
In-Silico Drug Toxicity and Interaction Prediction for Plant Complexes Based on Virtual Screening and Text Mining





Abstract
:1. Introduction
2. Results
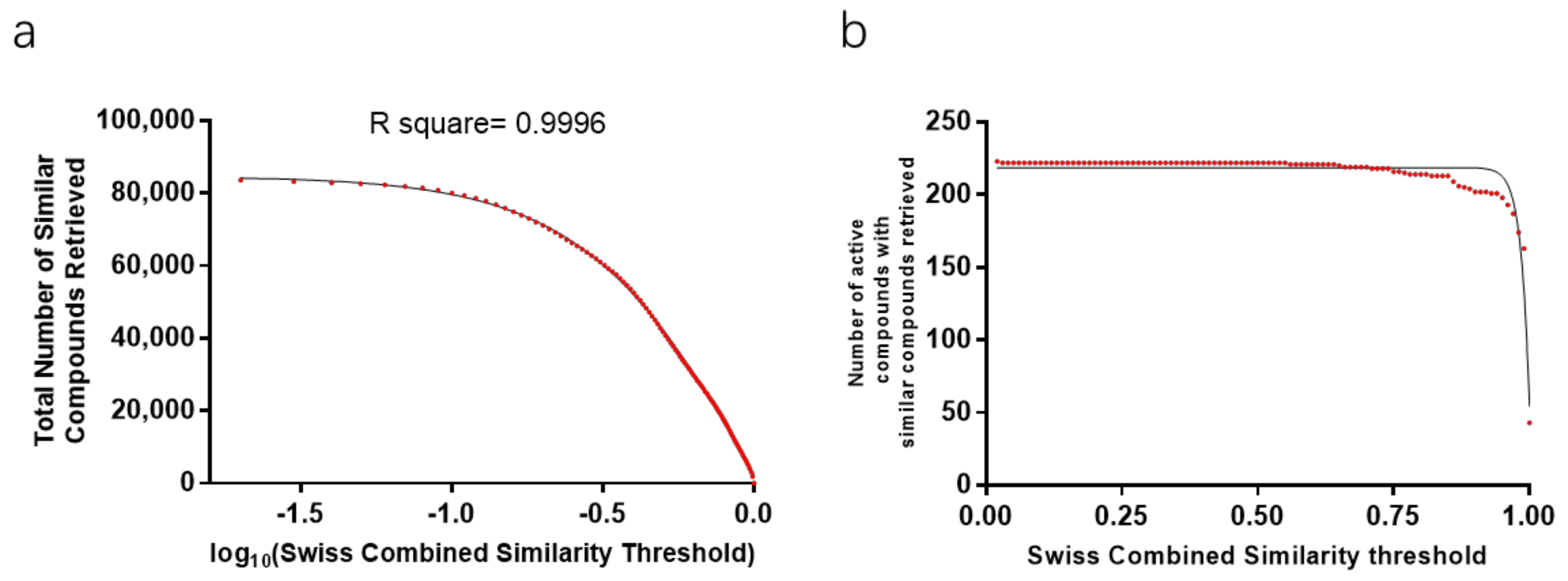
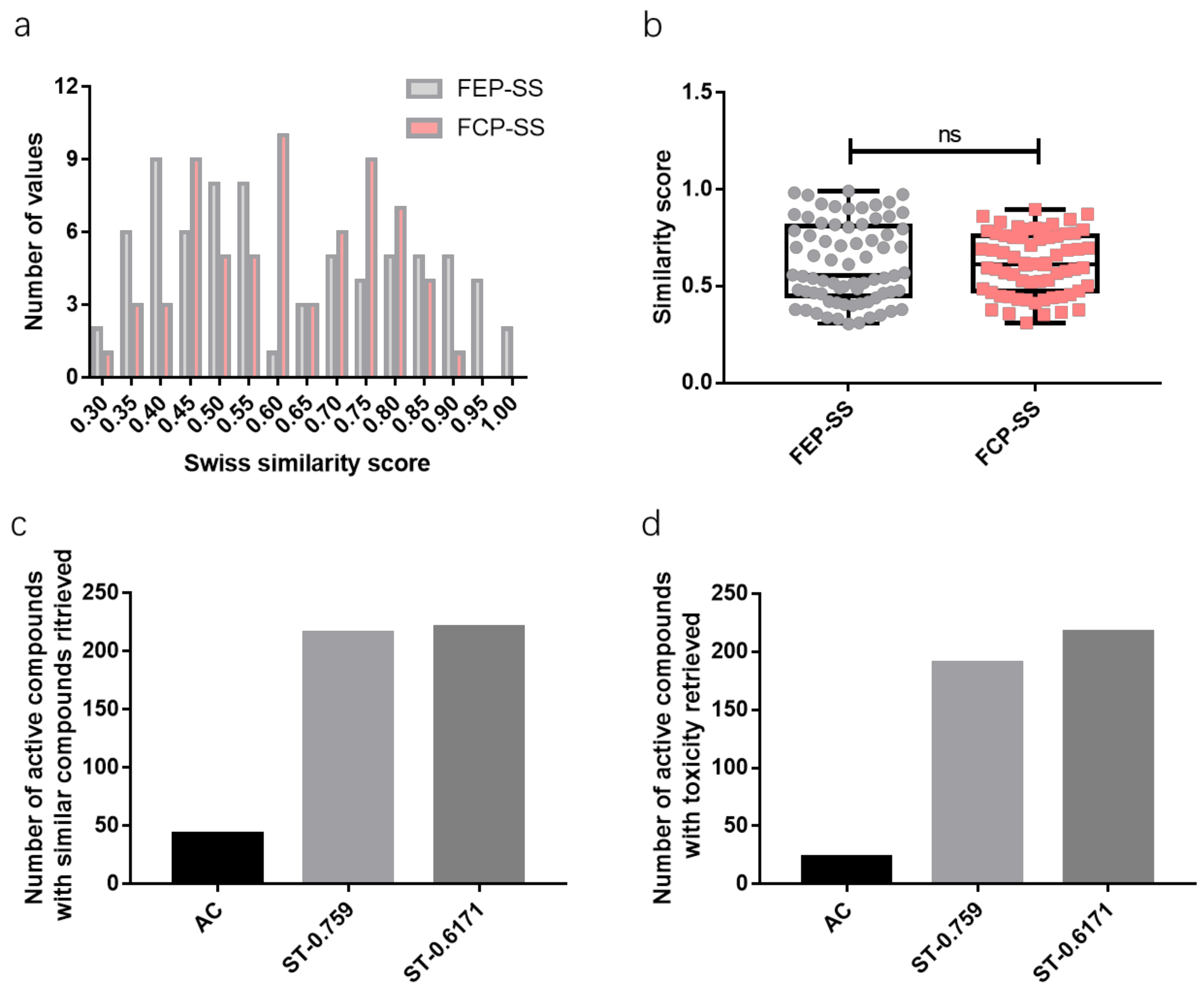
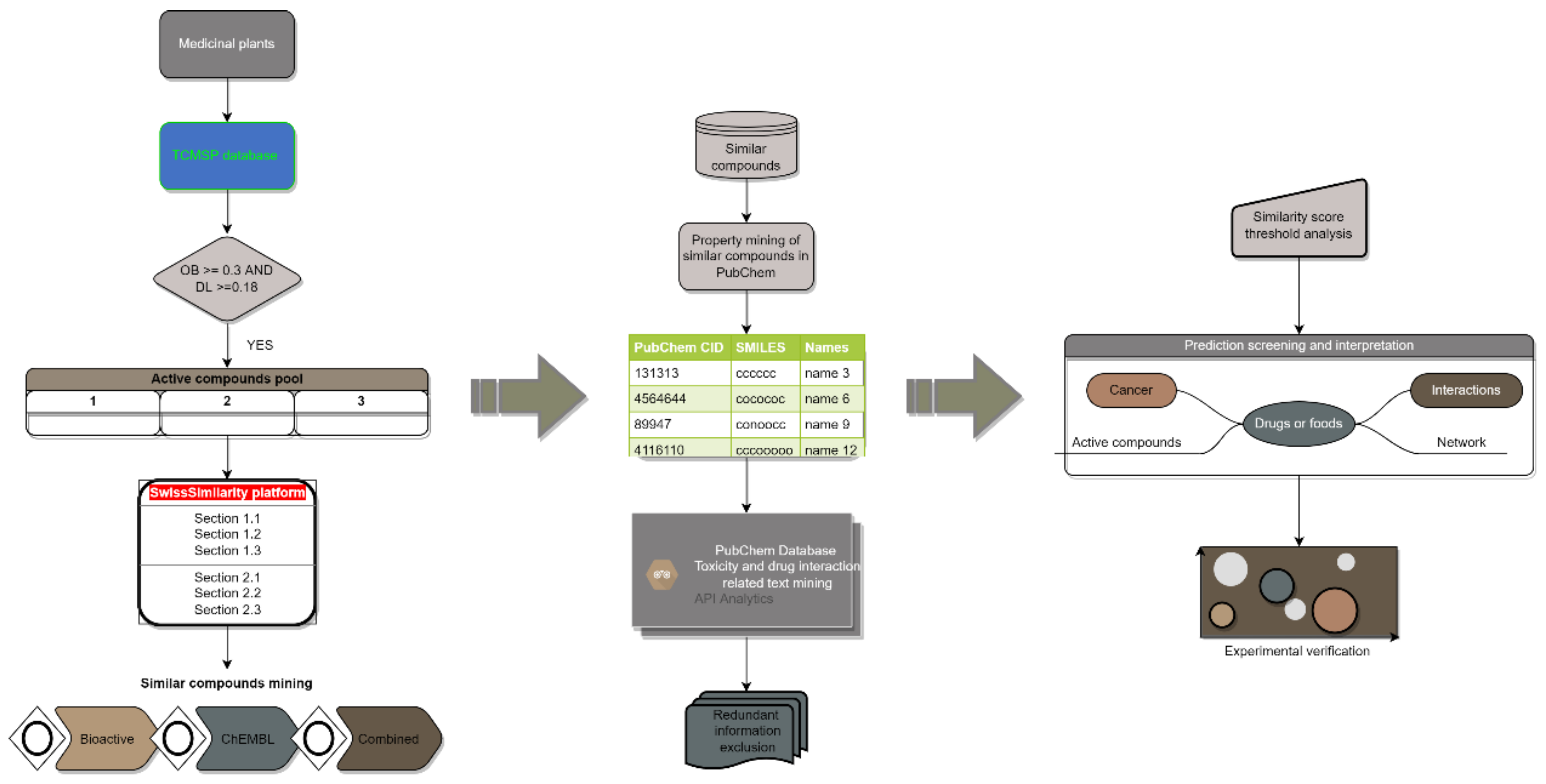
2.1. Similarity Score Threshold Analysis
2.2. Toxicity Prediction Interpretation
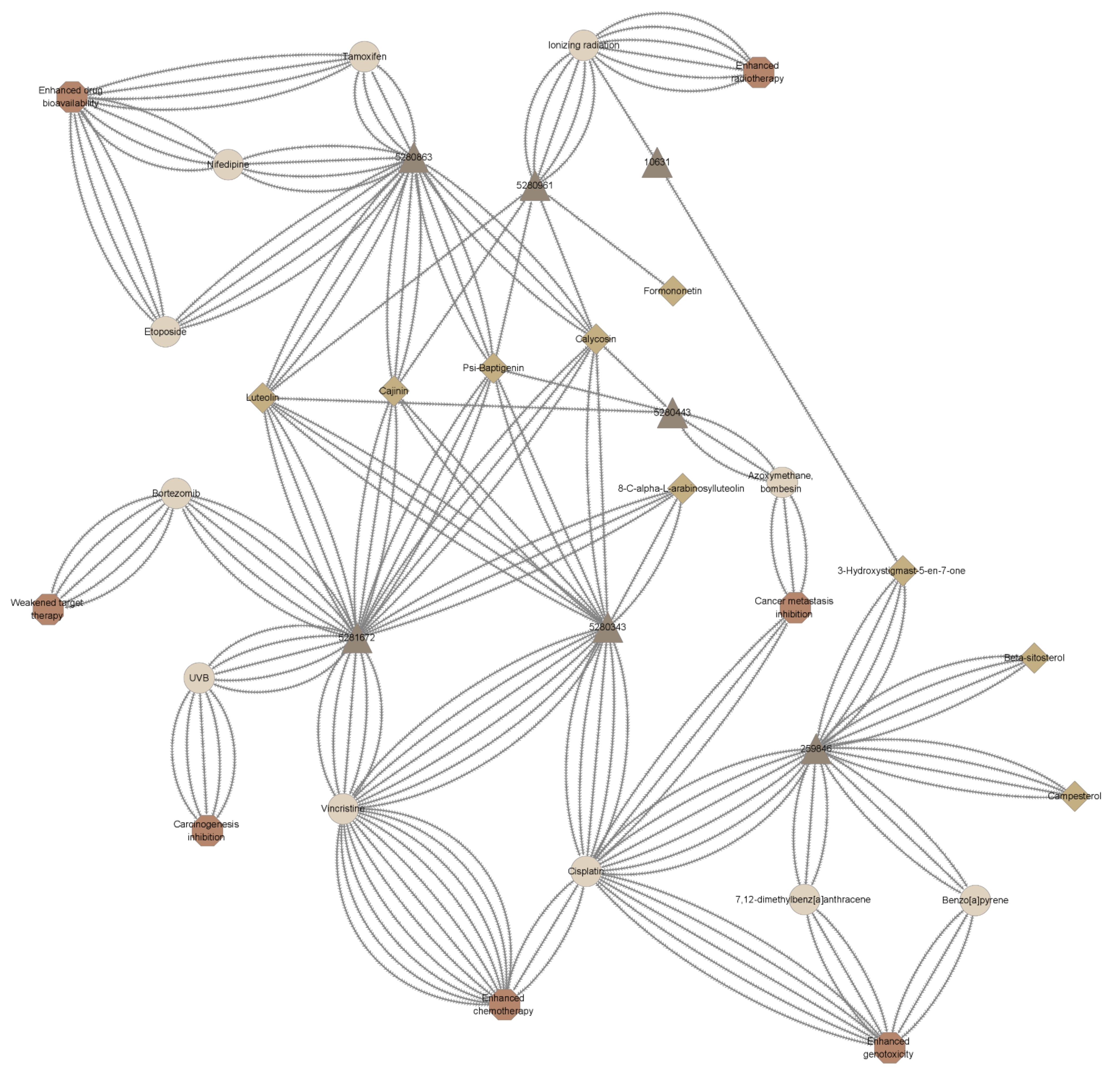
2.3. Drug Interaction Prediction Interpretation
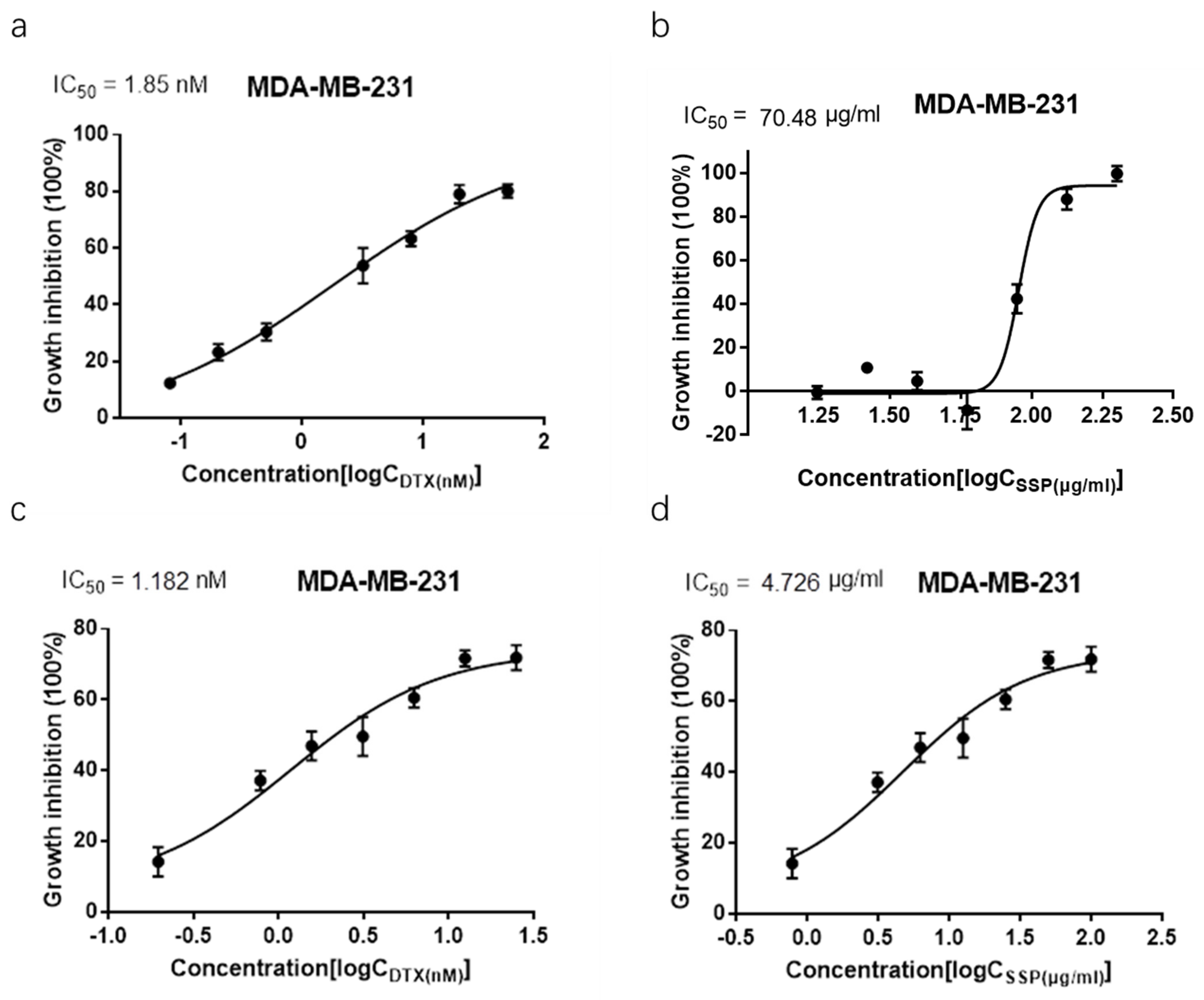
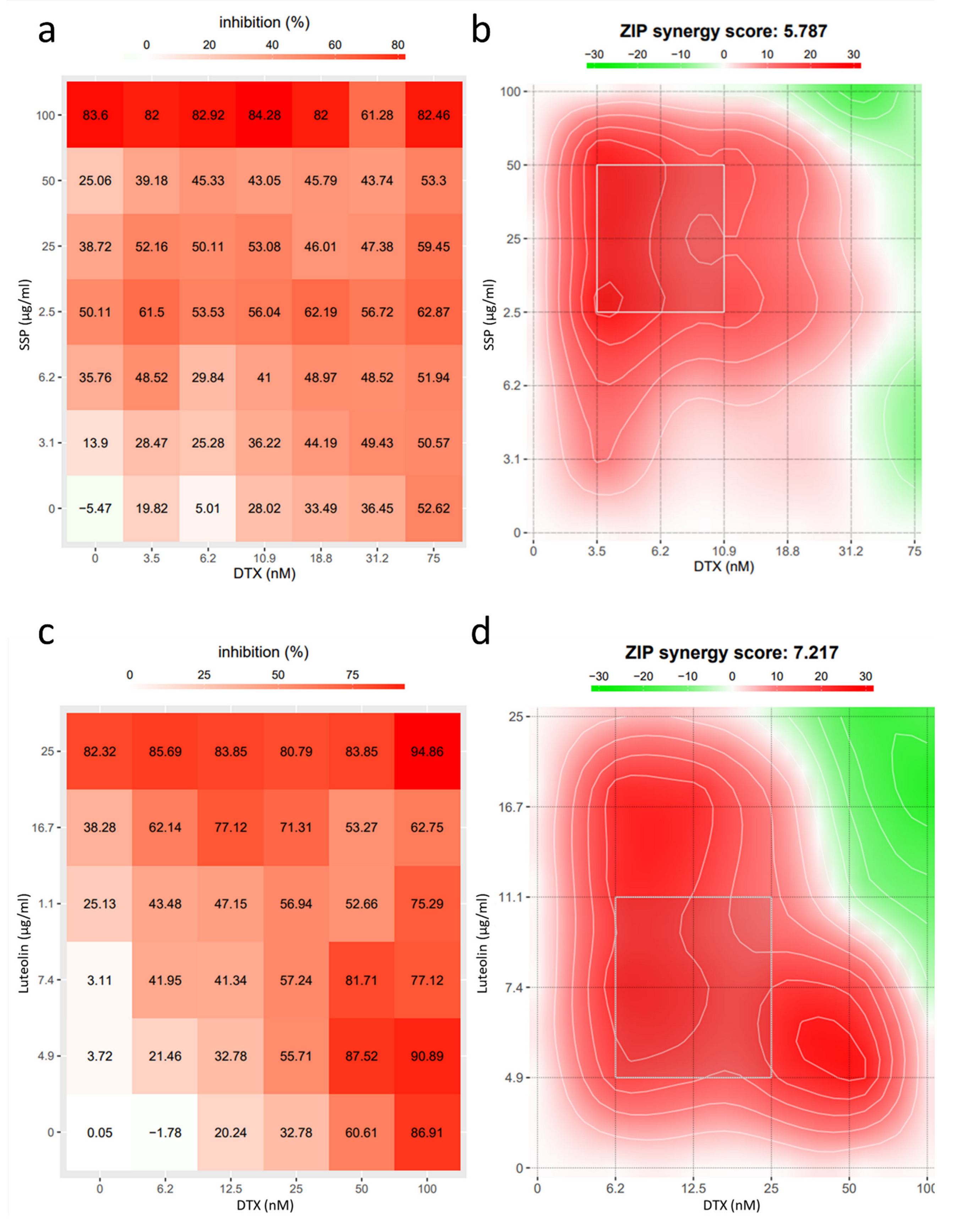
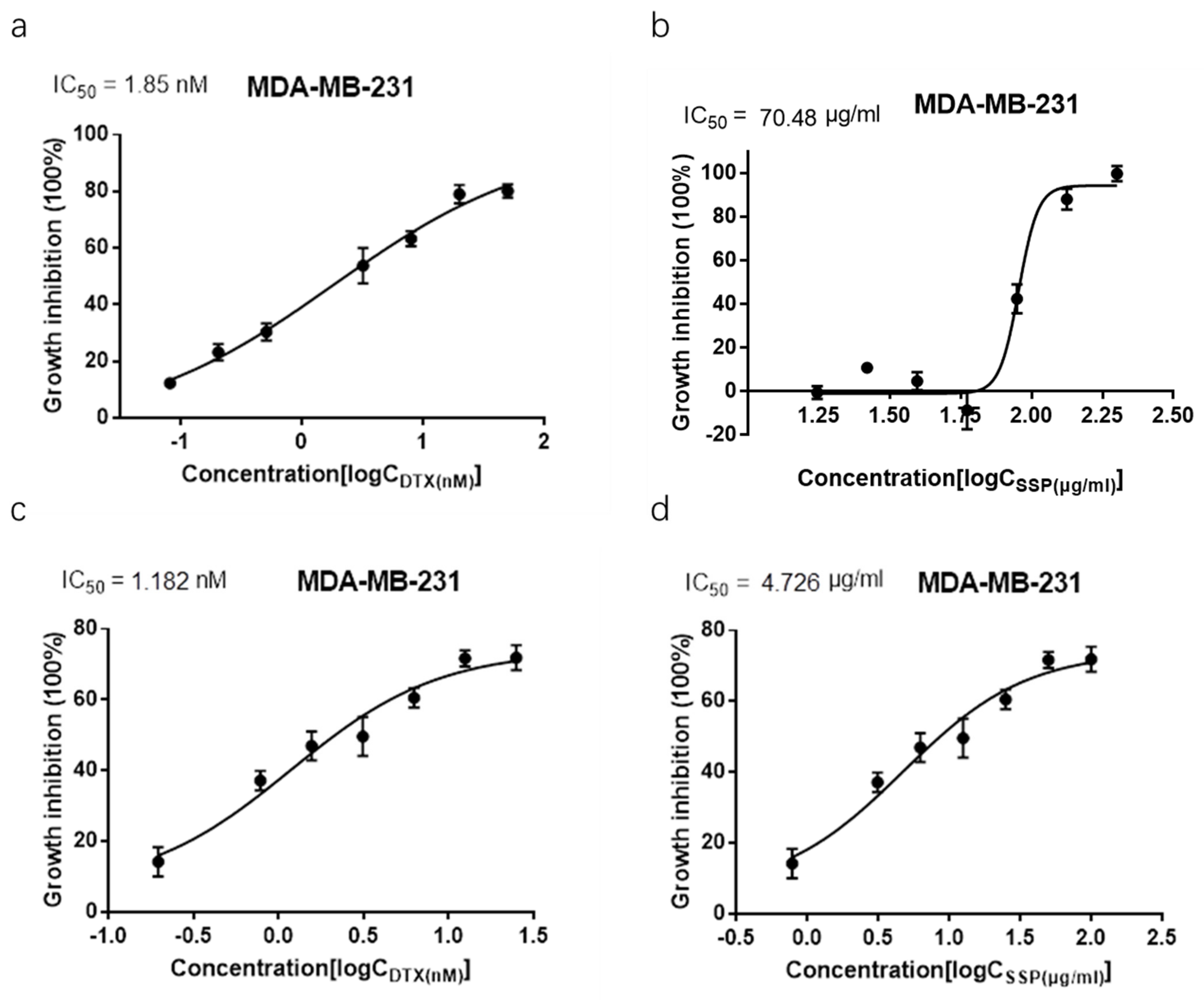
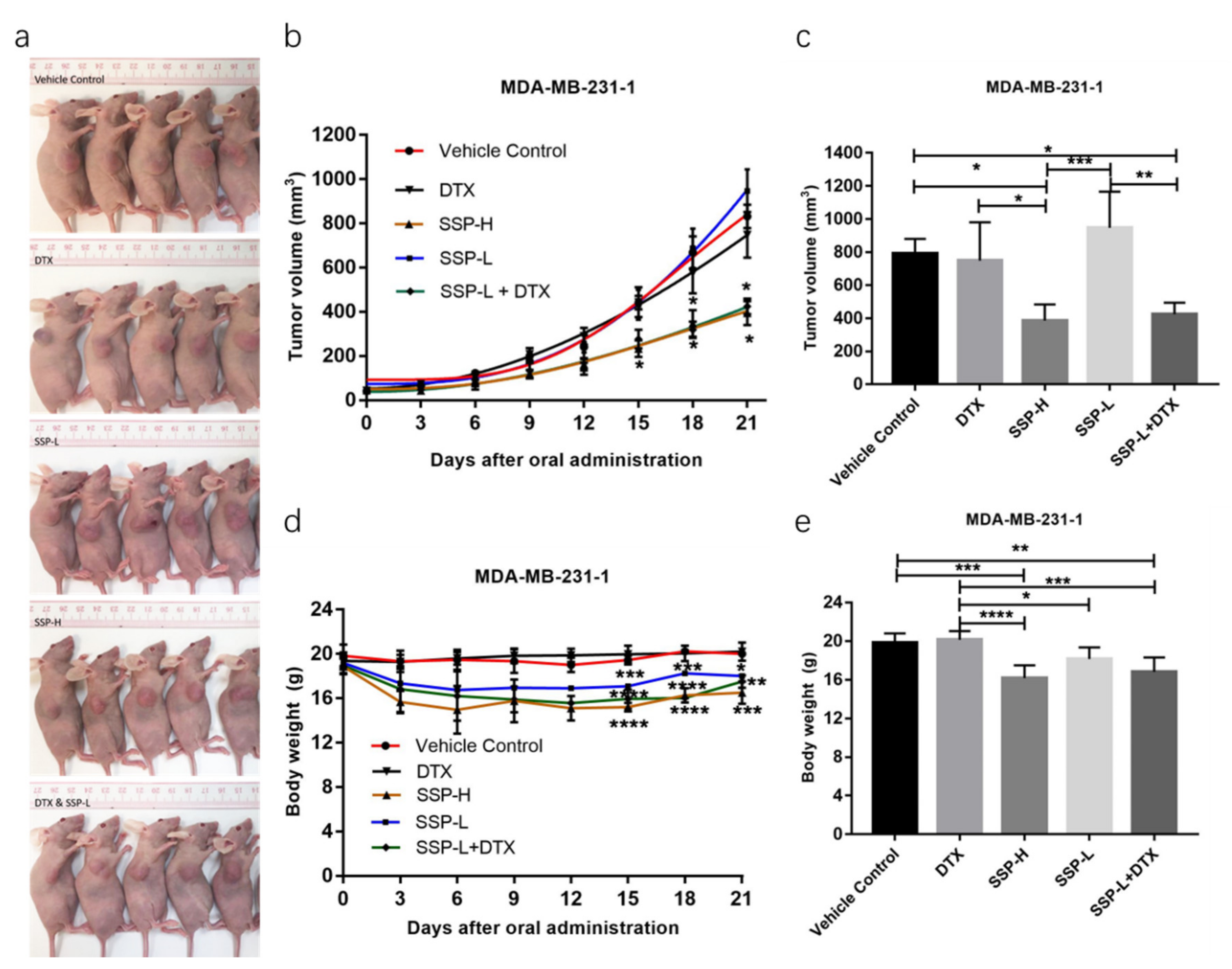
2.4. Synergism Detection
3. Discussion
4. Conclusions
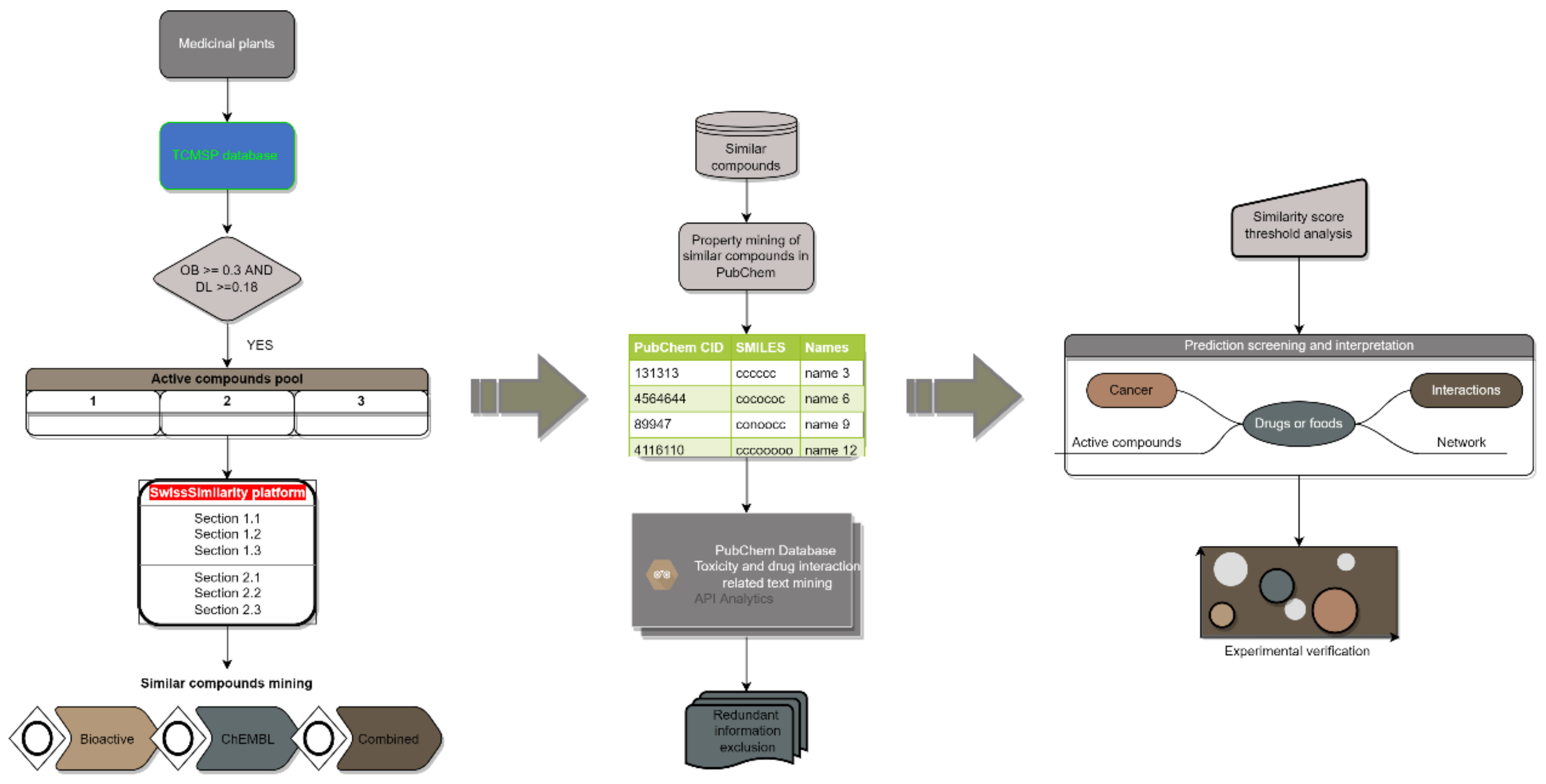
5. Materials and Methods
5.1. Dataset Assembly
5.2. Similar Compound Data Mining
5.3. Toxicity and Drug Interaction Information Mining
5.4. Prediction Interpretation
- 1.
- Active compoundThe active compounds of herbal medicine defined here, are the natural products documented in the TSCSP database for a certain herbal medicine, screened out based on the criteria (“Oral bioavailability” ≥ 0.3 and “Drug-likeness” ≥ 0.18).
- 2.
- Drug interactionsDrug interactions, in such a prediction system, include drug-food interactions, drug-drug interactions, and interactions of drugs with other factors such as carcinogens and ionizing radiation.
- 3.
- Prediction yieldIn such a prediction system, the prediction yield is defined as the number of active compounds of herbal medicines with at least one kind of toxicity or drug interaction information predicted.
- 4.
- First elusive prediction-similarity score (FEP-SS)Among all the information retrieved of similar compounds, for a certain toxicity prediction of an active compound based on such a system, as the similarity score decreases, the first elusive prediction-similarity score is the similarity score corresponding to the first elusive, arguable, or equivocal toxicity information retrieve compared to the toxicity information retrieve with the largest similarity score.
- 5.
- First contrast prediction-similarity score (FCP-SS)Among all the information retrieves of similar compounds, for a certain toxicity prediction of an active compound based on such a system, as the similarity score decreases, the first contrast prediction-similarity score is the similarity score corresponding to the first contrast toxicity information retrieve compared to the toxicity information retrieve with the largest similarity score.
5.5. Preparation of Spatholobus Suberectus Dunn-Percolation (SSP) Extract
5.6. Cell Culture and Treatment
5.7. Xenograft Model
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elghazaly, H.; Rugo, H.S.; Azim, H.A.; Swain, S.M.; Arun, B.; Aapro, M.; Perez, E.A.; Anderson, B.O.; Penault-Llorca, F.; Conte, P.; et al. Breast-Gynaecological & Immuno-Oncology International Cancer Conference (BGICC) Consensus and Recommendations for the Management of Triple-Negative Breast Cancer. Cancers 2021, 13, 2262. [Google Scholar]
- Miles, D.; Gligorov, J.; Andre, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; Chow, L.Q.M.; Burgio, M.A.; de Castro Carpeno, J.; Pluzanski, A.; Arrieta, O.; Frontera, O.A.; Chiari, R.; et al. Five-Year Outcomes from the Randomized, Phase III Trials CheckMate 017 and 057, Nivolumab Versus Docetaxel in Previously Treated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef]
- Zhao, M.Y.; Liu, P.; Sun, C.; Pei, L.J.; Huang, Y.G. Propofol Augments Paclitaxel-Induced Cervical Cancer Cell Ferroptosis In Vitro. Front. Pharmacol. 2022, 13, 816432. [Google Scholar] [CrossRef]
- Li, R.T.; Zhu, Y.D.; Li, W.Y.; Hou, Y.K.; Zou, Y.M.; Zhao, Y.H.; Zou, Q.; Zhang, W.H.; Chen, J.X. Synergistic photothermal-photodynamic-chemotherapy toward breast cancer based on a liposome-coated core-shell AuNS@NMOFs nanocomposite encapsulated with gambogic acid. J. Nanobiotechnol. 2022, 20, 212. [Google Scholar] [CrossRef]
- Date, T.; Kuche, K.; Chaudhari, D.; Ghadi, R.; Sahel, D.K.; Chitkara, D.; Jain, S. Hitting Multiple Cellular Targets in Triple-Negative Breast Cancer Using Dual-Action Cisplatin(IV) Prodrugs for Safer Synergistic Chemotherapy. ACS Biomater. Sci. Eng. 2022, 8, 2349–2362. [Google Scholar] [CrossRef]
- Wang, C.; Gao, H.; Huang, L.; Wang, Z.; Ding, X. Network Pharmacological Analysis and Experimental Study of the Antipharyngitis Mechanism of the Chaiqin Qingning Capsule. Biomed Res. Int. 2022, 2022, 5616942. [Google Scholar] [CrossRef]
- Li, X.; Ma, J.; Guo, L.; Dong, C.; Zhu, G.; Hong, W.; Chen, C.; Wang, H.; Wu, X. Identification of Bioactive Compounds and Potential Mechanisms of Kuntai Capsule in the Treatment of Polycystic Ovary Syndrome by Integrating Network Pharmacology and Bioinformatics. Oxid. Med. Cell. Longev. 2022, 2022, 3145938. [Google Scholar] [CrossRef]
- Crowther, N.R.; Holbrook, A.M.; Kenwright, R.; Kenwright, M. Drug interactions among commonly used medications. Chart simplifies data from critical literature review. Can. Fam. Physician 1997, 43, 1972–1976, 1979–1981. [Google Scholar]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do Structurally Similar Molecules Have Similar Biological Activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Fan, X.; Zhao, X.; Jin, Y.; Shen, X.; Liu, C. Network toxicology and its application to traditional Chinese medicine. Zhongguo Zhong Yao Za Zhi 2011, 36, 2920–2922. [Google Scholar]
- Ridings, J.E.; Barratt, M.D.; Cary, R.; Earnshaw, C.G.; Eggington, C.E.; Ellis, M.K.; Judson, P.N.; Langowski, J.J.; Marchant, C.A.; Payne, M.P.; et al. Computer prediction of possible toxic action from chemical structure: An update on the DEREK system. Toxicology 1996, 106, 267–279. [Google Scholar] [CrossRef]
- Jeliazkova, N.; Jeliazkov, V. AMBIT RESTful web services: An implementation of the OpenTox application programming interface. J. Cheminform. 2011, 3, 18. [Google Scholar] [CrossRef]
- Grulke, C.M.; Williams, A.J.; Thillanadarajah, I.; Richard, A.M. EPA’s DSSTox database: History of development of a curated chemistry resource supporting computational toxicology research. Comput. Toxicol. 2019, 12, 100096. [Google Scholar] [CrossRef]
- Langton, K.; Patlewicz, G.Y.; Long, A.; Marchant, C.A.; Basketter, D.A. Structure-activity relationships for skin sensitization: Recent improvements to Derek for Windows. Contact Dermat. 2006, 55, 342–347. [Google Scholar] [CrossRef]
- Greene, N.; Judson, P.N.; Langowski, J.J.; Marchant, C.A. Knowledge-based expert systems for toxicity and metabolism prediction: DEREK, StAR and METEOR. SAR QSAR Environ. Res. 1999, 10, 299–314. [Google Scholar] [CrossRef]
- Benfenati, E.; Gini, G. Computational predictive programs (expert systems) in toxicology. Toxicology 1997, 119, 213–225. [Google Scholar] [CrossRef]
- Poroikov, V.; Filimonov, D.; Lagunin, A.; Gloriozova, T.; Zakharov, A. PASS: Identification of probable targets and mechanisms of toxicity. SAR QSAR Environ. Res. 2007, 18, 101–110. [Google Scholar] [CrossRef]
- Cunningham, A.R.; Carrasquer, C.A.; Mattison, D.R. A categorical structure-activity relationship analysis of the developmental toxicity of antithyroid drugs. Int. J. Pediatr. Endocrinol. 2009, 2009, 936154. [Google Scholar] [CrossRef]
- Gallegos-Saliner, A.; Poater, A.; Jeliazkova, N.; Patlewicz, G.; Worth, A.P. Toxmatch—A chemical classification and activity prediction tool based on similarity measures. Regul. Toxicol. Pharmacol. 2008, 52, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Fumagalli, L.; Vistoli, G. The VEGA suite of programs: An versatile platform for cheminformatics and drug design projects. Bioinformatics 2021, 37, 1174–1175. [Google Scholar] [CrossRef] [PubMed]
- Tomasulo, P. ChemIDplus-super source for chemical and drug information. Med. Ref. Serv. Q. 2002, 21, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S. In Silico Models for Ecotoxicity of Pharmaceuticals. Methods Mol. Biol. 2016, 1425, 237–304. [Google Scholar]
- Kar, S.; Leszczynski, J. Exploration of Computational Approaches to Predict the Toxicity of Chemical Mixtures. Toxics 2019, 7, 15. [Google Scholar] [CrossRef]
- Prival, M.J. Evaluation of the TOPKAT system for predicting the carcinogenicity of chemicals. Environ. Mol. Mutagen. 2001, 37, 55–69. [Google Scholar] [CrossRef]
- Mombelli, E. An evaluation of the predictive ability of the QSAR software packages, DEREK, HAZARDEXPERT and TOPKAT, to describe chemically-induced skin irritation. Altern. Lab. Anim. 2008, 36, 15–24. [Google Scholar] [CrossRef]
- Lewi, D.F.; Bird, M.G.; Jacobs, M.N. Human carcinogens: An evaluation study via the COMPACT and HazardExpert procedures. Hum. Exp. Toxicol. 2002, 21, 115–122. [Google Scholar]
- Morita, T.; Shigeta, Y.; Kawamura, T.; Fujita, Y.; Honda, H.; Honma, M. In silico prediction of chromosome damage: Comparison of three (Q)SAR models. Mutagenesis 2019, 34, 91–100. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021, new data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.L. A probabilistic molecular fingerprint for big data settings. J. Cheminform. 2018, 10, 66. [Google Scholar] [CrossRef]
- Brown, B.P.; Vu, O.; Geanes, A.R.; Kothiwale, S.; Butkiewicz, M.; Lowe, E.W., Jr.; Mueller, R.; Pape, R.; Mendenhall, J.; Meiler, J. Introduction to the BioChemical Library (BCL): An Application-Based Open-Source Toolkit for Integrated Cheminformatics and Machine Learning in Computer-Aided Drug Discovery. Front. Pharmacol. 2022, 13, 833099. [Google Scholar] [CrossRef]
- Gobbi, A.; Poppinger, D. Genetic optimization of combinatorial libraries. Biotechnol. Bioeng. 1998, 61, 47–54. [Google Scholar] [CrossRef]
- Stiefl, N.; Watson, I.A.; Baumann, K.; Zaliani, A. ErG: 2D pharmacophore descriptions for scaffold hopping. J. Chem. Inf. Model. 2006, 46, 208–220. [Google Scholar] [CrossRef]
- Armstrong, M.S.; Finn, P.W.; Morris, G.M.; Richards, W.G. Improving the accuracy of ultrafast ligand-based screening: Incorporating lipophilicity into ElectroShape as an extra dimension. J. Comput.-Aided Mol. Des. 2011, 25, 785–790. [Google Scholar] [CrossRef]
- Bragina, M.E.; Daina, A.; Perez, M.A.S.; Michielin, O.; Zoete, V. The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int. J. Mol. Sci. 2022, 23, 811. [Google Scholar] [CrossRef]
- Axen, S.D.; Huang, X.P.; Caceres, E.L.; Gendelev, L.; Roth, B.L.; Keiser, M.J. A Simple Representation of Three-Dimensional Molecular Structure. J. Med. Chem. 2017, 60, 7393–7409. [Google Scholar] [CrossRef]
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef]
- Choi, Y.H.; Suh, J.H.; Lee, J.H.; Cho, I.H.; Lee, M.G. Effects of tesmilifene, a substrate of CYP3A and an inhibitor of P-glycoprotein, on the pharmacokinetics of intravenous and oral docetaxel in rats. J. Pharm. Pharmacol. 2010, 62, 1084–1088. [Google Scholar] [CrossRef]
- Van Waterschoot, R.A.; Lagas, J.S.; Wagenaar, E.; van der Kruijssen, C.M.; van Herwaarden, A.E.; Song, J.Y.; Rooswinkel, R.W.; van Tellingen, O.; Rosing, H.; Beijnen, J.H.; et al. Absence of both cytochrome P450 3A and P-glycoprotein dramatically increases docetaxel oral bioavailability and risk of intestinal toxicity. Cancer Res. 2009, 69, 8996–9002. [Google Scholar] [CrossRef]
- Van Zuylen, L.; Verweij, J.; Nooter, K.; Brouwer, E.; Stoter, G.; Sparreboom, A. Role of intestinal P-glycoprotein in the plasma and fecal disposition of docetaxel in humans. Clin. Cancer Res. 2000, 6, 2598–2603. [Google Scholar]
- National Comprehensive Cancer Network. NCCN Guidelines Version 4.2022 Breast Cancer. Available online: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed on 29 June 2022).
- Mayr, F.; Moller, G.; Garscha, U.; Fischer, J.; Rodriguez Castano, P.; Inderbinen, S.G.; Temml, V.; Waltenberger, B.; Schwaiger, S.; Hartmann, R.W.; et al. Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction. Int. J. Mol. Sci. 2020, 21, 7102. [Google Scholar] [CrossRef]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- Yadav, J.; Paragas, E.; Korzekwa, K.; Nagar, S. Time-dependent enzyme inactivation: Numerical analyses of in vitro data and prediction of drug-drug interactions. Pharmacol. Ther. 2020, 206, 107449. [Google Scholar] [CrossRef]
- Nozaki, Y.; Izumi, S. Recent advances in preclinical in vitro approaches towards quantitative prediction of hepatic clearance and drug-drug interactions involving organic anion transporting polypeptide (OATP) 1B transporters. Drug Metab. Pharmacokinet. 2020, 35, 56–70. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning improves prediction of drug-drug and drug-food interactions. Proc. Natl. Acad. Sci. USA 2018, 115, E4304–E4311. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef]
- D’Abreo, N.; Adams, S. Immune-checkpoint inhibition for metastatic triple-negative breast cancer: Safety first? Nat. Rev. Clin. Oncol. 2019, 16, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Sternschuss, M.; Yerushalmi, R.; Saleh, R.R.; Amir, E.; Goldvaser, H. Efficacy and safety of neoadjuvant immune checkpoint inhibitors in early-stage triple-negative breast cancer: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2021, 147, 3369–3379. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y.; et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminform. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, Q.; Ganesan, K.; Kewu, Z.; Shen, J.; Gang, F.; Luo, X.; Chen, J. The Antitriple Negative Breast cancer Efficacy of Spatholobus suberectus Dunn on ROS-Induced Noncanonical Inflammasome Pyroptotic Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 5187569. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genom. Proteom. Bioinform. 2022, in press. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Compound Name | Similarity Score | PubChem CID of Similar Compounds | Hepatotoxicity | Carcinogenicity |
|---|---|---|---|---|
| 3-Hydroxystigmast-5-en-7-one | 0.992 | 6010 | 0 | N.A. |
| 3-Hydroxystigmast-5-en-7-one | 0.986 | 10631 | 0 | N.A. |
| 3-Hydroxystigmast-5-en-7-one | 0.976 | 5997 | N.A. | −1 |
| 3-Hydroxystigmast-5-en-7-one | 0.917 | 6917715 | 0 | N.A. |
| 3-Hydroxystigmast-5-en-7-one | 0.540 | 54454 | 0 | N.A. |
| 3-Hydroxystigmast-5-en-7-one | 0.520 | 53232 | 0 | N.A. |
| 3-Hydroxystigmast-5-en-7-one | 0.498 | 5280453 | −1 | N.A. |
| 3-Hydroxystigmast-5-en-7-one | 0.302 | 445354 | 1 | N.A. |
| beta-sitosterol | 0.999 | 5997 | N.A. | −1 |
| beta-sitosterol | 0.804 | 5280453 | −1 | N.A. |
| beta-sitosterol | 0.588 | 445354 | 1 | N.A. |
| beta-sitosterol | 0.588 | 445354 | 1 | N.A. |
| campesterol | 0.999 | 5997 | N.A. | −1 |
| campesterol | 0.836 | 5280453 | −1 | N.A. |
| campesterol | 0.594 | 445354 | 1 | N.A. |
| Active Compound Name | PubChem CID of Similar Compound | FEP-SS (Hepatotoxicity) | FCP-SS (Hepatotoxicity) | FEP-SS (Carcinogenicity) | FCP-SS (Carcinogenicity) |
|---|---|---|---|---|---|
| (+)-catechin | 2369 | 0.472 | - | - | - |
| (20S)-Dammar-24-ene-3beta,20-diol 3-acetate | 5280453 | 0.31 | - | - | - |
| 18alpha-hydroxyglycyrrhetic acid | 10133 | 0.331 | - | - | - |
| 3,22-Dihydroxy-11-oxo-delta(12)-oleanene-27-alpha-methoxycarbonyl-29-oic acid | 5281004 | 0.265 | - | - | - |
| 3-Hydroxystigmast-5-en-7-one | 5280453 | 0.498 | - | - | - |
| DFV | 4764 | - | 0.558 | - | |
| Glabranin | 16078 | 0.703 | - | - | - |
| Glabrene | 3005573 | - | - | - | 0.353 |
| Kanzonol F | 441140 | 0.212 | - | - | - |
| Medicarpin | 441140 | - | 0.561 | - | - |
| Olitoriside | 54687/12560 | 0.337 | 0.449 | - | - |
| Psi-Baptigenin | 6237 | - | 0.176 | - | - |
| Stigmasterol | 445354 | - | 0.468 | - | - |
| Aloe-emodin | 42890/3059 | 0.463 | 0.413 | - | - |
| Beta-sitosterol | 445354 | - | 0.588 | - | - |
| Campesterol | 445354 | - | 0.594 | - | - |
| Hederagenin | 10133 | 0.486 | - | - | - |
| Liquiritin | 30323 | - | - | 0.379 | - |
| Naringenin | 5281576 | - | - | 0.477 | - |
| Sitosterol | 445354 | - | 0.588 | - | - |
| PubChem CID | Active Compound Name | AE | Is | AET | HTE | NHTE | CC | PSR | HT | EC | NHTV | OTS | NTPS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5280448 | Calycosin | - | + | + | + | + | - | - | - | - | - | - | - |
| 9064 | (+)-catechin | + | - | - | - | - | - | - | - | - | - | - | - |
| 222284 | Beta-sitosterol | + | - | - | - | - | - | - | - | - | - | - | - |
| 73299 | Hederagenin | + | - | - | - | - | - | - | - | - | - | - | - |
| 5280863 | Kaempferol | - | + | + | + | + | + | + | - | - | - | - | - |
| 5280445 | Luteolin | + | - | - | - | - | - | - | - | - | - | - | - |
| 5280343 | Quercetin | + | + | + | + | + | + | + | + | + | + | + | + |
| 5280794 | Stigmasterol | - | - | + | + | - | - | - | - | - | - | - | - |
| 439533 | Taxifolin | + | - | - | - | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Ganesan, K.; Li, Y.; Chen, J. In-Silico Drug Toxicity and Interaction Prediction for Plant Complexes Based on Virtual Screening and Text Mining. Int. J. Mol. Sci. 2022, 23, 10056. https://doi.org/10.3390/ijms231710056
Zhang F, Ganesan K, Li Y, Chen J. In-Silico Drug Toxicity and Interaction Prediction for Plant Complexes Based on Virtual Screening and Text Mining. International Journal of Molecular Sciences. 2022; 23(17):10056. https://doi.org/10.3390/ijms231710056
Chicago/Turabian StyleZhang, Feng, Kumar Ganesan, Yan Li, and Jianping Chen. 2022. "In-Silico Drug Toxicity and Interaction Prediction for Plant Complexes Based on Virtual Screening and Text Mining" International Journal of Molecular Sciences 23, no. 17: 10056. https://doi.org/10.3390/ijms231710056
APA StyleZhang, F., Ganesan, K., Li, Y., & Chen, J. (2022). In-Silico Drug Toxicity and Interaction Prediction for Plant Complexes Based on Virtual Screening and Text Mining. International Journal of Molecular Sciences, 23(17), 10056. https://doi.org/10.3390/ijms231710056