
Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics?


{kind=link}
{kind=link}
Abstract
1. Amyotrophic Lateral Sclerosis
1.1. Epidemiology
1.2. Genetics of ALS
1.3. Clinical Presentation
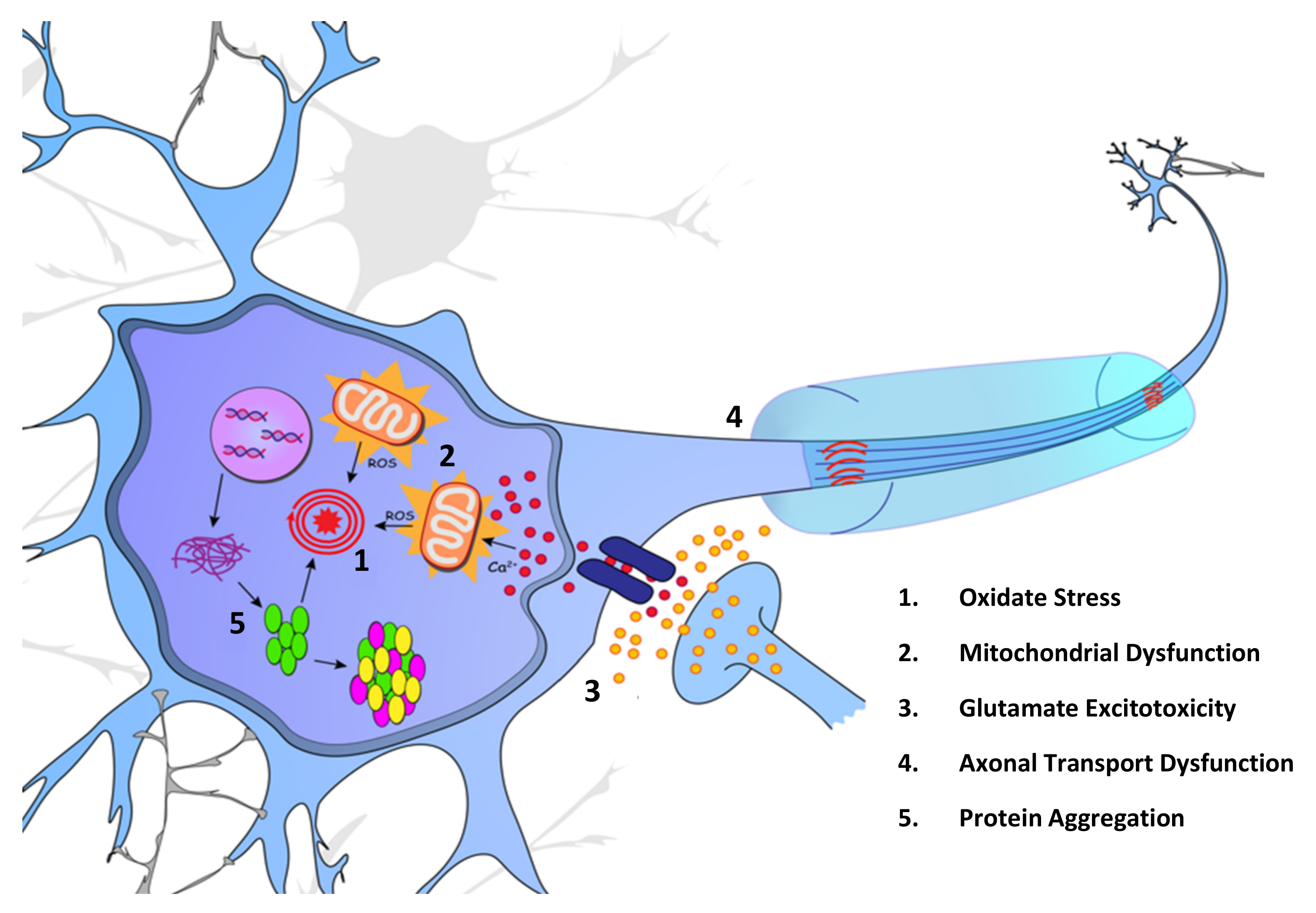
2. Molecular Mechanisms
2.1. Oxidative Stress and Mitochondrial Dysfunction
2.2. Glutamate Excitotoxicity
2.3. Dysfunction of Axonal Transport
2.4. Protein Misfolding and Aggregation
3. Alpha-Synuclein
3.1. Regulatory and Deregulatory Events
3.2. Genetics of Alpha-Synuclein
3.3. Environmental Effectors of Synucleinopathy Pathology
4. Is Amyotrophic Lateral Sclerosis a Synucleinopathy?
Alpha-Synuclein in the Motor Neuron
5. Current Treatment
5.1. Current Therapeutic Options for ALS
5.2. Pipeline Development for Antisense Therapies in ALS
5.3. Current Development and the Future of αSyn-Targeted Therapies
5.3.1. Reducing Uptake of Extracellular Alpha-Synuclein
5.3.2. Increasing Extracellular Alpha-Synuclein Degradation
5.3.3. Promoting Degradation of Intracellular Alpha-Synuclein Aggregates
5.3.4. Inhibiting Alpha-Synuclein Aggregation
5.3.5. Reducing Alpha-Synuclein Production
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Cappello, V.; Francolini, M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals, C. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chio, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Kukharsky, M.S.; Skvortsova, V.I.; Bachurin, S.O.; Buchman, V.L. In a search for efficient treatment for amyotrophic lateral sclerosis: Old drugs for new approaches. Med. Res. Rev. 2021, 41, 2804–2822. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef]
- García, J.; Bustos, R. The genetic diagnosis of neurodegenerative diseases and therapeutic perspectives. Brain Sci. 2018, 8, 222. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Theunissen, F.; Flynn, L.L.; Anderton, R.S.; Mastaglia, F.; Pytte, J.; Jiang, L.; Hodgetts, S.; Burns, D.K.; Saunders, A.; Fletcher, S.; et al. Structural variants may be a source of missing heritability in sALS. Front. Neurosci. 2020, 14, 47. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Dharmadasa, T.; Henderson, R.D.; Talman, P.S.; Macdonell, R.A.; Mathers, S.; Schultz, D.W.; Needham, M.; Zoing, M.; Vucic, S.; Kiernan, M.C. Motor neurone disease: Progress and challenges. Med. J. Aust. 2017, 206, 357–362. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Visscher, P.M. Motor neuron disease: Common genetic variants and the heritability of ALS. Nat. Rev. Neurol. 2014, 10, 549–550. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef]
- Roses, A. Polyallelic structural variants can provide accurate, highly informative genetic markers focused on diagnosis and therapeutic targets: Accuracy vs. Precision. Clin. Pharm. Therap. 2016, 99, 169–171. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Mis, M.S.C.; Brajkovic, S.; Tafuri, F.; Bresolin, N.; Comi, G.P.; Corti, S. Development of therapeutics for C9orf72 ALS/FTD-related disorders. Mol. Neurobiol. 2017, 54, 4466–4476. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H. Amyotrophic lateral sclerosis: An update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Kandler, K.; Witzel, S.; Eder, K.; Rothenbacher, D.; Nagel, G.; Peter, R.S.; Schuster, J.; Dorst, J.; Rosenbohm, A.; Ludolph, A.C. Phenotyping of the thoracic-onset variant of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Makkonen, T.; Ruottinen, H.; Puhto, R.; Helminen, M.; Palmio, J. Speech deterioration in amyotrophic lateral sclerosis (ALS) after manifestation of bulbar symptoms. Int. J. Lang Commun. Disord. 2018, 53, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Girish, K. Dysarthria and dysphagia in amyotrophic lateral sclerosis: A case study. J. Neurosci. Res. 2020, 10, 12–19. [Google Scholar]
- Perry, B.J.; Martino, R.; Yunusova, Y.; Plowman, E.K.; Green, J.R. Lingual and jaw kinematic abnormalities precede speech and swallowing impairments in ALS. Dysphagia 2018, 33, 840–847. [Google Scholar] [CrossRef]
- Hermann, W.; Langner, S.; Freigang, M.; Fischer, S.; Storch, A.; Günther, R.; Hermann, A. Affection of respiratory muscles in ALS and SMA. J. Clin. Med. 2022, 11, 1163. [Google Scholar] [CrossRef]
- Aoun, S.; Birks, C.; Hogden, A.; Mathers, S. Public policy in MND care: The australian perspective. In Public Policy in ALS/MND Care: An International Perspective; Blank, R.H., Kurent, J.E., Oliver, D., Eds.; Springer Singapore: Singapore, 2021; pp. 29–49. ISBN 978-981-15-5840-5. [Google Scholar]
- Tülek, Z.; Özakgül, A.; Alankaya, N.; Dik, A.; Kaya, A.; Ünalan, P.C.; Özaydin, A.N.; İdrisoğlu, H.A. Care burden and related factors among informal caregivers of patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2022, 1–8. [Google Scholar] [CrossRef]
- Chowdhury, A.; Mukherjee, A.; Sinharoy, U.; Pandit, A.; Biswas, A. Non-motor features of amyotrophic lateral sclerosis: A clinic-based study. Ann. Indian Acad. Neurol. 2021, 24, 745–753. [Google Scholar] [CrossRef]
- Crockford, C.; Newton, J.; Lonergan, K.; Chiwera, T.; Booth, T.; Chandran, S.; Colville, S.; Heverin, M.; Mays, I.; Pal, S.; et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018, 91, e1370–e1380. [Google Scholar] [CrossRef]
- Cavaleri, F. Review of amyotrophic lateral sclerosis, Parkinson’s and Alzheimer’s diseases helps further define pathology of the novel paradigm for Alzheimer’s with heavy metals as primary disease cause. Med. Hypotheses 2015, 85, 779–790. [Google Scholar] [CrossRef]
- Ferrari, R.; Kapogiannis, D.; D. Huey, E.; Momeni, P. FTD and ALS: A tale of two diseases. Curr. Alzheimer Res. 2011, 8, 273–294. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef]
- Richards, D.; Morren, J.A.; Pioro, E.P. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J. Neurol. Sci. 2020, 417, 117054. [Google Scholar] [CrossRef]
- Johnsen, B.; Pugdahl, K.; Fuglsang-Frederiksen, A.; Kollewe, K.; Paracka, L.; Dengler, R.; Camdessanché, J.; Nix, W.; Liguori, R.; Schofield, I. Diagnostic criteria for amyotrophic lateral sclerosis: A multicentre study of inter-rater variation and sensitivity. Clin. Neurophys. 2019, 130, 307–314. [Google Scholar] [CrossRef]
- Hannaford, A.; Pavey, N.; van den Bos, M.; Geevasinga, N.; Menon, P.; Shefner, J.M.; Kiernan, M.C.; Vucic, S. Diagnostic utility of gold coast criteria in amyotrophic lateral sclerosis. Ann. Neurol. 2021, 89, 979–986. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Shefner, J.M.; Kaji, R.; Burke, D. Amyotrophic lateral sclerosis: A new diagnostic paradigm. J. Neurol. Neurosurg. Psychiatry 2020, 91, 903–904. [Google Scholar] [CrossRef]
- Shen, D.; Yang, X.; Wang, Y.; He, D.; Sun, X.; Cai, Z.; Li, J.; Liu, M.; Cui, L. The Gold Coast criteria increases the diagnostic sensitivity for amyotrophic lateral sclerosis in a Chinese population. Transl. Neurodegener. 2021, 10, 28. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Proudfoot, M.; Jones, A.; Talbot, K.; Al-Chalabi, A.; Turner, M.R. The ALSFRS as an outcome measure in therapeutic trials and its relationship to symptom onset. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 414–425. [Google Scholar] [CrossRef]
- Wong, C.; Stavrou, M.; Elliott, E.; Gregory, J.M.; Leigh, N.; Pinto, A.A.; Williams, T.L.; Chataway, J.; Swingler, R.; Parmar, M.K.B.; et al. Clinical trials in amyotrophic lateral sclerosis: A systematic review and perspective. Brain Commun. 2021, 3, fcab242. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Benatar, M. Ensuring continued progress in biomarkers for amyotrophic lateral sclerosis. Muscle Nerve 2015, 51, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Traynor, B.; Zhang, H.; Shefner, J.; Schoenfeld, D.; Cudkowicz, M. Functional outcome measures as clinical trial endpoints in ALS. Neurology 2004, 63, 1933–1935. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Kaur, I.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Aleya, L.; Bungau, S. Connecting the dots between mitochondrial dysfunction and Parkinson’s disorder: Focus mitochondria-targeting therapeutic paradigm in mitigating the disease severity. Environ. Sci. Pollut. Res. 2021, 28, 37060–37081. [Google Scholar] [CrossRef]
- Niu, H.; Chen, X.; Fu, X.; Zhang, J.; Li, G.; Wang, Y.; Song, J.; Ma, X.; Hu, C.; Xu, X. SOD1G93A Induces a Unique PSAP-Dependent Mitochondrial Apoptosis Pathway via Bax–Bak Interaction. Biocell 2021, 45, 963. [Google Scholar] [CrossRef]
- Abati, E.; Bresolin, N.; Comi, G.; Corti, S. Silence superoxide dismutase 1 (SOD1): A promising therapeutic target for amyotrophic lateral sclerosis (ALS). Expert Opin. Ther. Targets 2020, 24, 295–310. [Google Scholar] [CrossRef]
- Forsberg, K.; Graffmo, K.; Pakkenberg, B.; Weber, M.; Nielsen, M.; Marklund, S.; Brännström, T.; Andersen, P.M. Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 861–869. [Google Scholar] [CrossRef]
- Trist, B.G.; Genoud, S.; Roudeau, S.; Rookyard, A.; Abdeen, A.; Cottam, V.; Hare, D.J.; White, M.; Altvater, J.; Fifita, J.A. Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord. Brain 2022, awac165. [Google Scholar] [CrossRef]
- Ince, P.G.; Tomkins, J.; Slade, J.Y.; Thatcher, N.M.; Shaw, P.J. Amyotrophic lateral sclerosis associated with genetic abnormalities in the gene encoding Cu/Zn superoxide dismutase: Molecular pathology of five new cases, and comparison with previous reports and 73 sporadic cases of ALS. J. Neuropathol. Exp. Neurol. 1998, 57, 895–904. [Google Scholar] [CrossRef]
- Dangoumau, A.; Verschueren, A.; Hammouche, E.; Papon, M.-A.; Blasco, H.; Cherpi-Antar, C.; Pouget, J.; Corcia, P.; Andres, C.R.; Vourc’h, P. A novel SOD1 mutation p. V31A identified with a slowly progressive form of amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 266.e1–266.e4. [Google Scholar] [CrossRef]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic lateral sclerosis (ALS): Stressed by dysfunctional mitochondria-endoplasmic reticulum contacts (MERCs). Cells 2021, 10, 1789. [Google Scholar] [CrossRef]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.-L.G. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef]
- Fontana, A.C. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef]
- Green, J.L.; Dos Santos, W.F.; Fontana, A.C.K. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem. Pharm. 2021, 193, 114786. [Google Scholar] [CrossRef]
- Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory amino acid transporters (EAATs): Glutamate transport and beyond. Int. J. Mol. Sci. 2019, 20, 5674. [Google Scholar] [CrossRef]
- Sundaram, R.S.; Gowtham, L.; Nayak, B.S. The role of excitatory neurotransmitter glutamate in brain physiology and pathology. Asian J. Pharm. Clin. Res. 2012, 5, 1–7. [Google Scholar]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Theunissen, F.; West, P.K.; Brennan, S.; Petrović, B.; Hooshmand, K.; Akkari, P.A.; Keon, M.; Guennewig, B. New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl. Neurodegener. 2021, 10, 46. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar] [CrossRef]
- Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 2012, 13, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, S.E. Protein misfolding toxicity and inclusion formation in cellular models of neurodegeneration. Transl. Neruodegener. 2021, 6, 7775. [Google Scholar] [CrossRef]
- Yang, X.; Ji, Y.; Wang, W.; Zhang, L.; Chen, Z.; Yu, M.; Shen, Y.; Ding, F.; Gu, X.; Sun, H. Amyotrophic Lateral Sclerosis: Molecular Mechanisms, Biomarkers, and Therapeutic Strategies. Antioxidants 2021, 10, 1012. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Yamamoto, A. Macroautophagy of aggregation-prone proteins in neurodegenerative disease. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2015; Volume 7, pp. 117–137. [Google Scholar]
- Gosset, P.; Camu, W.; Raoul, C.; Mezghrani, A. Prionoids in amyotrophic lateral sclerosis. Brain Commun. 2022, 4, fcac145. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef]
- Hasegawa, T.; Sugeno, N.; Kikuchi, A.; Baba, T.; Aoki, M. Membrane trafficking illuminates a path to Parkinson’s disease. Tohoku J. Exp. Med. 2017, 242, 63–76. [Google Scholar] [CrossRef][Green Version]
- Sugeno, N.; Jäckel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 2016, 6, 36328. [Google Scholar] [CrossRef]
- Bellani, S.; Sousa, V.L.; Ronzitti, G.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. The regulation of synaptic function by α-synuclein. Commun. Integr. Biol. 2010, 3, 106–109. [Google Scholar] [CrossRef]
- Parihar, A.; Parihar, P.; Solanki, I.; Parihar, M.S. Alpha synuclein and Parkinson’s disease. In Pathology, Prevention and Therapeutics of Neurodegenerative Disease, 1st ed.; Singh, S., Joshi, N., Eds.; Springer: Singapore, 2019; pp. 1–14. ISBN 978-981-13-0944-1. [Google Scholar]
- Marti, M.J.; Tolosa, E.; Campdelacreu, J. Clinical overview of the synucleinopathies. Mov. Disord. 2003, 18 (Suppl. 6), S21–S27. [Google Scholar] [CrossRef]
- Woerman, A.L.; Watts, J.C.; Aoyagi, A.; Giles, K.; Middleton, L.T.; Prusiner, S.B. α-Synuclein: Multiple System Atrophy Prions. Cold Spring Harb. Perspect. Med. 2018, 12, a024588. [Google Scholar] [CrossRef]
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: Apha-synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 28. [Google Scholar] [CrossRef]
- Barba, L.; Paolini Paoletti, F.; Bellomo, G.; Gaetani, L.; Halbgebauer, S.; Oeckl, P.; Otto, M.; Parnetti, L. Alpha and beta synucleins: From pathophysiology to clinical application as biomarkers. Mov. Disord. 2022, 37, 669–683. [Google Scholar] [CrossRef]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Prasad, S.; Katta, M.R.; Abhishek, S.; Sridhar, R.; Valisekka, S.S.; Hameed, M.; Kaur, J.; Walia, N. Recent advances in Lewy body dementia: A comprehensive review. Dis. Mon. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol 1998, 152, 879–884. [Google Scholar] [PubMed]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.P.; Thomas, A.; et al. Dementia with Lewy bodies: An update and outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Olrichs, N.K.; Gadella, B.M.; Kaloyanova, D.V.; Helms, J.B. Regulation of functional protein aggregation by multiple factors: Implications for the amyloidogenic behavior of the cap superfamily proteins. Int. J. Mol. Sci. 2020, 21, 6530. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Gubellini, P.; Picconi, B.; Di Filippo, M.; Calabresi, P. Downstream mechanisms triggered by mitochondrial dysfunction in the basal ganglia: From experimental models to neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 151–161. [Google Scholar] [CrossRef]
- Helferich, A.M.; Ruf, W.P.; Grozdanov, V.; Freischmidt, A.; Feiler, M.S.; Zondler, L.; Ludolph, A.C.; McLean, P.J.; Weishaupt, J.H.; Danzer, K.M. α-synuclein interacts with SOD1 and promotes its oligomerization. Mol. Neurodegener. 2015, 10, 66. [Google Scholar] [CrossRef]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef]
- Trist, B.G.; Fifita, J.A.; Freckleton, S.E.; Hare, D.J.; Lewis, S.J.G.; Halliday, G.M.; Blair, I.P.; Double, K.L. Accumulation of dysfunctional SOD1 protein in Parkinson’s disease is not associated with mutations in the SOD1 gene. Acta Neuropathol. 2018, 135, 155–156. [Google Scholar] [CrossRef]
- Golde, T.E.; Miller, V.M. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res. Ther. 2009, 1, 5. [Google Scholar] [CrossRef]
- Kikuchi, A.; Takeda, A.; Okamura, N.; Tashiro, M.; Hasegawa, T.; Furumoto, S.; Kobayashi, M.; Sugeno, N.; Baba, T.; Miki, Y. In vivo visualization of α-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl) ethenyl]-6-[2-(fluoro) ethoxy] benzoxazole positron emission tomography in multiple system atrophy. Brain 2010, 133, 1772–1778. [Google Scholar] [CrossRef]
- Han, W.; Liu, Y.; Mi, Y.; Zhao, J.; Liu, D.; Tian, Q. Alpha-synuclein (SNCA) polymorphisms and susceptibility to Parkinson’s disease: A meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet 2015, 168B, 123–134. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Ferese, R.; Modugno, N.; Campopiano, R.; Santilli, M.; Zampatti, S.; Giardina, E.; Nardone, A.; Postorivo, D.; Fornai, F.; Novelli, G. Four copies of SNCA responsible for autosomal dominant Parkinson’s disease in two Italian siblings. Parkinson’s Dis. 2015, 2015, 546462. [Google Scholar] [CrossRef]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef]
- Magistrelli, L.; Contaldi, E.; Comi, C. The impact of SNCA variations and its product alpha-synuclein on non-motor features of parkinson’s disease. Life 2021, 11, 804. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Cabin, D.E.; Gispert-Sanchez, S.; Murphy, D.; Auburger, G.; Myers, R.R.; Nussbaum, R.L. Exacerbated synucleinopathy in mice expressing A53T SNCA on a SNCA null background. Neurobiol. Aging 2005, 26, 25–35. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; de Silva, H.R.; Rosi, B.L.; Yamaoka, L.H.; Rimmler, J.B.; Pericak-Vance, M.A.; Roses, A.D.; Chen, X.; Masliah, E.; Deteresa, R. Genetic studies in Alzheimer’s disease with an NACP/α-synuclein polymorphism. Ann. Neurol. 1996, 40, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; De Marchi, F.; Tunesi, S.; Oggioni, G.D.; Carecchio, M.; Magistrelli, L.; Tesei, S.; Riboldazzi, G.; Di Fonzo, A.; Locci, C. The length of SNCA Rep1 microsatellite may influence cognitive evolution in Parkinson’s disease. Front. Neurol. 2018, 9, 213. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M.; Umbach, D.M.; Kamel, F.; Tanner, C.M. Head injury, α-synuclein Rep1 and Parkinson’s disease: A meta-analytic view of gene-environment interaction. Eur. J. Neurol. 2015, 22, e75. [Google Scholar] [CrossRef] [PubMed]
- Linnertz, C.; Lutz, M.W.; Ervin, J.F.; Allen, J.; Miller, N.R.; Welsh-Bohmer, K.A.; Roses, A.D.; Chiba-Falek, O. The genetic contributions of SNCA and LRRK2 genes to Lewy Body pathology in Alzheimer’s disease. Hum. Mol. Genet 2014, 23, 4814–4821. [Google Scholar] [CrossRef]
- Ng, A.S.; Tan, Y.J.; Zhao, Y.; Saffari, S.E.; Lu, Z.; Ng, E.Y.; Ng, S.Y.; Chia, N.S.; Setiawan, F.; Xu, Z. SNCA Rep1 promoter variability influences cognition in Parkinson’s disease. Mov. Disord. 2019, 34, 1232–1236. [Google Scholar] [CrossRef]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef]
- Chiba-Falek, O. Structural variants in SNCA gene and the implication to synucleinopathies. Curr. Opin. Genet. Dev. 2017, 44, 110–116. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurol. 2018, 12, 612. [Google Scholar] [CrossRef]
- Lai, B.; Marion, S.; Teschke, K.; Tsui, J. Occupational and environmental risk factors for Parkinson’s disease. Parkinsonism Relat. Disord. 2002, 8, 297–309. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease—The gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef]
- Innos, J.; Hickey, M.A. Using rotenone to model Parkinson’s disease in mice: A review of the role of pharmacokinetics. Chem. Res. Toxicol. 2021, 34, 1223–1239. [Google Scholar] [CrossRef]
- Mustapha, M.; Taib, C.N.M. MPTP-induced mouse model of Parkinson’s disease: A promising direction for therapeutic strategies. Bosn. J. Basic Med. Sci. 2021, 21, 422. [Google Scholar] [CrossRef]
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 2000, 11, 211–213. [Google Scholar] [CrossRef]
- Rodrigues, L.S.; Targa, A.D.; Noseda, A.C.D.; Aurich, M.F.; Da Cunha, C.; Lima, M.M. Olfactory impairment in the rotenone model of Parkinson’s disease is associated with bulbar dopaminergic D2 activity after REM sleep deprivation. Front. Cell Neurosci. 2014, 8, 383. [Google Scholar] [CrossRef]
- Cagac, A. Farming, well water consumption, rural living, and pesticide exposure in early life as the risk factors for Parkinson disease in Iğdır province. Neurosci. J. 2020, 25, 129–135. [Google Scholar] [CrossRef]
- Kori, R.K.; Singh, M.K.; Jain, A.K.; Yadav, R.S. Neurochemical and behavioral dysfunctions in pesticide exposed farm workers: A clinical outcome. Indian J. Clin. Biochem. 2018, 33, 372–381. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Z. Advances in Parkinson’s disease induced by α-synuclein transmitted through the gut-brain axis. Shengwu Gongcheng Xuebao/Chin. J. Biotechnol. 2022, 38, 2120–2127. [Google Scholar] [CrossRef]
- Esteves, A.R.; Munoz-Pinto, M.F.; Nunes-Costa, D.; Candeias, E.; Silva, D.F.; Magalhães, J.D.; Pereira-Santos, A.R.; Ferreira, I.L.; Alarico, S.; Tiago, I. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut 2021, 1–17. [Google Scholar] [CrossRef]
- Nunes-Costa, D.; Magalhães, J.D.; G-Fernandes, M.; Cardoso, S.M.; Empadinhas, N. Microbial BMAA and the pathway for Parkinson’s disease neurodegeneration. Front. Aging Neurosci. 2020, 12, 26. [Google Scholar] [CrossRef]
- Pierozan, P.; Piras, E.; Brittebo, E.; Karlsson, O. The cyanobacterial neurotoxin β-N-methylamino-l-alanine (BMAA) targets the olfactory bulb region. Arch Toxicol 2020, 94, 2799–2808. [Google Scholar] [CrossRef]
- Kuzuhara, S. Revisit to Kii ALS—The innovated concept of ALS-Parkinsonism-dementia complex, clinicopathological features, epidemiology and etiology. Brain Nerve 2007, 59, 1065–1074. [Google Scholar]
- Kokubo, Y.; Taniguchi, A.; Hasegawa, M.; Hayakawa, Y.; Morimoto, S.; Yoneda, M.; Hirokawa, Y.; Shiraishi, T.; Saito, Y.; Murayama, S. α-Synuclein pathology in the amyotrophic lateral sclerosis/parkinsonism dementia complex in the Kii Peninsula, Japan. J. Neuropathol. Exp. Neurol. 2012, 71, 625–630. [Google Scholar] [CrossRef]
- Duncan, M.W.; Steele, J.C.; Kopin, I.J.; Markey, S.P. 2-Amino-3-(methylamino)-propanoic acid (BMAA) in cycad flour: An unlikely cause of amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Neurology 1990, 40, 767. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.; Kopin, I.; Garruto, R.; Lavine, L.; Markey, S. 2-Amino-3 (methylamino)-propionic acid in cycad-derived foods is an unlikely cause of amyotrophic lateral sclerosis/parkinsonism. Lancet 1988, 332, 631–632. [Google Scholar] [CrossRef]
- Bradley, W.G.; Mash, D.C. Beyond Guam: The cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph. Lateral Scler. 2009, 10, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.A.; Mowrey, D.D.; Dokholyan, N.V. β-Methylamino-L-alanine substitution of serine in SOD1 suggests a direct role in ALS etiology. PLoS Comp. Biol. 2019, 15, e1007225. [Google Scholar] [CrossRef]
- Ghanem, S.S.; Majbour, N.K.; Vaikath, N.N.; Ardah, M.T.; Erskine, D.; Jensen, N.M.; Fayyad, M.; Sudhakaran, I.P.; Vasili, E.; Melachroinou, K. α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. USA 2022, 119, e2109617119. [Google Scholar] [CrossRef]
- Calvo, A.; Chio, A.; Pagani, M.; Cammarosano, S.; Dematteis, F.; Moglia, C.; Solero, L.; Manera, U.; Martone, T.; Brunetti, M.; et al. Parkinsonian traits in amyotrophic lateral sclerosis (ALS): A prospective population-based study. J. Neurol. 2019, 266, 1633–1642. [Google Scholar] [CrossRef]
- Noda, K.; Katayama, S.; Watanabe, C.; Yamamura, Y.; Nakamura, S.; Yonehara, S.; Inai, K. Pure autonomic failure with motor neuron disease: Report of a clinical study and postmortem examination of a patient. J. Neurol. Neurosurg. Psychiatry 1996, 60, 351–352. [Google Scholar] [CrossRef][Green Version]
- Doherty, M.J.; Bird, T.D.; Leverenz, J.B. α-synuclein in motor neuron disease: An immunohistologic study. Acta Neuropathol. 2004, 107, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Dehejia, A.; Harta, G.; Papp, M.I.; Polymeropoulos, M.H.; Brownstein, M.J. Alpha synuclein in neurodegenerative disorders: Murderer or accomplice? Nat. Med. 1998, 4, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Joo, K.M.; Kim, M.J.; Cha, C.I. Immunohistochemical study on the distribution of alpha-synuclein in the central nervous system of transgenic mice expressing a human Cu/Zn superoxide dismutase mutation. Neurosci. Lett. 2003, 342, 151–154. [Google Scholar] [CrossRef]
- Takei, Y.; Oguchi, K.; Koshihara, H.; Hineno, A.; Nakamura, A.; Ohara, S. α-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Hum. Pathol. 2013, 44, 1171–1176. [Google Scholar] [CrossRef]
- Yang, E.J.; Choi, S.-M. α-Synuclein modification in an ALS animal model. Evid. Based Complement. Altern. 2013, 2013, 259381. [Google Scholar] [CrossRef]
- Kim, S.H.; Jung, S.Y.; Lee, K.-W.; Lee, S.H.; Cai, M.; Choi, S.-M.; Yang, E.J. Bee venom effects on ubiquitin proteasome system in hSOD1G85R-expressing NSC34 motor neuron cells. BMC Comp. Altern. Med. 2013, 13, 179. [Google Scholar] [CrossRef]
- Koch, Y.; Helferich, A.M.; Steinacker, P.; Oeckl, P.; Walther, P.; Weishaupt, J.H.; Danzer, K.M.; Otto, M. Aggregated α-Synuclein increases SOD1 oligomerization in a mouse model of amyotrophic lateral sclerosis. Am. J. Clin. Pathol. 2016, 186, 2152–2161. [Google Scholar] [CrossRef]
- Guo, X.Y.; Chen, Y.P.; Song, W.; Zhao, B.; Cao, B.; Wei, Q.Q.; Ou, R.W.; Yang, Y.; Yuan, L.X.; Shang, H.-F. SNCA variants rs2736990 and rs356220 as risk factors for Parkinson’s disease but not for amyotrophic lateral sclerosis and multiple system atrophy in a Chinese population. Neurobiol. Aging 2014, 35, e2881–e2882. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, Q.Q.; Ou, R.; Cao, B.; Chen, X.; Zhao, B.; Guo, X.; Yang, Y.; Chen, K.; Wu, Y.; et al. Genetic variants of SNCA are associated with susceptibility to Parkinson’s disease but not amyotrophic lateral sclerosis or multiple system atrophy in a Chinese population. PLoS ONE 2015, 10, e0133776. [Google Scholar] [CrossRef]
- Vacchiano, V.; Bartoletti-Stella, A.; Rizzo, G.; Avoni, P.; Parchi, P.; Salvi, F.; Liguori, R.; Capellari, S. Frequency of Parkinson’s disease genes and role of PARK2 in amyotrophic lateral sclerosis: An NGS study. Genes 2022, 13, 1306. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Oda, S.; Sano, T.; Nishikawa, N.; Mikasa, M.; Takahashi, Y.; Takao, M. Amyotrophic lateral sclerosis with muscle weakness and dropped head during the course of Parkinson’s disease: An autopsy case. Clin. Neurol. 2021, 61, 373–377. [Google Scholar] [CrossRef]
- Yamada, T.; Itoh, K.; Matsuo, K.; Yamamoto, Y.; Hosokawa, Y.; Koizumi, T.; Shiga, K.; Mizuno, T.; Nakagawa, M.; Fushiki, S. Concomitant alpha-synuclein pathology in an autopsy case of amyotrophic lateral sclerosis presenting with orthostatic hypotension and cardiac arrests. Neuropath 2014, 34, 164–169. [Google Scholar] [CrossRef]
- Tian, T.; Huang, C.; Tong, J.; Yang, M.; Zhou, H.; Xia, X.-G. TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int. J. Biol. Sci. 2011, 7, 234–243. [Google Scholar] [CrossRef]
- Dhakal, S.; Wyant, C.E.; George, H.E.; Morgan, S.E.; Rangachari, V. Prion-like C-terminal domain of TDP-43 and α-Synuclein interact synergistically to generate neurotoxic hybrid fibrils. J. Mol. Biol. 2021, 433, 166953. [Google Scholar] [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.B. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef]
- Bridi, J.C.; Hirth, F. Mechanisms of α-synuclein induced synaptopathy in Parkinson’s disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef]
- Lin, M.; Mackie, P.M.; Shaerzadeh, F.; Gamble-George, J.; Miller, D.R.; Martyniuk, C.J.; Khoshbouei, H. In Parkinson’s patient-derived dopamine neurons, the triplication of α-synuclein locus induces distinctive firing pattern by impeding D2 receptor autoinhibition. Acta Neuropathol. Commun. 2021, 9, 107. [Google Scholar] [CrossRef]
- Courte, J.; Bousset, L.; Boxberg, Y.V.; Villard, C.; Melki, R.; Peyrin, J.-M. The expression level of alpha-synuclein in different neuronal populations is the primary determinant of its prion-like seeding. Sci. Rep. 2020, 10, 4895. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]
- Sommer, B.; Barbieri, S.; Hofele, K.; Wiederhold, K.-H.; Probst, A.; Mistl, C.; Danner, S.; Kauffmann, S.; Spooren, W.; Tolnay, M. Mouse models of α-synucleinopathy and Lewy pathology. Exp. Gerontol. 2000, 35, 1389–1403. [Google Scholar] [CrossRef]
- Van der Putten, H.; Wiederhold, K.-H.; Probst, A.; Barbieri, S.; Mistl, C.; Danner, S.; Kauffmann, S.; Hofele, K.; Spooren, W.P.; Ruegg, M.A. Neuropathology in mice expressing human α-synuclein. J. Neurosci. 2000, 20, 6021–6029. [Google Scholar] [CrossRef]
- Feng, G.-Y.; Liu, J.; Wang, Y.-C.; Wang, Z.-Y.; Hu, Y.; Xia, Q.-J.; Xu, Y.; Shang, F.-F.; Chen, M.-R.; Wang, F. Effects of alpha-synuclein on primary spinal cord neurons associated with apoptosis and CNTF expression. Cell. Mol. Neruobiol. 2017, 37, 817–829. [Google Scholar] [CrossRef]
- Sumikura, H.; Takao, M.; Hatsuta, H.; Ito, S.; Nakano, Y.; Uchino, A.; Nogami, A.; Saito, Y.; Mochizuki, H.; Murayama, S. Distribution of α-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol. Commun. 2015, 3, 57. [Google Scholar] [CrossRef]
- Caviness, J.N.; Smith, B.E.; Stevens, J.C.; Adler, C.H.; Caselli, R.J.; Reiners, C.A.; Hentz, J.G.; Muenter, M.D. Motor unit changes in sporadic idiopathic Parkinson’s disease. Mov. Disord. 2000, 15, 238–243. [Google Scholar] [CrossRef]
- Caviness, J.; Smith, B.; Stevens, J.C.; Adler, C.; Caselli, R.; Hentz, J.; Manfred, M.; Muenter, D. Motor unit number estimates in idiopathic Parkinson’s disease. Parkinsonism Relat. Disord. 2002, 8, 161–164. [Google Scholar] [CrossRef]
- Vivacqua, G.; Biagioni, F.; Yu, S.; Casini, A.; Bucci, D.; D’Este, L.; Fornai, F. Loss of spinal motor neurons and alteration of alpha-synuclein immunostaining in MPTP induced Parkinsonism in mice. J. Chem. Neuroanat. 2012, 44, 76–85. [Google Scholar] [CrossRef]
- Mendritzki, S.; Schmidt, S.; Sczepan, T.; Zhu, X.-R.; Segelcke, D.; Lübbert, H. Spinal cord pathology in alpha-synuclein transgenic mice. Parkinson’s Dis. 2010, 2010, 375462. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Shults, C.W.; Rockenstein, E.; Crews, L.; Adame, A.; Mante, M.; Larrea, G.; Hashimoto, M.; Song, D.; Iwatsubo, T.; Tsuboi, K. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human α-synuclein under oligodendrocyte promoter: Implications for multiple system atrophy. J. Neurosci. 2005, 25, 10689–10699. [Google Scholar] [CrossRef] [PubMed]
- Acsadi, G.; Li, X.; Murphy, K.J.; Swoboda, K.J.; Parker, G.C. Alpha-synuclein loss in spinal muscular atrophy. J. Mol. Neurosci. 2011, 43, 275–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kline, R.A.; Kaifer, K.A.; Osman, E.Y.; Carella, F.; Tiberi, A.; Ross, J.; Pennetta, G.; Lorson, C.L.; Murray, L.M. Comparison of independent screens on differentially vulnerable motor neurons reveals alpha-synuclein as a common modifier in motor neuron diseases. PLoS Genet. 2017, 13, e1006680. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S. Pain in amyotrophic lateral sclerosis: A narrative review. J. Yeungnam Med. Sci. 2022, 39, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, N.; Radakovic, R.; Boyce, E.; Peryer, G. Prevalence of pain in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 449–458. [Google Scholar] [CrossRef]
- Bedlack, R.S.; Joyce, N.; Carter, G.T.; Paganoni, S.; Karam, C. Complementary and alternative therapies in amyotrophic lateral sclerosis. Neurol. Clin. 2015, 33, 909–936. [Google Scholar] [CrossRef]
- Dalbello-Haas, V.; Florence, J.M.; Krivickas, L.S. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst. Rev. 2014, 5, 2013. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrere, B.; Couratier, P. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef]
- Aridegbe, T.; Kandler, R.; Walters, S.J.; Walsh, T.; Shaw, P.J.; McDermott, C.J. The natural history of motor neuron disease: Assessing the impact of specialist care. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 13–19. [Google Scholar] [CrossRef]
- Miller, R.G.; Jackson, C.E.; Kasarskis, E.J.; England, J.D.; Forshew, D.; Johnston, W.; Kalra, S.; Katz, J.S.; Mitsumoto, H.; Rosenfeld, J.; et al. Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009, 73, 1227–1233. [Google Scholar] [CrossRef]
- Wobst, H.J.; Mack, K.L.; Brown, D.G.; Brandon, N.J.; Shorter, J. The clinical trial landscape in amyotrophic lateral sclerosis-Past, present, and future. Med. Res. Rev. 2020, 40, 1352–1384. [Google Scholar] [CrossRef]
- Bellingham, M.C. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: What have we learned in the last decade? CNS Neurosci. Ther. 2011, 17, 4–31. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral. Scler. Front. Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Sawada, H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin. Pharm. 2017, 18, 735–738. [Google Scholar] [CrossRef]
- Tomar, S.; Gupta, S.; Singal, A.; Soni, R. Efficacy and safety of edaravone in amyotrophic lateral sclerosis patients in Indian population. J. Assoc. Physicians India 2022, 70, 11–12. [Google Scholar]
- Park, J.-M.; Park, D.; Kim, H.-J.; Park, J.-S. Long-term outcomes of edaravone in amyotrophic lateral sclerosis in South Korea: 72-week observational study. BMC Neurol. 2022, 22, 260. [Google Scholar] [CrossRef]
- Morgan, S.; Duguez, S.; Duddy, W. Personalized medicine and molecular interaction networks in amyotrophic lateral sclerosis (ALS): Current knowledge. J. Pers. Med. 2018, 8, 44. [Google Scholar] [CrossRef]
- Ly, C.V.; Miller, T.M. Emerging antisense oligonucleotide and viral therapies for amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2018, 31, 648–654. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2022. [Google Scholar] [CrossRef]
- Brettschneider, J.; Arai, K.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Lee, E.B.; Kuwabara, S.; Shibuya, K.; Irwin, D.J.; Fang, L. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol 2014, 128, 423–437. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Ojaimi, Y.A.; Dangoumau, A.; Alarcan, H.; Hergesheimer, R.; Vourc’h, P.; Corcia, P.; Lanznaster, D.; Blasco, H. TAR DNA-binding protein of 43 kDa (TDP-43) and amyotrophic lateral sclerosis (ALS): A promising therapeutic target. Expert Opin. Ther. Targets 2022, 26, 575–592. [Google Scholar] [CrossRef]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 86–95. [Google Scholar] [CrossRef]
- Polymenidou, M.; Cleveland, D.W. Biological spectrum of amyotrophic lateral sclerosis prions. Cold Spring Harb. Perspect. Med. 2017, 7, a024133. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Hatano, M.; Suzuki, N. RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci. Transl. Med. 2017, 9, eaah5436. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37, e98684. [Google Scholar] [CrossRef]
- Koyama, A.; Sugai, A.; Kato, T.; Ishihara, T.; Shiga, A.; Toyoshima, Y.; Koyama, M.; Konno, T.; Hirokawa, S.; Yokoseki, A.; et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016, 44, 5820–5836. [Google Scholar] [CrossRef]
- Bennett, C.F.; Kordasiewicz, H.B.; Cleveland, D.W. Antisense drugs make sense for neurological diseases. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 831–852. [Google Scholar] [CrossRef]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Melamed, Z.E.; López-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Krus, K.L.; Strickland, A.; Yamada, Y.; Devault, L.; Schmidt, R.E.; Bloom, A.J.; Milbrandt, J.; DiAntonio, A. Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell Rep. 2022, 39. [Google Scholar] [CrossRef]
- Klim, J.R.; Pintacuda, G.; Nash, L.A.; San Juan, I.G.; Eggan, K. Connecting TDP-43 pathology with neuropathy. Trends Neurosci. 2021, 44, 424–440. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Salmon, K.; Ross, J.P.; Bertone, V.; Gobbo, M.; Anoja, N.; Karamchandani, J.; Dion, P.A.; Rouleau, G.A.; Genge, A. The value of testing for ATXN2 intermediate repeat expansions in routine clinical practice for amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 2022. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.-W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Biogen and Ionis Announce Topline Phase 1 Study Results of Investigational Drug in C9orf72 Amyotrophic Lateral Sclerosis. 2022. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results (accessed on 9 July 2022).
- Tran, H.; Moazami, M.P.; Yang, H.; McKenna-Yasek, D.; Douthwright, C.L.; Pinto, C.; Metterville, J.; Shin, M.; Sanil, N.; Dooley, C.; et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022, 28, 117–124. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.P.; Dupre, N. From animal models to human disease: A genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 70. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Shaw, P.J.; Graham, D.; Fradette, S.; Houshyar, H.; Bennett, F.; Lane, R.M.; Nestorov, I.; Fanning, L. Safety, PK, PD, and exploratory efficacy in a single and multiple-dose study of a SOD1 antisense oligonucleotide (BIIB067) administered to participants with ALS. In Proceedings of the American Academy of Neruology 2019—71st Annual Meeting, Philadelphia, PA, USA, 4–10 May 2019. [Google Scholar]
- Miller, T.M.; Cudkowicz, M.E. Results from the Phase 3 VALOR study and its open-label extension: Evaluating the clinical efficacy and safety of Tofersen in adults with ALS and confirmed SOD1 mutation. In Proceedings of the American Neurological Association Annual Meeting, Virtual, 17–19 October 2021. [Google Scholar]
- Flynn, L.L.; Li, R.; Pitout, I.L.; Aung-Htut, M.T.; Larcher, L.M.; Cooper, J.A.L.; Greer, K.L.; Hubbard, A.; Griffiths, L.; Bond, C.S.; et al. Single stranded fully modified-phosphorothioate oligonucleotides can induce structured nuclear inclusions, alter nuclear protein localization and disturb the transcriptome in vitro. Front. Genet. 2022, 13, Art-791416. [Google Scholar] [CrossRef]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Léna, C. α-synuclein assemblies sequester neuronal α3-Na+/K+-ATP ase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423. [Google Scholar] [CrossRef] [PubMed]
- Brys, M.; Ellenbogen, A.; Fanning, L.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; Makh, S. Randomized, double-blind, placebo-controlled, single ascending dose study of anti-α-synuclein antibody BIIB054 in patients with Parkinson’s disease. Neurology 2018, 90, 1154–1163. [Google Scholar] [CrossRef]
- Arotcarena, M.-L.; Teil, M.; Dehay, B. Autophagy in synucleinopathy: The overwhelmed and defective machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.-C.; Van De Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Schapira, A.H. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in Parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Sardi, S.P.; Cheng, S.H.; Shihabuddin, L.S. Gaucher-related synucleinopathies: The examination of sporadic neurodegeneration from a rare (disease) angle. Prog. Neurobiol. 2015, 125, 47–62. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neuorol. 2020, 77, 427–434. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef]
- Krishnan, R.; Tsubery, H.; Proschitsky, M.Y.; Asp, E.; Lulu, M.; Gilead, S.; Gartner, M.; Waltho, J.P.; Davis, P.J.; Hounslow, A.M. A bacteriophage capsid protein provides a general amyloid interaction motif (GAIM) that binds and remodels misfolded protein assemblies. J. Mol. Biol. 2014, 426, 2500–2519. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, Y.; Li, Y.; Han, D.; Liu, T.; Yang, N.; Mi, X.; Hong, J.; Liu, K.; Song, Y. Inhibition of α-synuclein accumulation improves neuronal apoptosis and delayed postoperative cognitive recovery in aged mice. Oxid. Med. Cell. Longev. 2021, 2021, 5572899. [Google Scholar] [CrossRef]
- Somayaji, M.; Lanseur, Z.; Choi, S.J.; Sulzer, D.; Mosharov, E.V. Roles for α-synuclein in gene expression. Genes 2021, 12, 1166. [Google Scholar] [CrossRef]
- Liu, W.; Wang, G.; Wang, Z.; Wang, G.; Huang, J.; Liu, B. Repurposing small-molecule drugs for modulating toxic protein aggregates in neurodegenerative diseases. Drug Discov. 2022, 27, 1994–2007. [Google Scholar] [CrossRef]
- Alarcón-Arís, D.; Pavia-Collado, R.; Miquel-Rio, L.; Coppola-Segovia, V.; Ferrés-Coy, A.; Ruiz-Bronchal, E.; Galofré, M.; Paz, V.; Campa, L.; Revilla, R. Anti-α-synuclein ASO delivered to monoamine neurons prevents α-synuclein accumulation in a Parkinson’s disease-like mouse model and in monkeys. EBioMedicine 2020, 59, 102944. [Google Scholar] [CrossRef]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef]
- Boutros, S.W.; Raber, J.; Unni, V.K. Effects of alpha-synuclein targeted antisense oligonucleotides on Lewy body-like pathology and behavioral disturbances induced by injections of pre-formed fibrils in the mouse motor cortex. J. Parkinson’s Dis. 2021, 11, 1091. [Google Scholar] [CrossRef]
- Benatar, M.; Turner, M.R.; Wuu, J. Defining pre-symptomatic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2019, 20, 303–309. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, B.; Theunissen, F.; Mastaglia, F.L.; Akkari, P.A.; Flynn, L.L. Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics? Int. J. Mol. Sci. 2022, 23, 9364. https://doi.org/10.3390/ijms23169364
Roberts B, Theunissen F, Mastaglia FL, Akkari PA, Flynn LL. Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics? International Journal of Molecular Sciences. 2022; 23(16):9364. https://doi.org/10.3390/ijms23169364
Chicago/Turabian StyleRoberts, Bradley, Frances Theunissen, Francis L. Mastaglia, P. Anthony Akkari, and Loren L. Flynn. 2022. "Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics?" International Journal of Molecular Sciences 23, no. 16: 9364. https://doi.org/10.3390/ijms23169364
APA StyleRoberts, B., Theunissen, F., Mastaglia, F. L., Akkari, P. A., & Flynn, L. L. (2022). Synucleinopathy in Amyotrophic Lateral Sclerosis: A Potential Avenue for Antisense Therapeutics? International Journal of Molecular Sciences, 23(16), 9364. https://doi.org/10.3390/ijms23169364