
4-Phenylbutyric Acid (4-PBA) Derivatives Prevent SOD1 Amyloid Aggregation In Vitro with No Effect on Disease Progression in SOD1-ALS Mice

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
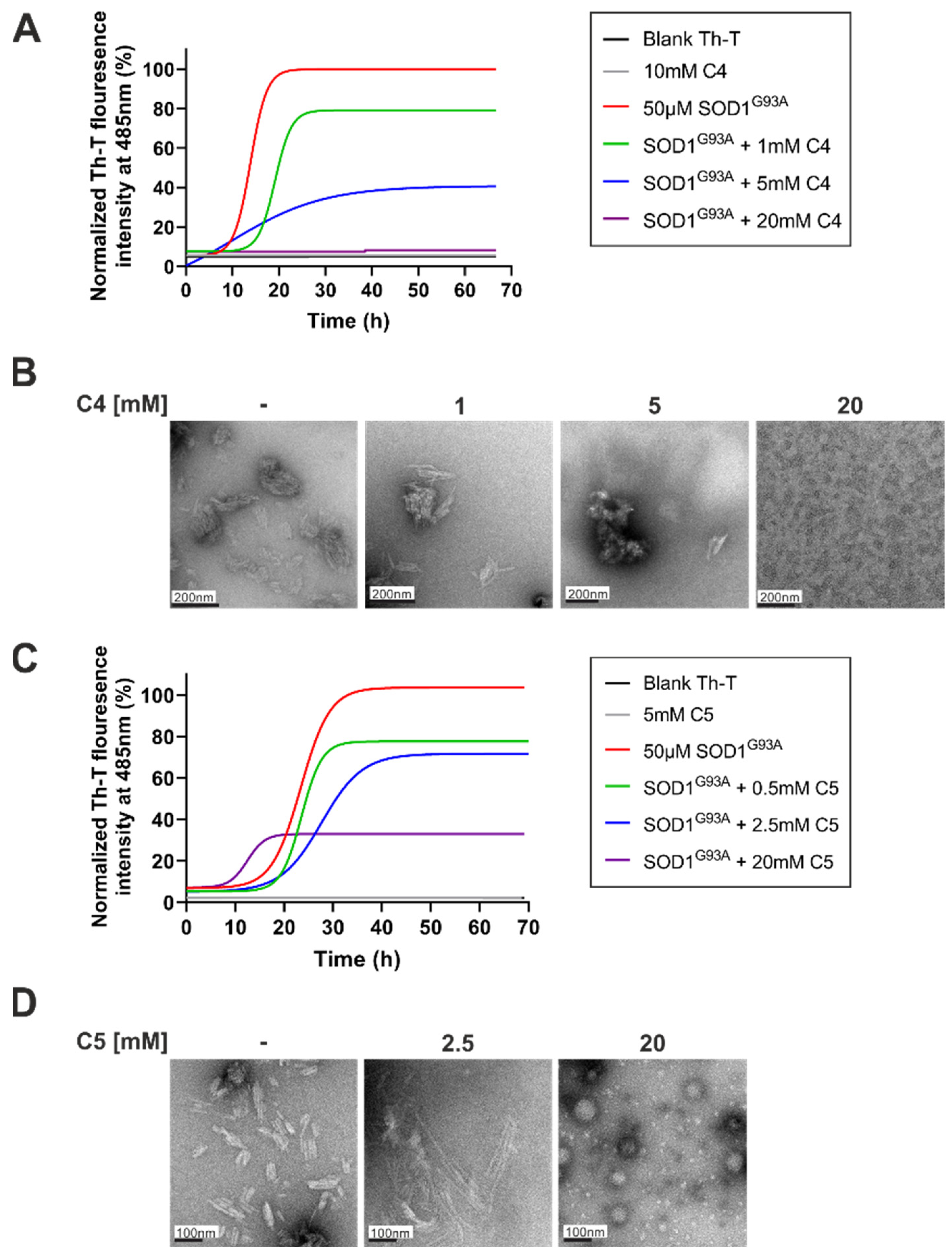
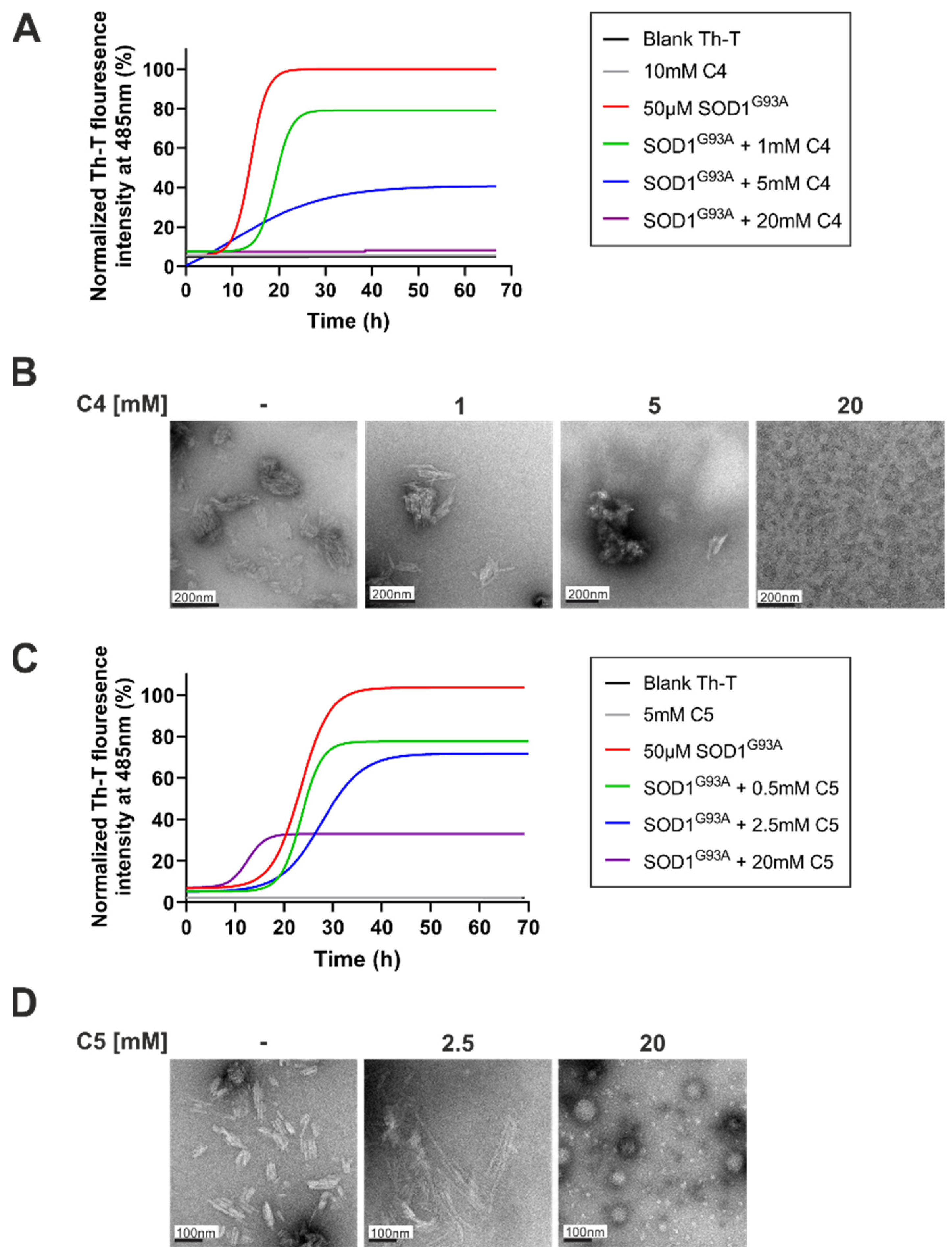
2.1. C4 and C5 Chemical Chaperones Strongly Inhibited the Formation of SOD1 Amyloid Aggregation In Vitro
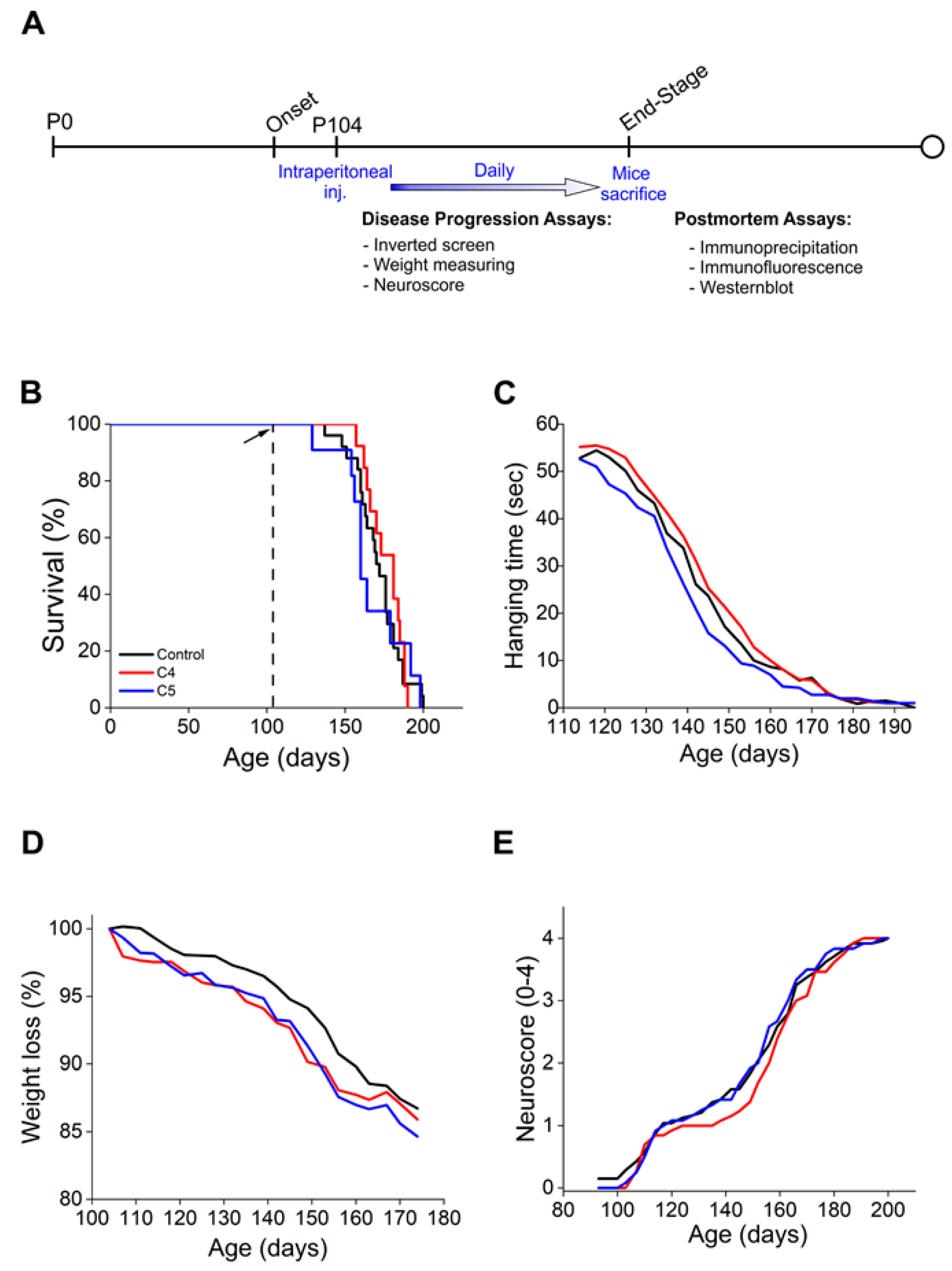
2.2. Daily Injection of C4 or C5 Chemical Chaperones Following Disease Onset Had No Effect on Disease Progression and Survival of Mutant SOD1G93A Mice
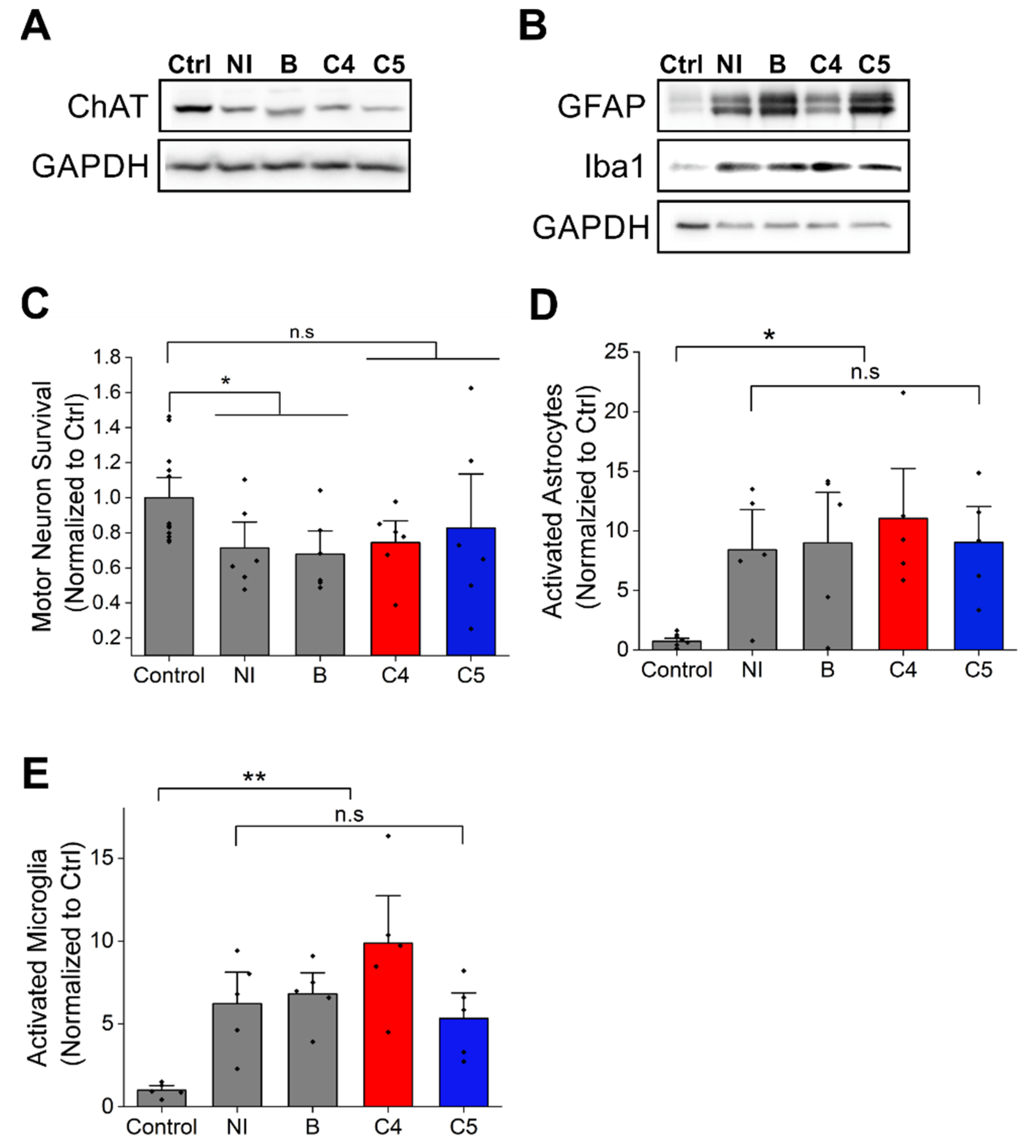
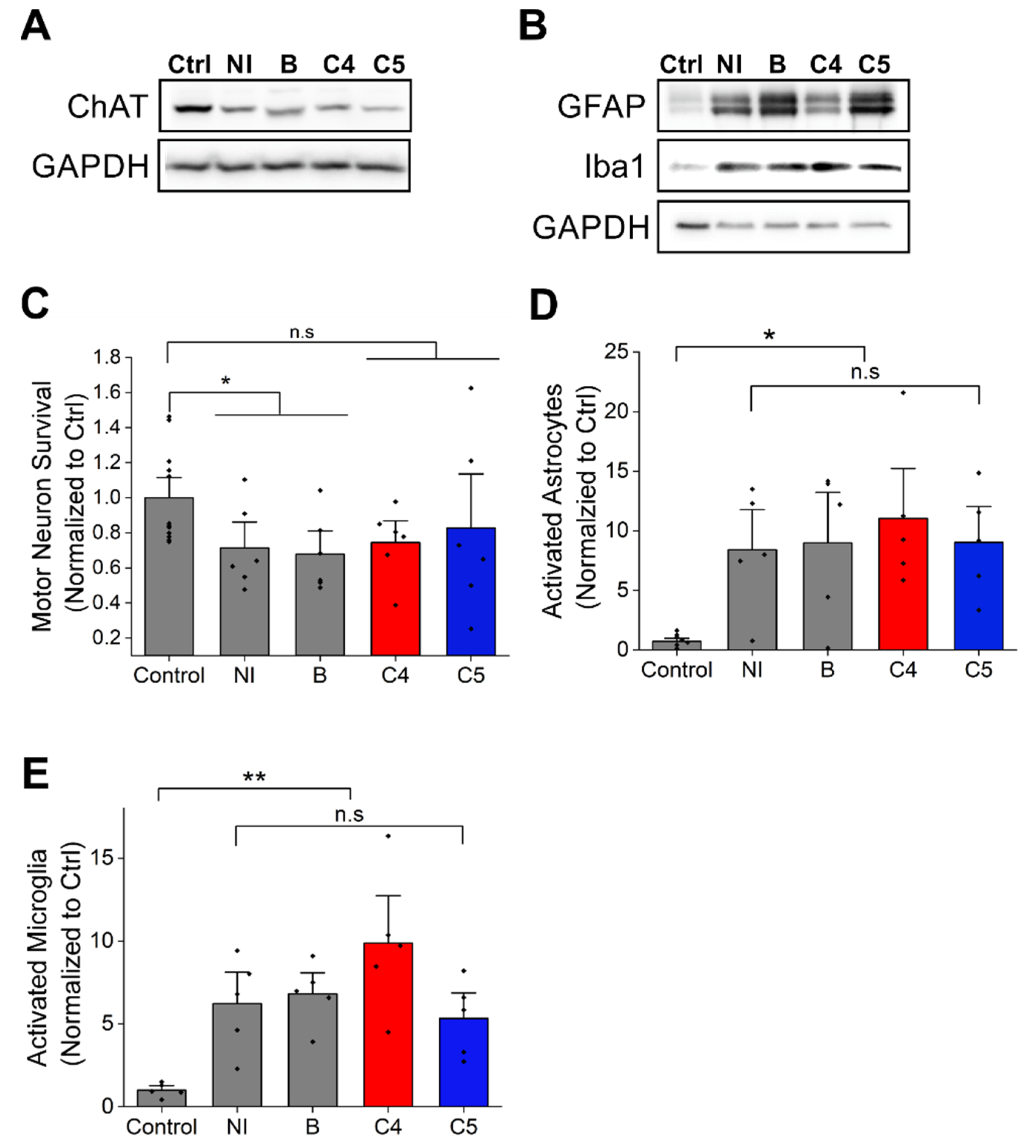
2.3. C4 or C5 Treatment Starting after Disease Onset Failed to Rescue the Motor Neurons and Reduce the Activation of Astrocytes and Microglia in the Spinal Cord of SOD1G93A Mice
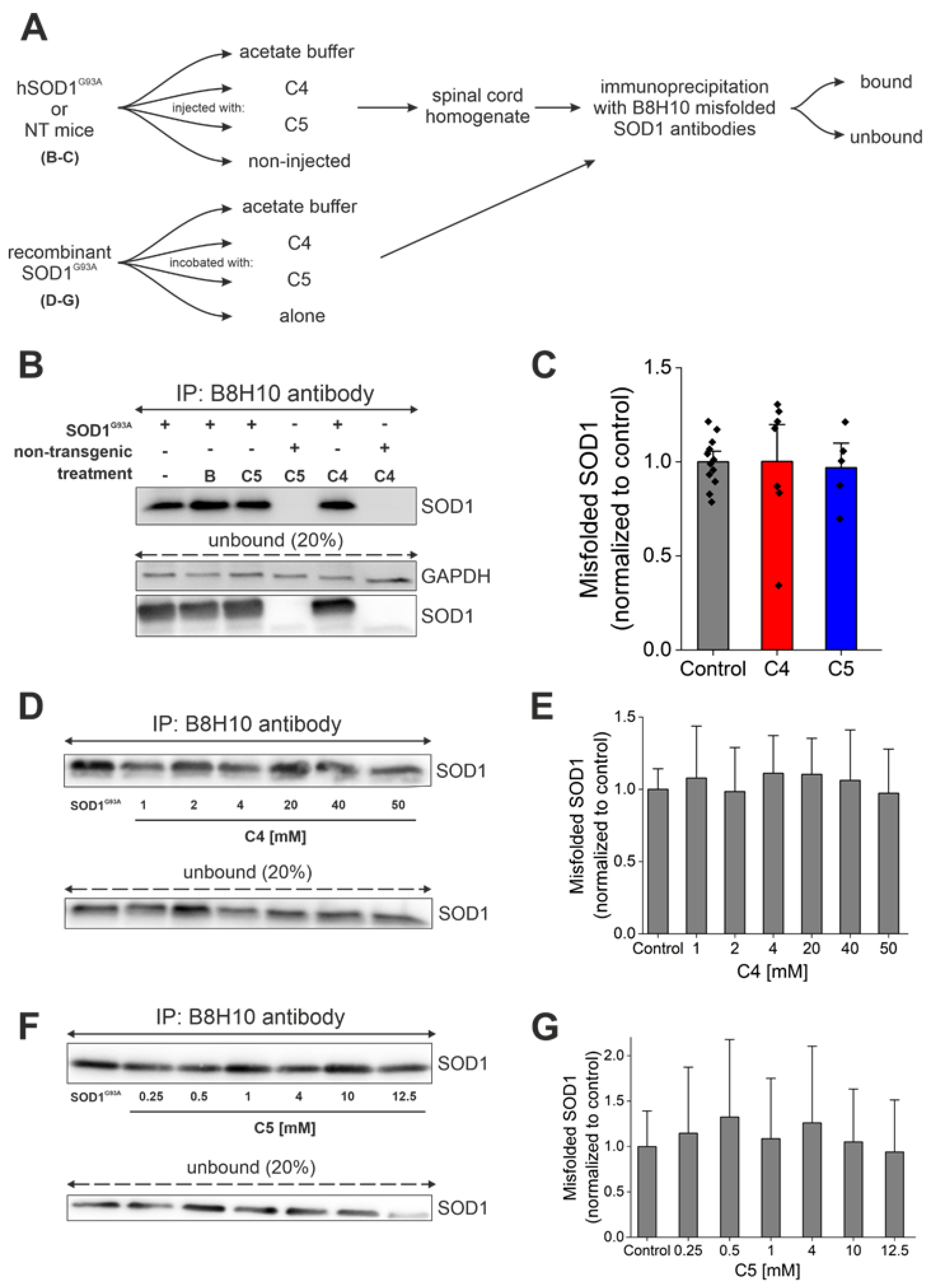
2.4. Daily Injection of C4 or C5 Chemical Chaperones Starting after Disease Onset Failed to Reduce the Accumulation of Misfolded SOD1
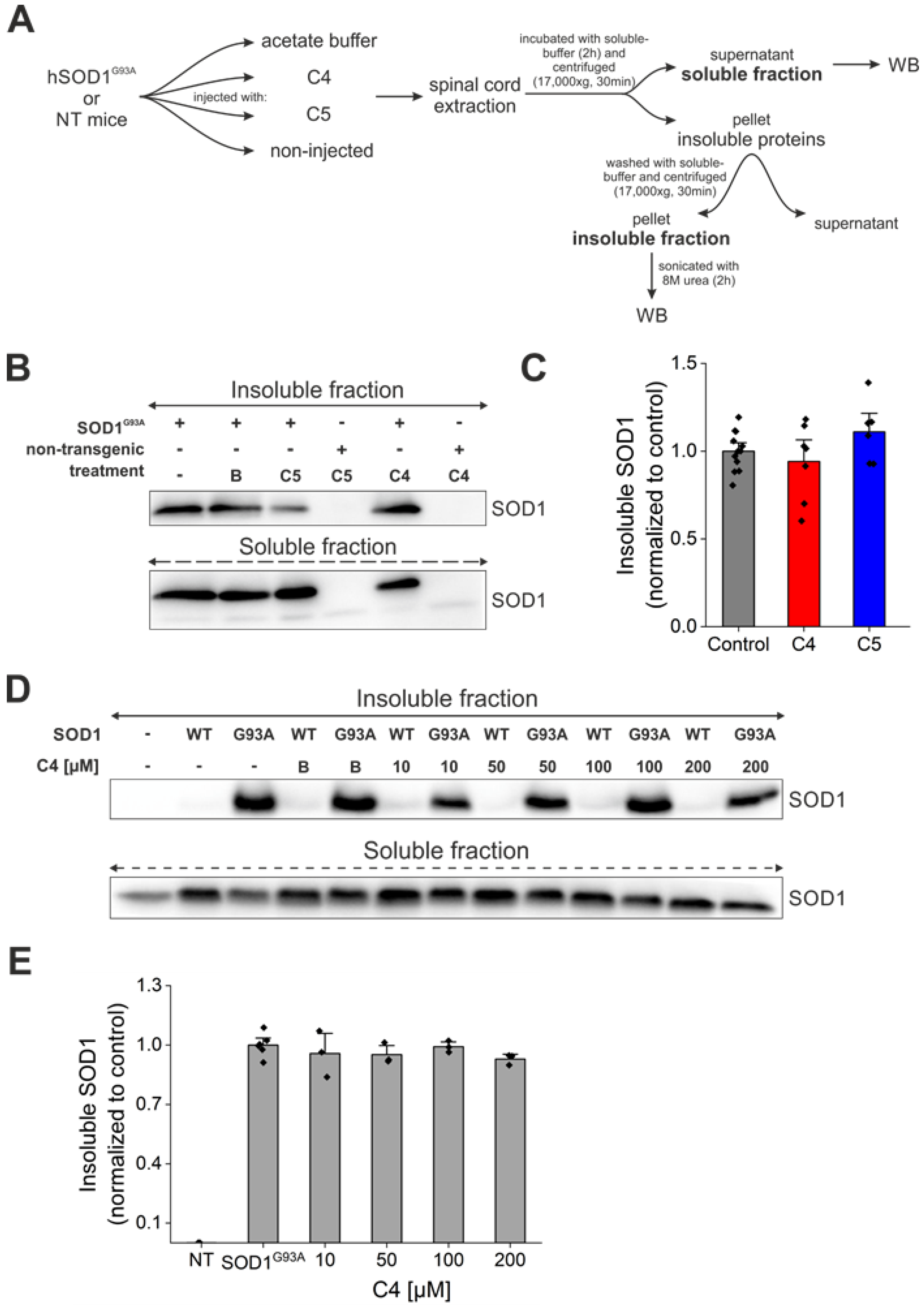
2.5. Daily Injection of C4 or C5 Chemical Chaperones Following Disease Onset Failed to Reduce Total Aggregate Formation of Mutant SOD1
2.6. High Levels of C4 and C5 Reached the Brain and Spinal Cord for a Short Period of Time
3. Discussion
4. Materials and Methods
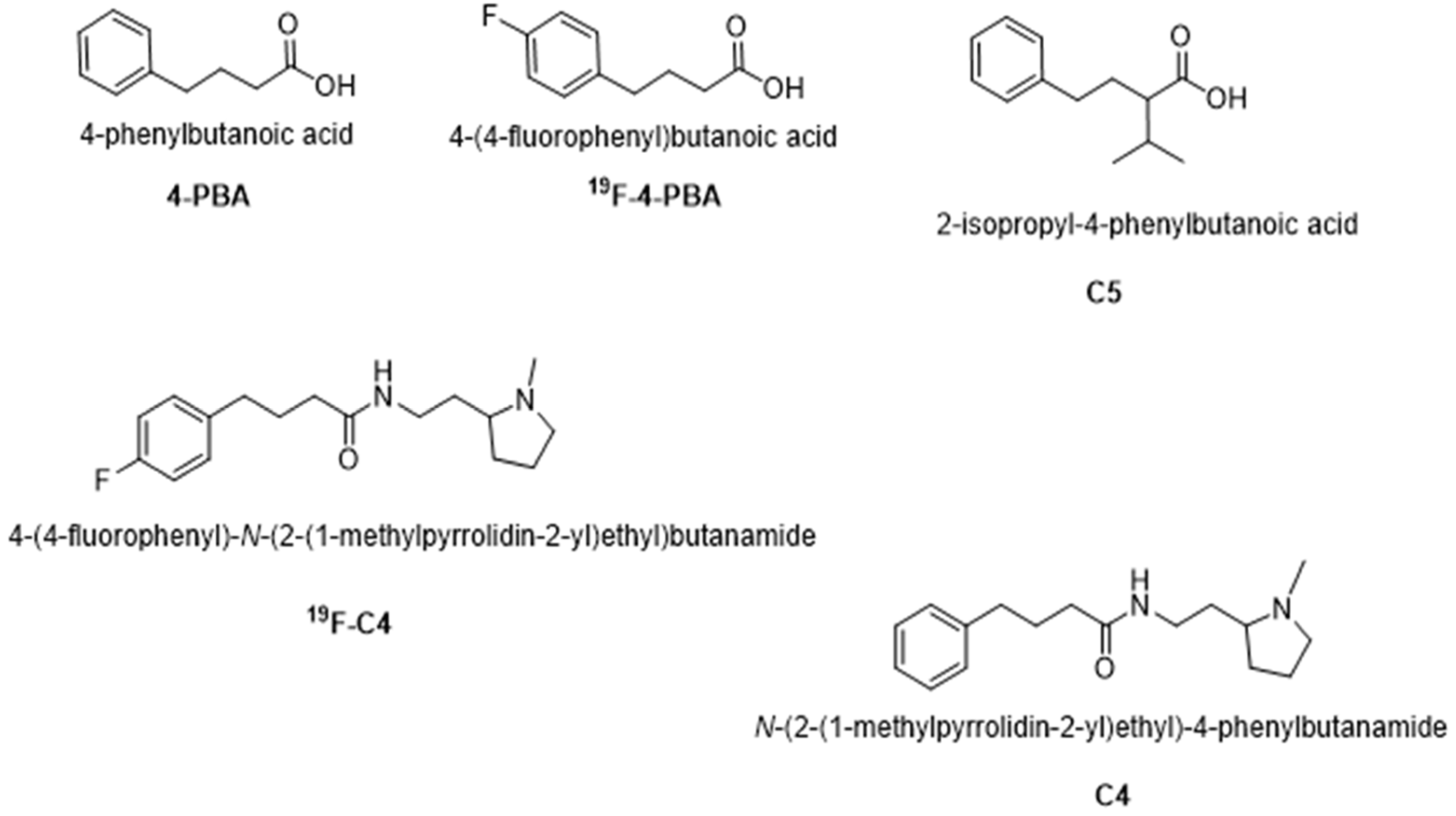
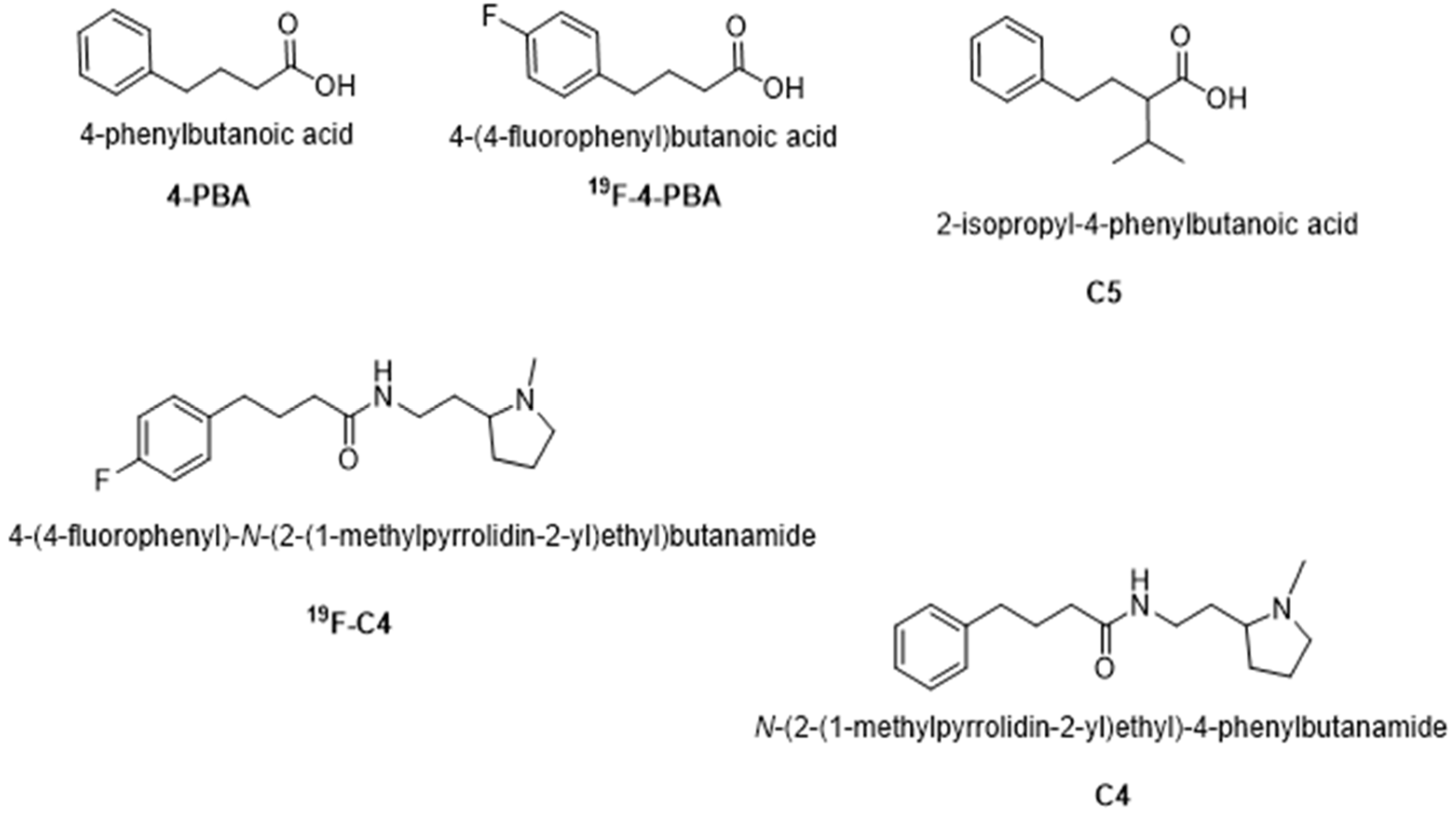
4.1. Chemical Chaperones Synthesis
4.2. Animals and Injection Protocol
4.3. 19F-NMR
4.4. SOD1G93A Protein Purification
4.5. Thioflavin-T (Th-T) Aggregation Assay
4.6. Transmission Electron Microscopy (TEM)
4.7. Cell Culture and Transfection
4.8. Cell Lysis and Protein Extraction
4.9. Tissue Harvesting and Protein Extraction
4.10. Immunoprecipitation (IP) Assay
4.11. Soluble-Insoluble Separation Assay
4.12. Immunoblotting
4.13. Pharmacokinetic (PK) Study
4.14. Immunofluorescence
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Israelson, A.; Ditsworth, D.; Sun, S.; Song, S.; Liang, J.; Hruska-Plochan, M.; McAlonis-Downes, M.; Abu-Hamad, S.; Zoltsman, G.; Shani, T.; et al. Macrophage Migration Inhibitory Factor as a Chaperone Inhibiting Accumulation of Misfolded SOD. Neuron 2015, 86, 218–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, V.; Shani, T.; Katzman, B.; Vyazmensky, M.; Papo, N.; Israelson, A.; Engel, S. Superoxide Dismutase 1 (SOD1)-Derived Peptide Inhibits Amyloid Aggregation of Familial Amyotrophic Lateral Sclerosis SOD1 Mutants. ACS Chem. Neurosci. 2016, 7, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the Mechanisms Involved in Motor Neuron Degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Siddique, T.; Deng, H.-X.; Hung, W.-Y.; Kato, T.; Asayama, K. Intense Superoxide Dismutase-1 Immunoreactivity in Intracytoplasmic Hyaline Inclusions of Familial Amyotrophic Lateral Sclerosis with Posterior Column Involvement. J. Neuropathol. Exp. Neurol. 1996, 55, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Dykes-Hoberg, M.; Cizewski Culotta, V.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological Evidence of Protein Aggregation in Mutant SOD1 Transgenic Mice and in Amyotrophic Lateral Sclerosis Neural Tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Kahn, J.; Leyton-Jaimes, M.F.; Rosenblatt, J.; Israelson, A. Misfolded SOD1 Accumulation and Mitochondrial Association Contribute to the Selective Vulnerability of Motor Neurons in Familial ALS: Correlation to Human Disease. ACS Chem. Neurosci. 2017, 8, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Johnson, J.L.; Agar, N.Y.R.; Agar, J.N. Protein Aggregation and Protein Instability Govern Familial Amyotrophic Lateral Sclerosis Patient Survival. PLoS Biol. 2008, 6, e170. [Google Scholar] [CrossRef] [Green Version]
- Mori, A.; Yamashita, S.; Uchino, K.; Suga, T.; Ikeda, T.; Takamatsu, K.; Ishizaki, M.; Koide, T.; Kimura, E.; Mita, S.; et al. Derlin-1 overexpression ameliorates mutant SOD1-induced endoplasmic reticulum stress by reducing mutant SOD1 accumulation. Neurochem. Int. 2010, 58, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.-X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.-Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magrì, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Boca, M.; Girotto, S.; Martinelli, M.; Valentine, J.S.; Vieru, M. SOD1 and Amyotrophic Lateral Sclerosis: Mutations and Oligomerization. PLoS ONE 2008, 3, e1677. [Google Scholar] [CrossRef] [Green Version]
- Elam, J.S.; Taylor, A.B.; Strange, R.; Antonyuk, S.; Doucette, P.A.; Rodriguez, J.A.; Hasnain, S.S.; Hayward, L.J.; Valentine, J.S.; Yeates, T.; et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Mol. Biol. 2003, 10, 461–467. [Google Scholar] [CrossRef]
- Durer, Z.A.O.; Cohlberg, J.A.; Dinh, P.; Padua, S.; Ehrenclou, K.; Downes, S.; Tan, J.K.; Nakano, Y.; Bowman, C.J.; Hoskins, J.L.; et al. Loss of Metal Ions, Disulfide Reduction and Mutations Related to Familial ALS Promote Formation of Amyloid-Like Aggregates from Superoxide Dismutase. PLoS ONE 2009, 4, e5004. [Google Scholar] [CrossRef] [Green Version]
- Shvil, N.; Banerjee, V.; Zoltsman, G.; Shani, T.; Kahn, J.; Abu-Hamad, S.; Papo, N.; Engel, S.; Bernhagen, J.; Israelson, A. MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: Implications for familial ALS. Cell Death Dis. 2018, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Graffmo, K.; Pakkenberg, B.; Weber, M.; Nielsen, M.; Marklund, S.; Brännström, T.; Andersen, P.M. Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [Green Version]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant Independent from Wild-Type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Xu, G.; Gonzales, V.; Coonfield, M.; Fromholt, D.; Copeland, N.G.; Jenkins, N.A.; Borchelt, D.R. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol. Dis. 2002, 10, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalmar, B.; Greensmith, L. Cellular Chaperones As Therapeutic Targets in ALS to Restore Protein Homeostasis and Improve Cellular Function. Front. Mol. Neurosci. 2017, 10, 251. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.M.; Valentão, P.; Andrade, P.B. Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef] [Green Version]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Ms, N.K.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2020, 63, 31–39. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, J.C.; Pettan-Brewer, C.; Ladiges, W.C. Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 2011, 10, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Browne, S.E.; Choi, D.-K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective Effects of Phenylbutyrate in the N171-82Q Transgenic Mouse Model of Huntington’s Disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Ginsburg, S.; Trobiani, L.; Setini, A.; Favaloro, F.L.; Giorda, E.; Jacob, A.; Hauschner, H.; Levy, L.; Cestra, G.; De Jaco, A.; et al. A Lipophilic 4-Phenylbutyric Acid Derivative That Prevents Aggregation and Retention of Misfolded Proteins. Chemistry 2019, 26, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Ginsburg, S.; Di Salvio, M.; Weitman, M.; Afri, M.; Ribeiro, S.; Ebbinghaus, S.; Cestra, G.; Gruzman, A. Chemical chaperones targeted to the endoplasmic reticulum (ER) and lysosome prevented neurodegeneration in a C9orf72 repeat expansion drosophila amyotrophic lateral sclerosis (ALS) model. Pharmacol. Rep. 2021, 73, 536–550. [Google Scholar] [CrossRef]
- Lee, N.-Y.; Kang, Y.-S. In Vivo and In Vitro Evidence for Brain Uptake of 4-Phenylbutyrate by the Monocarboxylate Transporter 1 (MCT1). Pharm. Res. 2016, 33, 1711–1722. [Google Scholar] [CrossRef]
- Chen, H.; Kankel, M.W.; Su, S.C.; Han, S.W.S.; Ofengeim, D. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018, 25, 648–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Harten, A.C.; Phatnani, H.; Przedborski, S. Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Leyton-Jaimes, M.F.; Benaim, C.; Abu-Hamad, S.; Kahn, J.; Guetta, A.; Bucala, R.; Israelson, A. Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2016, 113, 10198–10203. [Google Scholar] [CrossRef] [Green Version]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc. Natl. Acad. Sci. USA 2019, 116, 14755–14760. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, N.K.; Srivastava, A.; Katyal, N.; Jain, N.; Khan, M.A.I.; Kundu, B.; Deep, S. Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim. Biophys. Acta 2015, 1854, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef]
- Carija, A.; Navarro, S.; de Groot, N.S.; Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 2017, 12, 699–711. [Google Scholar] [CrossRef]
- Stefani, M. Protein misfolding and aggregation: New examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Monsellier, E.; Ramazzotti, M.; Taddei, N.; Chiti, F. Aggregation Propensity of the Human Proteome. PLoS Comput. Biol. 2008, 4, e1000199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassler, J.S.; Skuodas, S.; Weeks, D.L.; Phillips, B.T. Protein Aggregation and Disaggregation in Cells and Development. J. Mol. Biol. 2021, 433, 167215. [Google Scholar] [CrossRef]
- Saad, S.; Cereghetti, G.; Feng, Y.; Picotti, P.; Peter, M.; DeChant, R. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat. Cell Biol. 2017, 19, 1202–1213. [Google Scholar] [CrossRef]
- Hnath, B.; Dokholyan, N.V. Toxic SOD1 trimers are off-pathway in the formation of amyloid-like fibrils in ALS. Biophys. J. 2022, 121, 2084–2095. [Google Scholar] [CrossRef]
- Zhu, C.; Beck, M.V.; Griffith, J.D.; Deshmukh, M.; Dokholyan, N.V. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 4661–4665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef]
- Del Signore, S.J.; Amante, D.; Kim, J.; Stack, E.C.; Goodrich, S.; Cormier, K.; Smith, K.; Cudkowicz, M.E.; Ferrante, R.J. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph. Lateral Scler. 2009, 10, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2018, 39, 733–748. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V.; The ALS/Riluzole Study Group. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Takei, K.; Watanabe, K.; Yuki, S.; Akimoto, M.; Sakata, T.; Palumbo, J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, M.K. Riluzole But Not Melatonin Ameliorates Acute Motor Neuron Degeneration and Moderately Inhibits SOD1-Mediated Excitotoxicity Induced Disrupted Mitochondrial Ca2+ Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2017, 10, 295. [Google Scholar] [CrossRef] [Green Version]
- Bellingham, M.C. A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis: What have we Learned in the Last Decade? CNS Neurosci. Ther. 2011, 17, 4–31. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Hogg, M.; Halang, L.; Woods, I.; Coughlan, K.S.; Prehn, J.H.M. Riluzole does not improve lifespan or motor function in three ALS mouse models. Amyotroph. Lateral Scler. Front. Degener. 2017, 19, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 2006, 7, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Nomura, E.; Shang, J.; Feng, T.; Huang, Y.; Liu, X.; Shi, X.; Nakano, Y.; Hishikawa, N.; Sato, K.; et al. Enhanced oxidative stress and the treatment by edaravone in mice model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2018, 97, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Deacon, R.M. Measuring the Strength of Mice. J. Vis. Exp. 2013, 76, e2610. [Google Scholar] [CrossRef]
- Hatzipetros, T.; Kidd, J.D.; Moreno, A.J.; Thompson, K.; Gill, A.; Vieira, F.G. A Quick Phenotypic Neurological Scoring System for Evaluating Disease Progression in the SOD1-G93A Mouse Model of ALS. J. Vis. Exp. 2015, 104, e53257. [Google Scholar] [CrossRef] [Green Version]
- Bakavayev, S.; Chetrit, N.; Zvagelsky, T.; Mansour, R.; Vyazmensky, M.; Barak, Z.; Israelson, A.; Engel, S. Cu/Zn-superoxide dismutase and wild-type like fALS SOD1 mutants produce cytotoxic quantities of H2O2 via cysteine-dependent redox short-circuit. Sci. Rep. 2019, 9, 10826. [Google Scholar] [CrossRef]
- Sheffield, P.; Garrard, S.; Derewenda, Z. Overcoming Expression and Purification Problems of RhoGDI Using a Family of “Parallel” Expression Vectors. Protein Expr. Purif. 1999, 15, 34–39. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfahel, L.; Argueti-Ostrovsky, S.; Barel, S.; Ali Saleh, M.; Kahn, J.; Azoulay-Ginsburg, S.; Rothstein, A.; Ebbinghaus, S.; Gruzman, A.; Israelson, A. 4-Phenylbutyric Acid (4-PBA) Derivatives Prevent SOD1 Amyloid Aggregation In Vitro with No Effect on Disease Progression in SOD1-ALS Mice. Int. J. Mol. Sci. 2022, 23, 9403. https://doi.org/10.3390/ijms23169403
Alfahel L, Argueti-Ostrovsky S, Barel S, Ali Saleh M, Kahn J, Azoulay-Ginsburg S, Rothstein A, Ebbinghaus S, Gruzman A, Israelson A. 4-Phenylbutyric Acid (4-PBA) Derivatives Prevent SOD1 Amyloid Aggregation In Vitro with No Effect on Disease Progression in SOD1-ALS Mice. International Journal of Molecular Sciences. 2022; 23(16):9403. https://doi.org/10.3390/ijms23169403
Chicago/Turabian StyleAlfahel, Leenor, Shirel Argueti-Ostrovsky, Shir Barel, Mahmood Ali Saleh, Joy Kahn, Salome Azoulay-Ginsburg, Ayelet Rothstein, Simon Ebbinghaus, Arie Gruzman, and Adrian Israelson. 2022. "4-Phenylbutyric Acid (4-PBA) Derivatives Prevent SOD1 Amyloid Aggregation In Vitro with No Effect on Disease Progression in SOD1-ALS Mice" International Journal of Molecular Sciences 23, no. 16: 9403. https://doi.org/10.3390/ijms23169403
APA StyleAlfahel, L., Argueti-Ostrovsky, S., Barel, S., Ali Saleh, M., Kahn, J., Azoulay-Ginsburg, S., Rothstein, A., Ebbinghaus, S., Gruzman, A., & Israelson, A. (2022). 4-Phenylbutyric Acid (4-PBA) Derivatives Prevent SOD1 Amyloid Aggregation In Vitro with No Effect on Disease Progression in SOD1-ALS Mice. International Journal of Molecular Sciences, 23(16), 9403. https://doi.org/10.3390/ijms23169403