
Evaluation of a Serum-Free Medium for Human Epithelial and Stromal Cell Culture



Abstract
:1. Introduction
2. Results
2.1. Microscopic Appearance Assessment
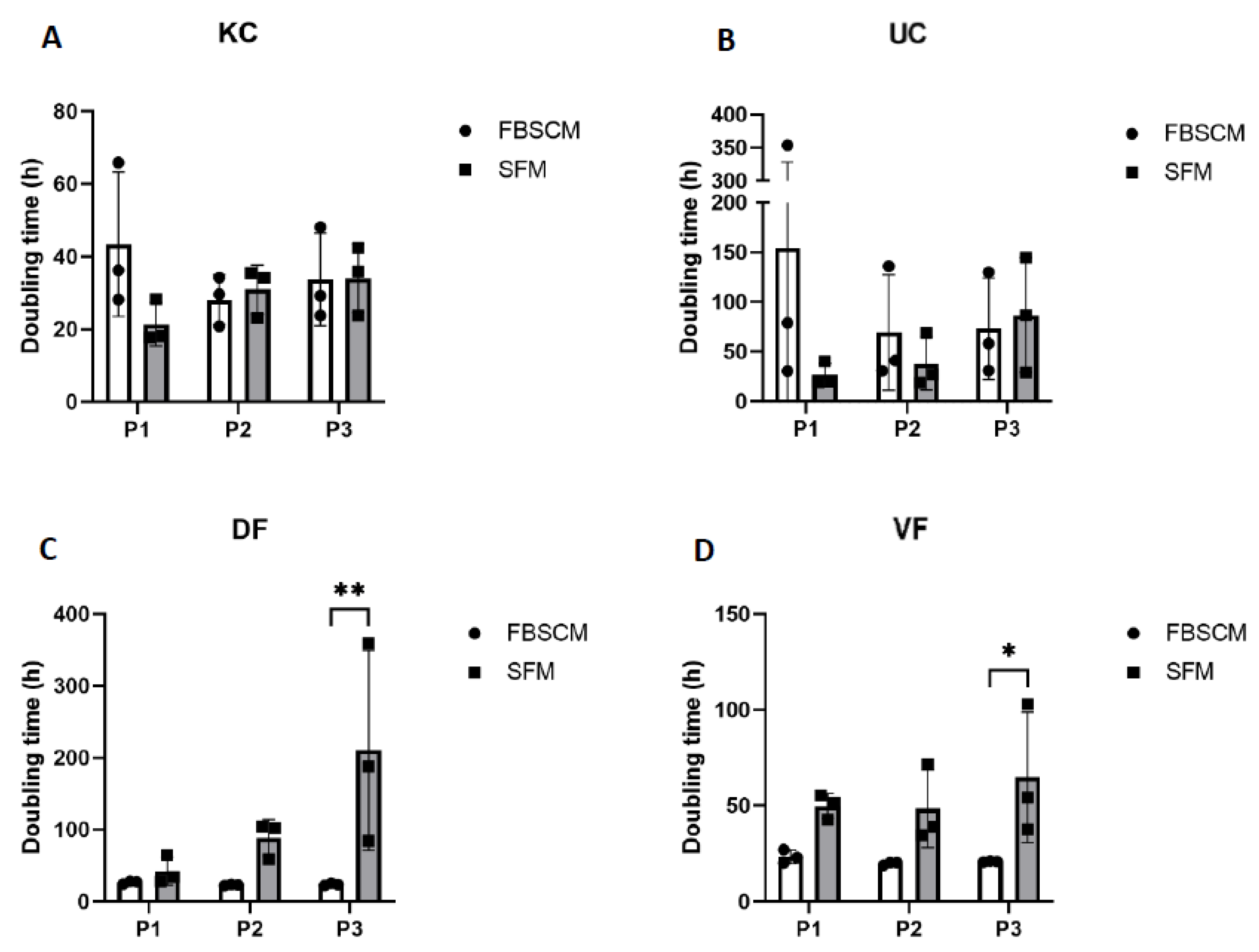
2.2. Doubling Time Measurement
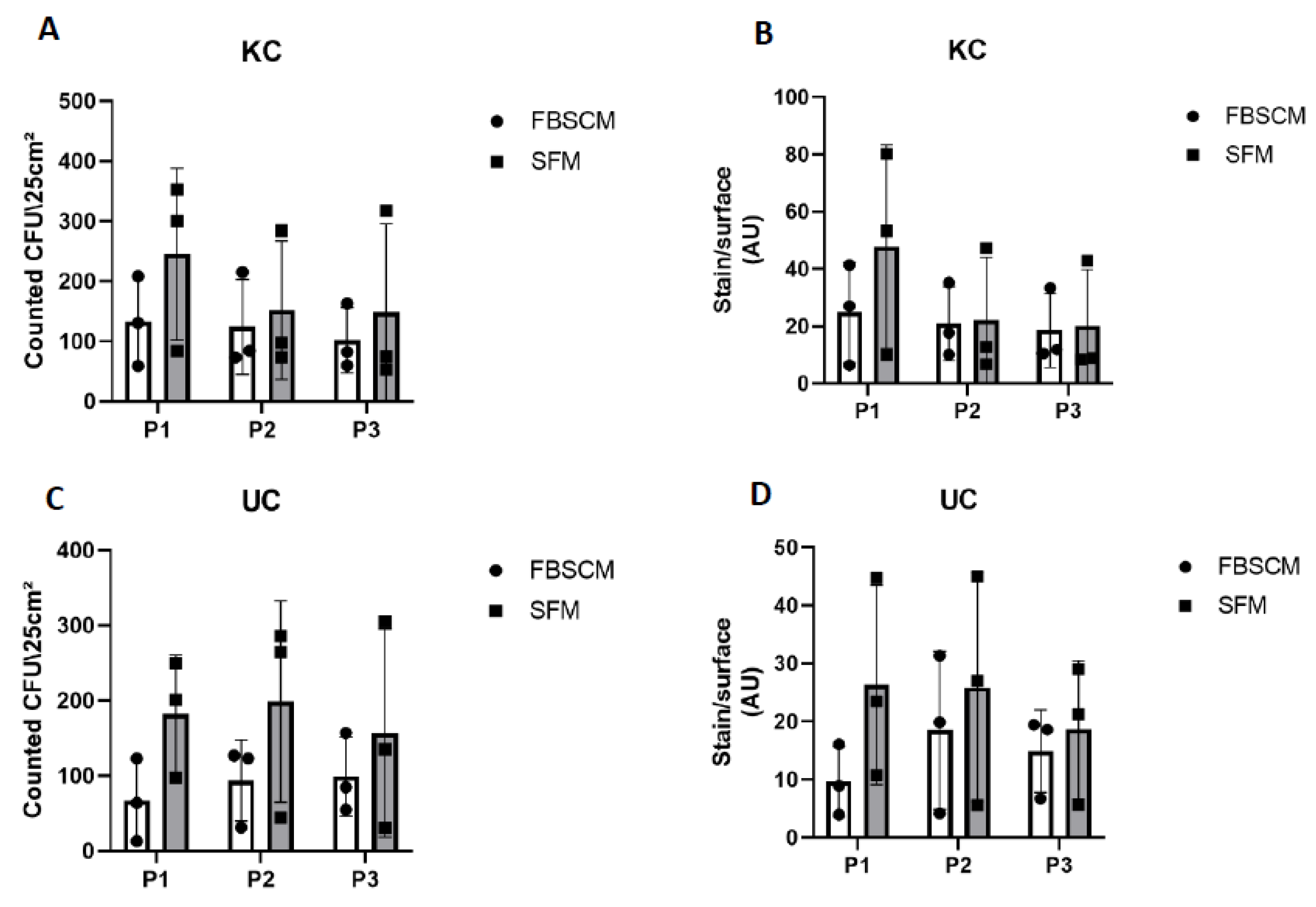
2.3. Epithelial Clonogenicity Evaluation
2.4. Metabolism Evaluation
2.5. SFM and FBSCM Cost Evaluation
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Isolation and Culture
4.3. Constitution of the Serum-Free Medium (SFM)
4.4. Proliferation and Size Determination
4.5. Circularity Index Measurement
4.6. Clonogenicity Determination
4.7. Metabolism Evaluation
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karnieli, O.; Friedner, O.M.; Allickson, J.G.; Zhang, N.; Jung, S.; Fiorentini, D.; Abraham, E.; Eaker, S.S.; Yong, T.K.; Chan, A.; et al. A consensus introduction to serum replacements and serum-free media for cellular therapies. Cytotherapy 2017, 19, 155–169. [Google Scholar] [CrossRef]
- Brindley, D.A.; Davie, N.L.; Culme-Seymour, E.J.; Mason, C.; Smith, D.W.; Rowley, J.A. Peak serum: Implications of serum supply for cell therapy manufacturing. Regen. Med. 2012, 7, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gstraunthaler, G.; Lindl, T.; van der Valk, J. A plea to reduce or replace fetal bovine serum in cell culture media. Cytotechnology 2013, 65, 791–793. [Google Scholar] [CrossRef]
- Boone, C.W.; Mantel, N.; Caruso, T.D., Jr.; Kazam, E.; Stevenson, R.E. Quality control studies on fetal bovine serum used in tissue culture. In Vitro 1971, 7, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.E.A.; van der Valk, J.B.F.; Stafleu, F.R.; Baumans, V. The Use of Fetal Bovine Serum: Ethical or Scientific Problem? Altern. Lab. Anim. 2002, 30, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Gstraunthaler, G. Alternatives to the use of fetal bovine serum: Serum-free cell culture. Altex 2003, 20, 275–281. [Google Scholar] [CrossRef]
- Afzal, E.; Pakzad, M.; Nouri, M.; Moghadasali, R.; Zarrabi, M. Human umbilical cord serum as an alternative to fetal bovine serum for in vitro expansion of umbilical cord mesenchymal stromal cells. Cell Tissue Bank. 2022. [Google Scholar] [CrossRef]
- Naskou, M.C.; Sumner, S.M.; Chocallo, A.; Kemelmakher, H.; Thoresen, M.; Copland, I.; Galipeau, J.; Peroni, J.F. Platelet lysate as a novel serum-free media supplement for the culture of equine bone marrow-derived mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 75. [Google Scholar] [CrossRef]
- Palombella, S.; Orfei, C.P.; Castellini, G.; Gianola, S.; Lopa, S.; Mastrogiacomo, M.; Moretti, M.; de Girolamo, L. Systematic review and meta-analysis on the use of human platelet lysate for mesenchymal stem cell cultures: Comparison with fetal bovine serum and considerations on the production protocol. Stem Cell Res. Ther. 2022, 13, 142. [Google Scholar] [CrossRef]
- Kocaoemer, A.; Kern, S.; Kluter, H.; Bieback, K. Human AB serum and thrombin-activated platelet-rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells 2007, 25, 1270–1278. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kang, M.; Choi, J.; Chun, Y.; Jang, J.; Yang, Y.; Lee, J.; Kim, S.; Park, S. Comparative analysis of FBS containing media and serum free chemically defined media for adipose derived stem cells production. Cytotherapy 2019, 21, S82. [Google Scholar] [CrossRef]
- Radtke, S.; Giebel, B.; Wagner, W.; Horn, P.A. Platelet lysates and their role in cell therapy. ISBT Sci. Ser. 2014, 9, 193–197. [Google Scholar] [CrossRef]
- Lohmann, M.; Walenda, G.; Hemeda, H.; Joussen, S.; Drescher, W.; Jockenhoevel, S.; Hutschenreuter, G.; Zenke, M.; Wagner, W. Donor age of human platelet lysate affects proliferation and differentiation of mesenchymal stem cells. PLoS ONE 2012, 7, e37839. [Google Scholar] [CrossRef] [PubMed]
- Angello, J.C.; Pendergrass, W.R.; Norwood, T.H.; Prothero, J. Proliferative potential of human fibroblasts: An inverse dependence on cell size. J. Cell. Physiol. 1987, 132, 125–130. [Google Scholar] [CrossRef]
- De Paiva, C.S.; Pflugfelder, S.C.; Li, D.Q. Cell size correlates with phenotype and proliferative capacity in human corneal epithelial cells. Stem Cells 2006, 24, 368–375. [Google Scholar] [CrossRef]
- Barrandon, Y.; Green, H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Natl. Acad. Sci. USA 1987, 84, 2302–2306. [Google Scholar] [CrossRef]
- Chase, L.G.; Lakshmipathy, U.; Solchaga, L.A.; Rao, M.S.; Vemuri, M.C. A novel serum-free medium for the expansion of human mesenchymal stem cells. Stem Cell Res. Ther. 2010, 1, 8. [Google Scholar] [CrossRef]
- Taihi, I.; Pilon, C.; Cohen, J.; Berdal, A.; Gogly, B.; Nassif, A.; Fournier, B.P. Efficient isolation of human gingival stem cells in a new serum-free medium supplemented with platelet lysate and growth hormone for osteogenic differentiation enhancement. Stem Cell Res. Ther. 2022, 13, 125. [Google Scholar] [CrossRef]
- Darby, I.A.; Hewitson, T.D. Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 2007, 257, 143–179. [Google Scholar] [CrossRef]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–408, 410–412. [Google Scholar] [CrossRef]
- Mitra, M.; Ho, L.D.; Coller, H.A. An In Vitro Model of Cellular Quiescence in Primary Human Dermal Fibroblasts. Methods Mol. Biol. 2018, 1686, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, M.A.; Cassano, A.; Vinella, A.; Vincenti, A.; Fumarulo, R.; Lo Muzio, L.; Maiorano, E.; Ribatti, D.; Favia, G. Enhancement of fibroblast proliferation, collagen biosynthesis and production of growth factors as a result of combining sodium hyaluronate and aminoacids. Int. J. Immunopathol. Pharmacol. 2009, 22, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Mast, B.A.; Diegelmann, R.F.; Krummel, T.M.; Cohen, I.K. Hyaluronic acid modulates proliferation, collagen and protein synthesis of cultured fetal fibroblasts. Matrix 1993, 13, 441–446. [Google Scholar] [CrossRef]
- David-Raoudi, M.; Tranchepain, F.; Deschrevel, B.; Vincent, J.C.; Bogdanowicz, P.; Boumediene, K.; Pujol, J.P. Differential effects of hyaluronan and its fragments on fibroblasts: Relation to wound healing. Wound Repair Regen. 2008, 16, 274–287. [Google Scholar] [CrossRef]
- Devireddy, L.R.; Myers, M.; Screven, R.; Liu, Z.; Boxer, L. A serum-free medium formulation efficiently supports isolation and propagation of canine adipose-derived mesenchymal stem/stromal cells. PLoS ONE 2019, 14, e0210250. [Google Scholar] [CrossRef]
- Mattinger, C.; Nyugen, T.; Schafer, D.; Hormann, K. Evaluation of serum-free culture conditions for primary human nasal epithelial cells. Int. J. Hyg. Environ. Health 2002, 205, 235–238. [Google Scholar] [CrossRef]
- Bhat, S.; Viswanathan, P.; Chandanala, S.; Prasanna, S.J.; Seetharam, R.N. Expansion and characterization of bone marrow derived human mesenchymal stromal cells in serum-free conditions. Sci. Rep. 2021, 11, 3403. [Google Scholar] [CrossRef]
- Chabaud, S.; Saba, I.; Baratange, C.; Boiroux, B.; Leclerc, M.; Rousseau, A.; Bouhout, S.; Bolduc, S. Urothelial cell expansion and differentiation are improved by exposure to hypoxia. J. Tissue Eng. Regen. Med. 2017, 11, 3090–3099. [Google Scholar] [CrossRef]
- Smolina, N.; Bruton, J.; Kostareva, A.; Sejersen, T. Assaying Mitochondrial Respiration as an Indicator of Cellular Metabolism and Fitness. Methods Mol. Biol. 2017, 1601, 79–87. [Google Scholar] [CrossRef]
- Chaudhry, R.; Varacallo, M. Biochemistry, Glycolysis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Nicholls, D.G. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem. Soc. Trans. 2009, 37, 1385–1388. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K.; Fernstrom, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Hojlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Tigchelaar, W.; De Jong, A.M.; van Gilst, W.H.; De Boer, R.A.; Sillje, H.H. In EXOG-depleted cardiomyocytes cell death is marked by a decreased mitochondrial reserve capacity of the electron transport chain. Bioessays 2016, 38 (Suppl. S1), S136–S145. [Google Scholar] [CrossRef]
- Pellerin, E.; Chabaud, S.; Pouliot, F.; Pelletier, M.; Bolduc, S. Bisphenol A Alters the Energy Metabolism of Stromal Cells and Could Promote Bladder Cancer Progression. Cancers 2021, 13, 5461. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Caneparo, C.; Baratange, C.; Chabaud, S.; Bolduc, S. Conditioned medium produced by fibroblasts cultured in low oxygen pressure allows the formation of highly structured capillary-like networks in fibrin gels. Sci. Rep. 2020, 10, 9291. [Google Scholar] [CrossRef]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFbeta Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef]
- Goulet, C.R.; Bernard, G.; Chabaud, S.; Couture, A.; Langlois, A.; Neveu, B.; Pouliot, F.; Bolduc, S. Tissue-engineered human 3D model of bladder cancer for invasion study and drug discovery. Biomaterials 2017, 145, 233–241. [Google Scholar] [CrossRef]
- Auger, F.A.; Valle, C.A.L.; Guignard, R.; Tremblay, N.; Noel, B.; Goulet, F.; Germain, L. Skin equivalent produced with human collagen. Vitr. Cell. Dev. Biol. Anim. 1995, 31, 432–439. [Google Scholar] [CrossRef]
- Bisson, F.; Rochefort, E.; Lavoie, A.; Larouche, D.; Zaniolo, K.; Simard-Bisson, C.; Damour, O.; Auger, F.A.; Guerin, S.L.; Germain, L. Irradiated human dermal fibroblasts are as efficient as mouse fibroblasts as a feeder layer to improve human epidermal cell culture lifespan. Int. J. Mol. Sci. 2013, 14, 4684–4704. [Google Scholar] [CrossRef] [PubMed]
- Auxenfans, C.; Thépot, A.; Justin, V.; Hautefeuille, A.; Shahabeddin, L.; Damour, O.; Hainaut, P. Characterisation of human fibroblasts as keratinocyte feeder layer using p63 isoforms status. Biomed. Mater. Eng. 2009, 19, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Pouyani, T.; Ronfard, V.; Scott, P.G.; Dodd, C.M.; Ahmed, A.; Gallo, R.L.; Parenteau, N.L. De novo synthesis of human dermis in vitro in the absence of a three-dimensional scaffold. Vitr. Cell. Dev. Biol. Anim. 2009, 45, 430–441. [Google Scholar] [CrossRef]
- Pellerin, F.A.; Caneparo, C.; Pellerin, E.; Chabaud, S.; Pelletier, M.; Bolduc, S. Heat-Inactivation of Fetal and Newborn Sera Did Not Impair the Expansion and Scaffold Engineering Potentials of Fibroblasts. Bioengineering 2021, 8, 184. [Google Scholar] [CrossRef]
- Pelletier, M.; Billingham, L.K.; Ramaswamy, M.; Siegel, R.M. Extracellular flux analysis to monitor glycolytic rates and mitochondrial oxygen consumption. Methods Enzymol. 2014, 542, 125–149. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P1 | P2 | P3 | ||||
|---|---|---|---|---|---|---|
| FBSCM | SFM | FBSCM | SFM | FBSCM | SFM | |
| KC | 0.888 ± 0.064 | 0.860 ± 0.065 | 0.840 ± 0.037 | 0.855 ± 0.055 | 0.834 ± 0.062 | 0.870 ± 0.059 |
| UC | 0.838 ± 0.073 | 0.845 ± 0.065 | 0.814 ± 0.057 | 0.826 ± 0.071 | 0.832 ± 0.052 | 0.872 ± 0.065 |
| DF | 0.224 ± 0.049 | 0.283 ± 0.061 | 0.229 ± 0.062 | 0.243 ± 0.032 | 0.267 ± 0.069 | 0.249 ± 0.071 |
| VF | 0.235 ± 0.064 | 0.305 ± 0.066 | 0.230 ± 0.066 | 0.202 ± 0.040 | 0.239 ± 0.087 | 0.236 ± 0.054 |
| KC | UC | DF | VF | ||
|---|---|---|---|---|---|
| ECAR | Basal | → | ↗ | ↘ | → |
| Maximal | → | ↗ | → | → | |
| OCR | Basal | → | ↗ | ↗ | ↗ |
| Maximal | ↗ | ↗ | ↗ | → |
| Components | Price for a 500 mL Bottle (USD) | |
|---|---|---|
| SFM | 3:1 mix of Dulbecco–Vogt modification of Eagle’s and Ham’s F12 | 0.20 |
| L-Glutamax | 6.42 | |
| Epidermal Growth Factor | 3.25 | |
| Hydrocortisone | 0.06 | |
| Insulin-Transferin-Selenium (ITS) | 34.22 | |
| Lipid concentrate | 4.56 | |
| Triiodothyronine | <0.01 | |
| Ethanolamine (M) | <0.01 | |
| Isoproterenol | <0.01 | |
| O-phosphoryl-ethanolamine (mM) | 0.04 | |
| Bovine Serum Albumin (BSA) | 2.82 | |
| L-proline | <0.01 | |
| Glycline | <0.01 | |
| Antibiotics:penicillin | <0.01 | |
| Antibiotics:gentamycin | 0.12 | |
| TOTAL | 51.50 | |
| FBSCM for stromal cells | Dulbecco–Vogt modification of Eagle’s medium | 0.21 |
| Antibiotics:penicillin | <0.01 | |
| Antibiotics:gentamycin | 0.12 | |
| FBS | 75.60 | |
| TOTAL | 76.05 | |
| FBSCM for epithelial cells | 3:1 mix of Dulbecco–Vogt modification of Eagle’s medium and Ham’s F12 | 0.20 |
| Epidermal Growth Factor | 3.25 | |
| Hydrocortisone | 0.06 | |
| Isoproterenol | <0.01 | |
| Insulin | 9.94 | |
| Antibiotics:penicillin | <0.01 | |
| Antibiotics:gentamycin | 0.12 | |
| Fetal Bovine Serum (FBS) | 42.20 | |
| TOTAL | 55.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caneparo, C.; Chabaud, S.; Fradette, J.; Bolduc, S. Evaluation of a Serum-Free Medium for Human Epithelial and Stromal Cell Culture. Int. J. Mol. Sci. 2022, 23, 10035. https://doi.org/10.3390/ijms231710035
Caneparo C, Chabaud S, Fradette J, Bolduc S. Evaluation of a Serum-Free Medium for Human Epithelial and Stromal Cell Culture. International Journal of Molecular Sciences. 2022; 23(17):10035. https://doi.org/10.3390/ijms231710035
Chicago/Turabian StyleCaneparo, Christophe, Stéphane Chabaud, Julie Fradette, and Stéphane Bolduc. 2022. "Evaluation of a Serum-Free Medium for Human Epithelial and Stromal Cell Culture" International Journal of Molecular Sciences 23, no. 17: 10035. https://doi.org/10.3390/ijms231710035
APA StyleCaneparo, C., Chabaud, S., Fradette, J., & Bolduc, S. (2022). Evaluation of a Serum-Free Medium for Human Epithelial and Stromal Cell Culture. International Journal of Molecular Sciences, 23(17), 10035. https://doi.org/10.3390/ijms231710035