhibitors (SGLT2is), glucose-lowering anti-diabetic drugs that directly interfere with renal glucose reabsorption. Despite the absence of the sodium-glucose co-transporter-2 in the heart, there is accumulating clinical evidence that SGLT2is have marked beneficial effects in the setting of both HFrEF and HFpEF. It still remains to be determined if the beneficial outcome is related to their effects on systemic metabolism, cardiomyocyte metabolism or that of other cell types in the heart, due to off-target effects, or to a combination of those [216].
6. Concluding Remarks and Future Perspectives
So far, changes in cardiac metabolism in the failing heart have been primarily considered from the perspective of the consequences for the amount of ATP needed to maintain cardiac pump function. As discussed above, there is accumulating evidence that, similar to cancer cells, the different cell types constituting the cardiac muscle make use of metabolic rewiring to adjust their phenotype and function. Consequently, in the context of the diseased heart, metabolic rewiring provides a mechanism to adjust cellular phenotype and function and, as such, is likely to play a role in many, if not all, aspects of cardiac structural remodeling, that is cardiac hypertrophy, fibrosis, angiogenesis and inflammation.
Unfortunately, current research on cardiac metabolism does not allow one to discriminate between different cell types in the heart. When using tracers to monitor metabolism in vivo or ex vivo using isolated heart preparations, the cardiac muscle is studied as one entity and it is assumed that the measurements reflect the metabolism of contracting cardiomyocytes. For a large part, this assumption is correct given that cardiomyocytes constitute the largest volume fraction of the myocardium and are the main energy-consuming units of the cardiac muscle. Nonetheless, it is equally important to assess how the other cell types in cardiac muscle tissue adjust their metabolism during the development of cardiac hypertrophy and failure, and how these cells respond to metabolic therapies. Single-cell RNA-sequencing analyses of the different cell populations in the healthy and diseased heart are already providing useful information about the metabolic setup of the different cell types under these conditions. Additionally, in vitro strategies such as cellular metabolic flux analysis and live cell imaging will assist in unraveling the exact nature of the relationship between metabolic rewiring and cellular phenotype. In the near future, mass spectrometry imaging might provide a useful tool to look at, quite literally, the metabolism of different cell types in the heart in situ, especially since technical developments already allow a resolution in the micrometer range of the acquired metabolic images [217]. Undoubtedly, the implementation of such novel techniques will deepen our understanding of the significance of metabolic rewiring in cardiac disease.
In summary, in the present review we provide an alternative point of view on the significance of metabolic remodeling in the diseased heart, diverging somewhat from the traditional concept that adjustments in cardiac metabolism merely serve to secure ATP production as much as possible and to maintain cardiac pump function at an adequate level. Specific points that we would like to present as “substrate for thought” are:
- Heart failure pathogenesis is associated with marked alterations in cardiac substrate and energy metabolism, the relevance of which is still incompletely understood.
- Similar to cancer cells, metabolic rewiring not only regulates energy balance but also affects the phenotype and function of the principal cell types of the cardiovascular system.
- Cardiac lactate production does not necessarily reflect a shortage in oxygen supply. It can also be due to the Warburg effect.
- Glycolytic enzymes have multiple biological functions, exceeding their role as catalyzers of chemical reactions in the glycolytic pathway.
- Metabolic signaling is a still underdeveloped area of research that is likely to have marked implications for our understanding of cardiac pathogenesis
- The switch toward enhanced glycolysis in cardiac hypertrophy and the polyploidization and binucleation of cardiomyocytes may be related events.
- The various cell types in the heart have different substrate demands and respond differently to disease- or pharmacologically-induced changes in metabolism. This should be taken into account when designing metabolic therapies aimed at improving cardiac function.
To conclude, particularly developments in research areas outside the cardiovascular arena force us to take a look at disease-associated changes in cardiac substrate metabolism from other angles. After decades of research, perhaps this change in paradigm may eventually lead to a breakthrough in elucidating if metabolic remodeling is adaptive or maladaptive and in deciding which metabolic therapies may have long-term favorable effects on the failing heart.
Author Contributions
Conceptualization, M.v.B. and A.-R.K.; writing—original draft preparation, A.-R.K. and M.v.B.; writing—review and editing, M.v.B. and A.-R.K.; visualization, A.-R.K.; supervision, M.v.B.; project administration, M.v.B.; funding acquisition, M.v.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the European Research Area Network Cardiovascular Disease [Hartstichting ERA-CVD LYMIT-DIS 2016T091].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Blanche Schroen for the critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ponikowski, P.; Anker, S.D.; AlHabib, K.F.; Cowie, M.R.; Force, T.L.; Hu, S.; Jaarsma, T.; Krum, H.; Rastogi, V.; Rohde, L.E.; et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail 2014, 1, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Heggermont, W.A.; Papageorgiou, A.P.; Heymans, S.; van Bilsen, M. Metabolic support for the heart: Complementary therapy for heart failure? Eur. J. Heart Fail 2016, 18, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- van Bilsen, M.; Smeets, P.J.; Gilde, A.J.; van der Vusse, G.J. Metabolic remodelling of the failing heart: The cardiac burn-out syndrome? Cardiovasc. Res. 2004, 61, 218–226. [Google Scholar] [CrossRef]
- Conway, M.A.; Allis, J.; Ouwerkerk, R.; Niioka, T.; Rajagopalan, B.; Radda, G.K. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 1991, 338, 973–976. [Google Scholar] [CrossRef]
- Neubauer, S.; Krahe, T.; Schindler, R.; Horn, M.; Hillenbrand, H.; Entzeroth, C.; Mader, H.; Kromer, E.P.; Riegger, G.A.; Lackner, K.; et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation 1992, 86, 1810–1818. [Google Scholar] [CrossRef]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschutz, W.; Lipke, C.; Kostler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Karlstaedt, A.; Schiffer, W.; Taegtmeyer, H. Actionable Metabolic Pathways in Heart Failure and Cancer-Lessons From Cancer Cell Metabolism. Front. Cardiovasc. Med. 2018, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Duran, R.V.; Boulahbel, H.; Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 2009, 30, 1269–1280. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Rosenblatt-Velin, N.; Montessuit, C.; Papageorgiou, I.; Terrand, J.; Lerch, R. Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc. Res. 2001, 52, 407–416. [Google Scholar] [CrossRef]
- Gibbs, C.L.; Loiselle, D.S. Cardiac basal metabolism. Jpn. J. Physiol. 2001, 51, 399–426. [Google Scholar] [CrossRef]
- Zebrowski, D.C.; Engel, F.B. The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 67–96. [Google Scholar] [CrossRef]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef]
- Sukonina, V.; Ma, H.; Zhang, W.; Bartesaghi, S.; Subhash, S.; Heglind, M.; Foyn, H.; Betz, M.J.; Nilsson, D.; Lidell, M.E.; et al. FOXK1 and FOXK2 regulate aerobic glycolysis. Nature 2019, 566, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar]
- Morris, M.R.; Maina, E.; Morgan, N.V.; Gentle, D.; Astuti, D.; Moch, H.; Kishida, T.; Yao, M.; Schraml, P.; Richards, F.M.; et al. Molecular genetic analysis of FIH-1, FH, and SDHB candidate tumour suppressor genes in renal cell carcinoma. J. Clin. Pathol. 2004, 57, 706–711. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Pan, Y.; Mansfield, K.D.; Bertozzi, C.C.; Rudenko, V.; Chan, D.A.; Giaccia, A.J.; Simon, M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 2007, 27, 912–925. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Holness, M.J.; Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef]
- Sena, J.A.; Wang, L.; Heasley, L.E.; Hu, C.J. Hypoxia regulates alternative splicing of HIF and non-HIF target genes. Mol. Cancer Res. 2014, 12, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Babapoor-Farrokhran, S.; Rodrigues, M.; Deshpande, M.; Puchner, B.; Kashiwabuchi, F.; Hassan, S.J.; Asnaghi, L.; Handa, J.T.; Merbs, S.; et al. Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget 2016, 7, 7816–7828. [Google Scholar] [CrossRef]
- Robciuc, M.R.; Skrobuk, P.; Anisimov, A.; Olkkonen, V.M.; Alitalo, K.; Eckel, R.H.; Koistinen, H.A.; Jauhiainen, M.; Ehnholm, C. Angiopoietin-like 4 mediates PPAR delta effect on lipoprotein lipase-dependent fatty acid uptake but not on beta-oxidation in myotubes. PLoS ONE 2012, 7, e46212. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Lionetti, V.; Young, M.E.; Chandler, M.P.; d’Agostino, C.; Kang, E.; Altarejos, M.; Matsuo, K.; Hintze, T.H.; Stanley, W.C.; et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J. Mol. Cell Cardiol. 2004, 36, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Rempel, A.; Bannasch, P.; Mayer, D. Differences in expression and intracellular distribution of hexokinase isoenzymes in rat liver cells of different transformation stages. Biochim. Biophys. Acta 1994, 1219, 660–668. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Riddle, S.R.; Ahmad, A.; Ahmad, S.; Deeb, S.S.; Malkki, M.; Schneider, B.K.; Allen, C.B.; White, C.W. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L407–L416. [Google Scholar] [CrossRef]
- Chiara, F.; Castellaro, D.; Marin, O.; Petronilli, V.; Brusilow, W.S.; Juhaszova, M.; Sollott, S.J.; Forte, M.; Bernardi, P.; Rasola, A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE 2008, 3, e1852. [Google Scholar] [CrossRef]
- Pantic, B.; Trevisan, E.; Citta, A.; Rigobello, M.P.; Marin, O.; Bernardi, P.; Salvatori, S.; Rasola, A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis. 2013, 4, e858. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Liu, X.; Yan, N.; Li, S.; Cao, G.; Cheng, Q.; Xia, Q.; Wang, H. Hypoxia-inducible transcription factor-1alpha promotes hypoxia-induced A549 apoptosis via a mechanism that involves the glycolysis pathway. BMC Cancer 2006, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, T.; Funasaka, T.; Tsutsumi, S.; Hu, H.; Watanabe, H.; Raz, A. Regulation of phosphoglucose isomerase/autocrine motility factor activities by the poly(ADP-ribose) polymerase family-14. Cancer Res. 2007, 67, 8682–8689. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Li, L.; Albrecht, T.; Johnson, J.D.; Kojic, L.D.; Nabi, I.R. Autocrine motility factor/phosphoglucose isomerase regulates ER stress and cell death through control of ER calcium release. Cell Death Differ. 2011, 18, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Guimaraes-Camboa, N.; Stowe, J.; Aneas, I.; Sakabe, N.; Cattaneo, P.; Henderson, L.; Kilberg, M.S.; Johnson, R.S.; Chen, J.; McCulloch, A.D.; et al. HIF1alpha Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev. Cell 2015, 33, 507–521. [Google Scholar] [CrossRef]
- Peng, M.; Yang, D.; Hou, Y.; Liu, S.; Zhao, M.; Qin, Y.; Chen, R.; Teng, Y.; Liu, M. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death Dis. 2019, 10, 228. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Jean, J.C.; Rich, C.B.; Joyce-Brady, M. Hypoxia results in an HIF-1-dependent induction of brain-specific aldolase C in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L950–L956. [Google Scholar] [CrossRef] [PubMed]
- Gess, B.; Hofbauer, K.H.; Deutzmann, R.; Kurtz, A. Hypoxia up-regulates triosephosphate isomerase expression via a HIF-dependent pathway. Pflugers Arch. 2004, 448, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Garcin, E.D. GAPDH as a model non-canonical AU-rich RNA binding protein. Semin. Cell Dev. Biol. 2019, 86, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007, 26, 2606–2620. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Lu, Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy 2017, 13, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.; Roux, D.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006, 66, 3688–3698. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, N.; Sun, W.; Li, X.; Liu, B.; Xie, Z.; Qu, J.; Xu, J.; Yang, X.; Su, Y.; et al. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene 2017, 36, 2900–2909. [Google Scholar] [CrossRef]
- Principe, M.; Ceruti, P.; Shih, N.Y.; Chattaragada, M.S.; Rolla, S.; Conti, L.; Bestagno, M.; Zentilin, L.; Yang, S.H.; Migliorini, P.; et al. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget 2015, 6, 11098–11113. [Google Scholar] [CrossRef]
- Gao, S.; Li, H.; Feng, X.J.; Li, M.; Liu, Z.P.; Cai, Y.; Lu, J.; Huang, X.Y.; Wang, J.J.; Li, Q.; et al. alpha-Enolase plays a catalytically independent role in doxorubicin-induced cardiomyocyte apoptosis and mitochondrial dysfunction. J. Mol. Cell Cardiol. 2015, 79, 92–103. [Google Scholar] [CrossRef]
- Entelis, N.; Brandina, I.; Kamenski, P.; Krasheninnikov, I.A.; Martin, R.P.; Tarassov, I. A glycolytic enzyme, enolase, is recruited as a cofactor of tRNA targeting toward mitochondria in Saccharomyces cerevisiae. Genes Dev. 2006, 20, 1609–1620. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, H.; Lu, W.; Huang, P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. Biophys. Acta 2009, 1787, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal 2009, 2, ra73. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Rouzeau, J.D.; Farhadian, F.; Wisnewsky, C.; Marotte, F.; Lamande, N.; Samuel, J.L.; Schwartz, K.; Lazar, M.; Lucas, M. Differential expression of alpha- and beta-enolase genes during rat heart development and hypertrophy. Am. J. Physiol. 1995, 269, H1843–H1851. [Google Scholar] [CrossRef]
- Zhu, L.A.; Fang, N.Y.; Gao, P.J.; Jin, X.; Wang, H.Y. Differential expression of alpha-enolase in the normal and pathological cardiac growth. Exp. Mol. Pathol. 2009, 87, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.L.; Subramaniam, J.; Li, Y.; Hamilton, D.J.; Frazier, O.H.; Taegtmeyer, H. A PKM2 signature in the failing heart. Biochem. Biophys. Res. Commun. 2015, 459, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Kenney, J.W.; Manousopoulou, A.; Johnston, H.E.; Kamei, M.; Woelk, C.H.; Xie, J.; Schwarzer, M.; Garbis, S.D.; Proud, C.G. Quantitative Non-canonical Amino Acid Tagging (QuaNCAT) Proteomics Identifies Distinct Patterns of Protein Synthesis Rapidly Induced by Hypertrophic Agents in Cardiomyocytes, Revealing New Aspects of Metabolic Remodeling. Mol. Cell Proteom. 2016, 15, 3170–3189. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II Positively Regulates Glucose Starvation-Induced Autophagy through TORC1 Inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef]
- Wu, R.; Smeele, K.M.; Wyatt, E.; Ichikawa, Y.; Eerbeek, O.; Sun, L.; Chawla, K.; Hollmann, M.W.; Nagpal, V.; Heikkinen, S.; et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ. Res. 2011, 108, 60–69. [Google Scholar] [CrossRef]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Hecker, P.A.; Lionetti, V.; Ribeiro, R.F., Jr.; Rastogi, S.; Brown, B.H.; O’Connell, K.A.; Cox, J.W.; Shekar, K.C.; Gamble, D.M.; Sabbah, H.N.; et al. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ. Heart Fail. 2013, 6, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 2010, 3, 420–430. [Google Scholar] [CrossRef] [PubMed]
- McCommis, K.S.; Douglas, D.L.; Krenz, M.; Baines, C.P. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J. Am. Heart Assoc. 2013, 2, e000355. [Google Scholar] [CrossRef]
- Gupte, S.A.; Levine, R.J.; Gupte, R.S.; Young, M.E.; Lionetti, V.; Labinskyy, V.; Floyd, B.C.; Ojaimi, C.; Bellomo, M.; Wolin, M.S.; et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell Cardiol. 2006, 41, 340–349. [Google Scholar] [CrossRef]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; de Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; de Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef]
- Pham, L.V.; Bryant, J.L.; Mendez, R.; Chen, J.; Tamayo, A.T.; Xu-Monette, Z.Y.; Young, K.H.; Manyam, G.C.; Yang, D.; Medeiros, L.J.; et al. Targeting the hexosamine biosynthetic pathway and O-linked N-acetylglucosamine cycling for therapeutic and imaging capabilities in diffuse large B-cell lymphoma. Oncotarget 2016, 7, 80599–80611. [Google Scholar] [CrossRef]
- Taparra, K.; Wang, H.; Malek, R.; Lafargue, A.; Barbhuiya, M.A.; Wang, X.; Simons, B.W.; Ballew, M.; Nugent, K.; Groves, J.; et al. O-GlcNAcylation is required for mutant KRAS-induced lung tumorigenesis. J. Clin. Investig. 2018, 128, 4924–4937. [Google Scholar] [CrossRef]
- Durand, P.; Golinelli-Pimpaneau, B.; Mouilleron, S.; Badet, B.; Badet-Denisot, M.-A. Highlights of glucosamine-6P synthase catalysis. Arch. Biochem. Biophys. 2008, 474, 302–317. [Google Scholar] [CrossRef]
- Chaiyawat, P.; Netsirisawan, P.; Svasti, J.; Champattanachai, V. Aberrant O-GIcNAcylated proteins: New perspectives in breast and colorectal cancer. Front. Endocrinol. 2014, 5, 193. [Google Scholar] [CrossRef]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., 3rd; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.Y.; Dang, C.V.; Hart, G.W. Glycosylation of the c-Myc transactivation domain. Proc. Natl. Acad. Sci. USA 1995, 92, 4417–4421. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Correa, G.A.; Jin, W.; Wang, Z.; Zhong, X.; Gao, W.D.; Dias, W.B.; Vecoli, C.; Hart, G.W.; Murphy, A.M. O-linked GlcNAc modification of cardiac myofilament proteins: A novel regulator of myocardial contractile function. Circ. Res. 2008, 103, 1354–1358. [Google Scholar] [CrossRef]
- Gelinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef]
- Dassanayaka, S.; Brainard, R.E.; Watson, L.J.; Long, B.W.; Brittian, K.R.; DeMartino, A.M.; Aird, A.L.; Gumpert, A.M.; Audam, T.N.; Kilfoil, P.J.; et al. Cardiomyocyte Ogt limits ventricular dysfunction in mice following pressure overload without affecting hypertrophy. Basic Res. Cardiol. 2017, 112, 23. [Google Scholar] [CrossRef]
- Lunde, I.G.; Aronsen, J.M.; Kvaloy, H.; Qvigstad, E.; Sjaastad, I.; Tonnessen, T.; Christensen, G.; Gronning-Wang, L.M.; Carlson, C.R. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genom. 2012, 44, 162–172. [Google Scholar] [CrossRef]
- Watson, L.J.; Facundo, H.T.; Ngoh, G.A.; Ameen, M.; Brainard, R.E.; Lemma, K.M.; Long, B.W.; Prabhu, S.D.; Xuan, Y.T.; Jones, S.P. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 17797–17802. [Google Scholar] [CrossRef]
- Jones, S.P.; Zachara, N.E.; Ngoh, G.A.; Hill, B.G.; Teshima, Y.; Bhatnagar, A.; Hart, G.W.; Marban, E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 2008, 117, 1172–1182. [Google Scholar] [CrossRef]
- Lehmann, L.H.; Jebessa, Z.H.; Kreusser, M.M.; Horsch, A.; He, T.; Kronlage, M.; Dewenter, M.; Sramek, V.; Oehl, U.; Krebs-Haupenthal, J.; et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 2018, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.H.; May, H.I.; Li, Q.; Luo, X.; Huang, J.; Zhang, G.; Niewold, E.; Wang, X.; Gillette, T.G.; Deng, Y.; et al. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat. Commun. 2020, 11, 1771. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619. [Google Scholar] [CrossRef]
- Lauzier, B.; Vaillant, F.; Merlen, C.; Gelinas, R.; Bouchard, B.; Rivard, M.E.; Labarthe, F.; Dolinsky, V.W.; Dyck, J.R.; Allen, B.G.; et al. Metabolic effects of glutamine on the heart: Anaplerosis versus the hexosamine biosynthetic pathway. J. Mol. Cell Cardiol. 2013, 55, 92–100. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.H.; Parikh, K.; Ryan, J.J.; Toth, P.T.; Archer, S.L. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J. Mol. Med. 2013, 91, 1185–1197. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef]
- Langston, P.K.; Shibata, M.; Horng, T. Metabolism Supports Macrophage Activation. Front. Immunol. 2017, 8, 61. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Nahrendorf, M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc. Res. 2014, 102, 240–248. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Rodriguez-Prados, J.C.; Traves, P.G.; Cuenca, J.; Rico, D.; Aragones, J.; Martin-Sanz, P.; Cascante, M.; Bosca, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Fukuzumi, M.; Shinomiya, H.; Shimizu, Y.; Ohishi, K.; Utsumi, S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect. Immun. 1996, 64, 108–112. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; Prange, K.H.M.; van Weeghel, M.; van der Velden, S.; Ryan, D.G.; Wust, R.C.I.; Neele, A.E.; Speijer, D.; Denis, S.W.; et al. A Defective Pentose Phosphate Pathway Reduces Inflammatory Macrophage Responses during Hypercholesterolemia. Cell Rep. 2018, 25, 2044–2052.e5. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. Alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Lemmens, K.; Vrints, C.J.; Van Craenenbroeck, E.M. Targeting Endothelial Function to Treat Heart Failure with Preserved Ejection Fraction: The Promise of Exercise Training. Oxid. Med. Cell Longev. 2017, 2017, 4865756. [Google Scholar] [CrossRef]
- Brutsaert, D.L. Cardiac endothelial-myocardial signaling: Its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003, 83, 59–115. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef]
- Vizan, P.; Sanchez-Tena, S.; Alcarraz-Vizan, G.; Soler, M.; Messeguer, R.; Pujol, M.D.; Lee, W.N.; Cascante, M. Characterization of the metabolic changes underlying growth factor angiogenic activation: Identification of new potential therapeutic targets. Carcinogenesis 2009, 30, 946–952. [Google Scholar] [CrossRef]
- Federici, M.; Menghini, R.; Mauriello, A.; Hribal, M.L.; Ferrelli, F.; Lauro, D.; Sbraccia, P.; Spagnoli, L.G.; Sesti, G.; Lauro, R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation 2002, 106, 466–472. [Google Scholar] [CrossRef]
- Luo, B.; Soesanto, Y.; McClain, D.A. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arter. Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Fang, L.H.; Choi, S.H.; Baek, J.S.; Liu, C.; Almazan, F.; Ulrich, F.; Wiesner, P.; Taleb, A.; Deer, E.; Pattison, J.; et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013, 498, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.S.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.L.; Lin, C.J.; Fu, W.M. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol. Pharmacol. 2008, 73, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef]
- Stone, O.A.; El-Brolosy, M.; Wilhelm, K.; Liu, X.; Romao, A.M.; Grillo, E.; Lai, J.K.H.; Gunther, S.; Jeratsch, S.; Kuenne, C.; et al. Loss of pyruvate kinase M2 limits growth and triggers innate immune signaling in endothelial cells. Nat. Commun. 2018, 9, 4077. [Google Scholar] [CrossRef]
- Lee, L.Y.; Oldham, W.M.; He, H.; Wang, R.; Mulhern, R.; Handy, D.E.; Loscalzo, J. Interferon-gamma Impairs Human Coronary Artery Endothelial Glucose Metabolism by Tryptophan Catabolism and Activates Fatty Acid Oxidation. Circulation 2021, 144, 1612–1628. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.J. Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscle. Am. J. Physiol. 1983, 244, C399–C409. [Google Scholar] [CrossRef]
- Yang, L.; Gao, L.; Nickel, T.; Yang, J.; Zhou, J.; Gilbertsen, A.; Geng, Z.; Johnson, C.; Young, B.; Henke, C.; et al. Lactate Promotes Synthetic Phenotype in Vascular Smooth Muscle Cells. Circ. Res. 2017, 121, 1251–1262. [Google Scholar] [CrossRef]
- Barron, J.T.; Kopp, S.J.; Tow, J.; Parrillo, J.E. Fatty acid, tricarboxylic acid cycle metabolites, and energy metabolism in vascular smooth muscle. Am. J. Physiol. 1994, 267, H764–H769. [Google Scholar] [CrossRef]
- Nef, H.M.; Mollmann, H.; Joseph, A.; Troidl, C.; Voss, S.; Vogt, A.; Weber, M.; Hamm, C.W.; Elsasser, A. Effects of 2-deoxy-D-glucose on proliferation of vascular smooth muscle cells and endothelial cells. J. Int. Med. Res. 2008, 36, 986–991. [Google Scholar] [CrossRef]
- Hall, J.L.; Matter, C.M.; Wang, X.; Gibbons, G.H. Hyperglycemia inhibits vascular smooth muscle cell apoptosis through a protein kinase C-dependent pathway. Circ. Res. 2000, 87, 574–580. [Google Scholar] [CrossRef]
- Hall, J.L.; Chatham, J.C.; Eldar-Finkelman, H.; Gibbons, G.H. Upregulation of glucose metabolism during intimal lesion formation is coupled to the inhibition of vascular smooth muscle cell apoptosis. Role of GSK3beta. Diabetes 2001, 50, 1171–1179. [Google Scholar] [CrossRef]
- Perez, J.; Hill, B.G.; Benavides, G.A.; Dranka, B.P.; Darley-Usmar, V.M. Role of cellular bioenergetics in smooth muscle cell proliferation induced by platelet-derived growth factor. Biochem. J. 2010, 428, 255–267. [Google Scholar] [CrossRef]
- Heiss, E.H.; Schachner, D.; Donati, M.; Grojer, C.S.; Dirsch, V.M. Increased aerobic glycolysis is important for the motility of activated VSMC and inhibited by indirubin-3′-monoxime. Vascul. Pharmacol. 2016, 83, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zhou, Q.; Chen, J.; Rexius-Hall, M.L.; Rehman, J.; Zhou, G. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat. Commun. 2018, 9, 3850. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.V.; Fagard, R.H.; Lijnen, P.J. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension 2002, 39, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Miteva, K.; Tschope, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef]
- Ding, H.; Jiang, L.; Xu, J.; Bai, F.; Zhou, Y.; Yuan, Q.; Luo, J.; Zen, K.; Yang, J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Renal. Physiol. 2017, 313, F561–F575. [Google Scholar] [CrossRef]
- Shamhart, P.E.; Luther, D.J.; Adapala, R.K.; Bryant, J.E.; Petersen, K.A.; Meszaros, J.G.; Thodeti, C.K. Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can. J. Physiol. Pharmacol. 2014, 92, 598–604. [Google Scholar] [CrossRef]
- Fowlkes, V.; Clark, J.; Fix, C.; Law, B.A.; Morales, M.O.; Qiao, X.; Ako-Asare, K.; Goldsmith, J.G.; Carver, W.; Murray, D.B.; et al. Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci. 2013, 92, 669–676. [Google Scholar] [CrossRef]
- Li, M.; Riddle, S.; Zhang, H.; D’Alessandro, A.; Flockton, A.; Serkova, N.J.; Hansen, K.C.; Moldvan, R.; McKeon, B.A.; Frid, M.; et al. Metabolic Reprogramming Regulates the Proliferative and Inflammatory Phenotype of Adventitial Fibroblasts in Pulmonary Hypertension Through the Transcriptional Corepressor C-Terminal Binding Protein-1. Circulation 2016, 134, 1105–1121. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef]
- Bernard, K.; Logsdon, N.J.; Ravi, S.; Xie, N.; Persons, B.P.; Rangarajan, S.; Zmijewski, J.W.; Mitra, K.; Liu, G.; Darley-Usmar, V.M.; et al. Metabolic Reprogramming Is Required for Myofibroblast Contractility and Differentiation. J. Biol. Chem. 2015, 290, 25427–25438. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.; Choi, H.; Hsieh, M.H.; Neugent, M.L.; Ahn, J.M.; Hayenga, H.N.; Singh, P.K.; Shackelford, D.B.; Lee, I.K.; Shulaev, V.; et al. Targeting Hypoxia-Inducible Factor-1alpha/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 216–231. [Google Scholar] [CrossRef]
- Guido, C.; Whitaker-Menezes, D.; Lin, Z.; Pestell, R.G.; Howell, A.; Zimmers, T.A.; Casimiro, M.C.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.E.; et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012, 3, 798–810. [Google Scholar] [CrossRef]
- Han, D.C.; Isono, M.; Hoffman, B.B.; Ziyadeh, F.N. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: Mediation by autocrine activation of TGF-beta. J. Am. Soc. Nephrol. 1999, 10, 1891–1899. [Google Scholar] [CrossRef]
- Aguilar, H.; Fricovsky, E.; Ihm, S.; Schimke, M.; Maya-Ramos, L.; Aroonsakool, N.; Ceballos, G.; Dillmann, W.; Villarreal, F.; Ramirez-Sanchez, I. Role for high-glucose-induced protein O-GlcNAcylation in stimulating cardiac fibroblast collagen synthesis. Am. J. Physiol. Cell Physiol. 2014, 306, C794–C804. [Google Scholar] [CrossRef]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 2012, 143, 1319–1329.e11. [Google Scholar] [CrossRef]
- Zhao, X.; Psarianos, P.; Ghoraie, L.S.; Yip, K.; Goldstein, D.; Gilbert, R.; Witterick, I.; Pang, H.; Hussain, A.; Lee, J.H.; et al. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat. Metab. 2019, 1, 147–157. [Google Scholar] [CrossRef]
- Donthi, R.V.; Ye, G.; Wu, C.; McClain, D.A.; Lange, A.J.; Epstein, P.N. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J. Biol. Chem. 2004, 279, 48085–48090. [Google Scholar] [CrossRef]
- Gibb, A.A.; Lorkiewicz, P.K.; Zheng, Y.T.; Zhang, X.; Bhatnagar, A.; Jones, S.P.; Hill, B.G. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem. J. 2017, 474, 2785–2801. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Byrne, N.J.; Kim, T.T.; Levasseur, J.; Masson, G.; Boisvenue, J.J.; Febbraio, M.; Dyck, J.R.B. Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2017, 312, H552–H560. [Google Scholar] [CrossRef] [PubMed]
- Schisler, J.C.; Grevengoed, T.J.; Pascual, F.; Cooper, D.E.; Ellis, J.M.; Paul, D.S.; Willis, M.S.; Patterson, C.; Jia, W.; Coleman, R.A. Cardiac energy dependence on glucose increases metabolites related to glutathione and activates metabolic genes controlled by mechanistic target of rapamycin. J. Am. Heart Assoc. 2015, 4, e001136. [Google Scholar] [CrossRef]
- Pascual, F.; Schisler, J.C.; Grevengoed, T.J.; Willis, M.S.; Coleman, R.A. Modeling the Transition From Decompensated to Pathological Hypertrophy. J. Am. Heart Assoc. 2018, 7, e008293. [Google Scholar] [CrossRef]
- Haynie, K.R.; Vandanmagsar, B.; Wicks, S.E.; Zhang, J.; Mynatt, R.L. Inhibition of carnitine palymitoyltransferase1b induces cardiac hypertrophy and mortality in mice. Diabetes Obes. Metab. 2014, 16, 757–760. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Hasek, L.Y.; Harris, K.L.; Berman, A.G.; Damen, F.W.; Goergen, C.J.; Ellis, J.M. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem. 2017, 292, 18443–18456. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; de Mattos, A.B.; Shao, D.; Li, T.; Nabben, M.; Kim, M.; Wang, W.; Tian, R.; Kolwicz, S.C., Jr. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J. Mol. Cell Cardiol. 2016, 100, 64–71. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Olson, D.P.; Marney, L.C.; Garcia-Menendez, L.; Synovec, R.E.; Tian, R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 2012, 111, 728–738. [Google Scholar] [CrossRef]
- Pereira, R.O.; Wende, A.R.; Olsen, C.; Soto, J.; Rawlings, T.; Zhu, Y.; Riehle, C.; Abel, E.D. GLUT1 deficiency in cardiomyocytes does not accelerate the transition from compensated hypertrophy to heart failure. J. Mol. Cell Cardiol. 2014, 72, 95–103. [Google Scholar] [CrossRef]
- Pereira, R.O.; Wende, A.R.; Olsen, C.; Soto, J.; Rawlings, T.; Zhu, Y.; Anderson, S.M.; Abel, E.D. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J. Am. Heart Assoc. 2013, 2, e000301. [Google Scholar] [CrossRef]
- Abel, E.D.; Kaulbach, H.C.; Tian, R.; Hopkins, J.C.; Duffy, J.; Doetschman, T.; Minnemann, T.; Boers, M.E.; Hadro, E.; Oberste-Berghaus, C.; et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Investig. 1999, 104, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Caggiano, M.; Kamynina, A.; Francois, A.A.; Prysyazhna, O.; Eykyn, T.R.; Krasemann, S.; Crespo-Leiro, M.G.; Vieites, M.G.; Bianchi, K.; Morales, V.; et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2020, 2, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Taufalele, P.V.; Cochran, J.D.; Robillard-Frayne, I.; Marx, J.M.; Soto, J.; Rauckhorst, A.J.; Tayyari, F.; Pewa, A.D.; Gray, L.R.; et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat. Metab. 2020, 2, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Jeoung, N.H.; Burgess, S.C.; Rosaaen-Stowe, K.A.; Inagaki, T.; Latif, S.; Shelton, J.M.; McAnally, J.; Bassel-Duby, R.; Harris, R.A.; et al. Overexpression of pyruvate dehydrogenase kinase 4 in heart perturbs metabolism and exacerbates calcineurin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H936–H943. [Google Scholar] [CrossRef]
- Li, J.M.; Poolman, R.A.; Brooks, G. Role of G1 phase cyclins and cyclin-dependent kinases during cardiomyocyte hypertrophic growth in rats. Am. J. Physiol. 1998, 275, H814–H822. [Google Scholar] [CrossRef]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1α and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kashihara, D.; Ichimura, A.; Kimura, I.; Tsujimoto, G.; Hirasawa, A. Role of free fatty acid receptors in the regulation of energy metabolism. Biochim. Biophys. Acta 2014, 1841, 1292–1300. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens. 2007, 20, 1209–1215. [Google Scholar] [CrossRef]
- Aguiar, C.J.; Rocha-Franco, J.A.; Sousa, P.A.; Santos, A.K.; Ladeira, M.; Rocha-Resende, C.; Ladeira, L.O.; Resende, R.R.; Botoni, F.A.; Barrouin Melo, M.; et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun. Signal 2014, 12, 78. [Google Scholar] [CrossRef]
- van Bilsen, M.; van Nieuwenhoven, F.A.; van der Vusse, G.J. Metabolic remodelling of the failing heart: Beneficial or detrimental? Cardiovasc. Res. 2009, 81, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Diakos, N.A.; Navankasattusas, S.; Abel, E.D.; Rutter, J.; McCreath, L.; Ferrin, P.; McKellar, S.H.; Miller, D.V.; Park, S.Y.; Richardson, R.S.; et al. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart: Implications for Cardiac Reloading and Conditioning. JACC Basic Transl. Sci. 2016, 1, 432–444. [Google Scholar] [CrossRef] [PubMed]
- McHugh, K.; DeVore, A.D.; Wu, J.; Matsouaka, R.A.; Fonarow, G.C.; Heidenreich, P.A.; Yancy, C.W.; Green, J.B.; Altman, N.; Hernandez, A.F. Heart Failure With Preserved Ejection Fraction and Diabetes: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Sorop, O.; Heinonen, I.; van Kranenburg, M.; van de Wouw, J.; de Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; van Duin, R.W.B.; Stam, K.; van Geuns, R.J.; et al. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Dabkowski, E.R.; Ribeiro, R.F., Jr.; O’Connell, K.A. Dietary fat and heart failure: Moving from lipotoxicity to lipoprotection. Circ. Res. 2012, 110, 764–776. [Google Scholar] [CrossRef]
- Dyck, J.R.B.; Sossalla, S.; Hamdani, N.; Coronel, R.; Weber, N.C.; Light, P.E.; Zuurbier, C.J. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: Evidence for potential off-target effects. J. Mol. Cell Cardiol. 2022, 167, 17–31. [Google Scholar] [CrossRef]
- Niehaus, M.; Soltwisch, J.; Belov, M.E.; Dreisewerd, K. Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at subcellular resolution. Nat. Methods 2019, 16, 925–931. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).