
A Uniquely Stable Trimeric Model of SARS-CoV-2 Spike Transmembrane Domain

 , , , and
, , , and 
Abstract
1. Introduction
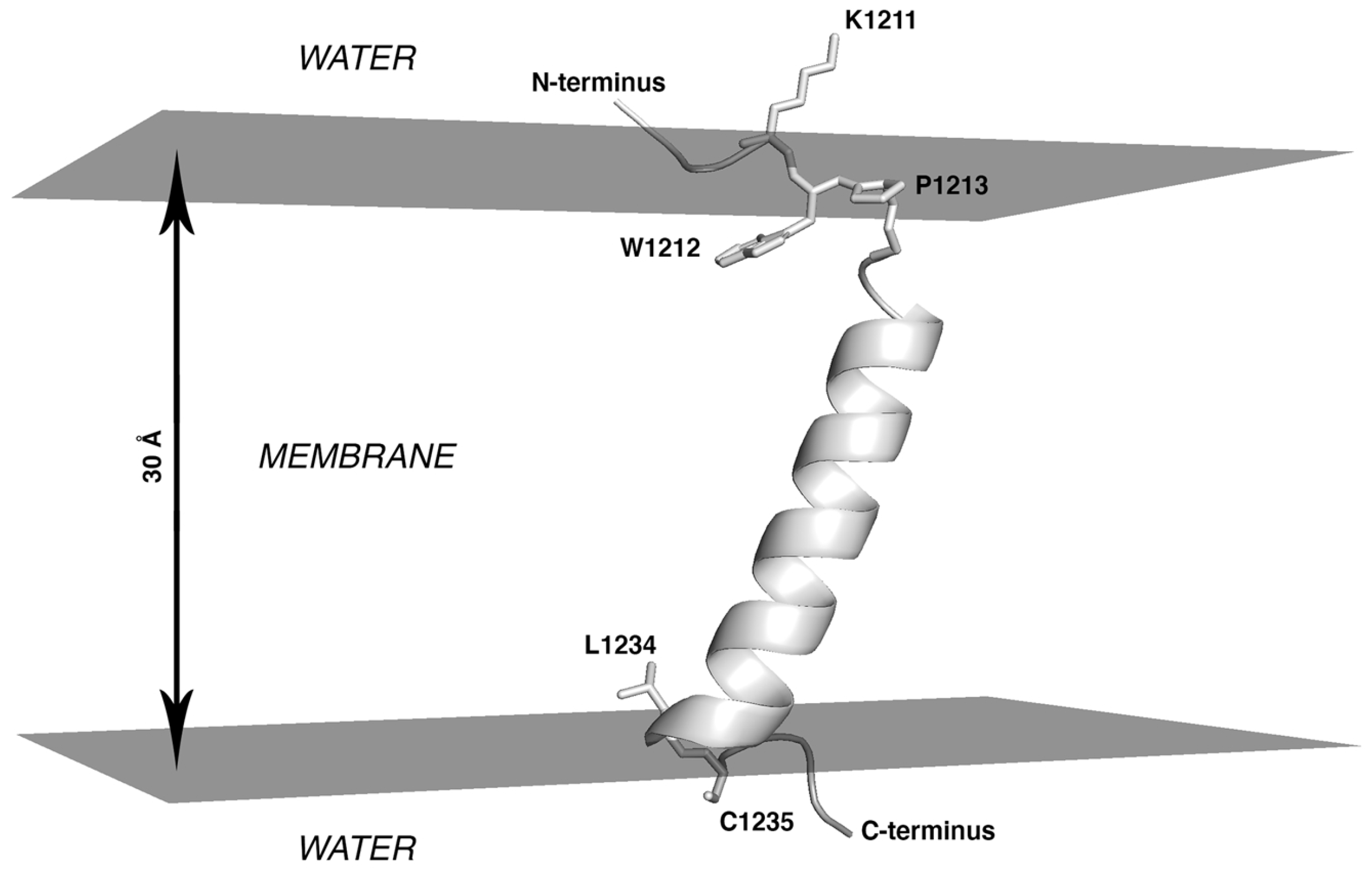
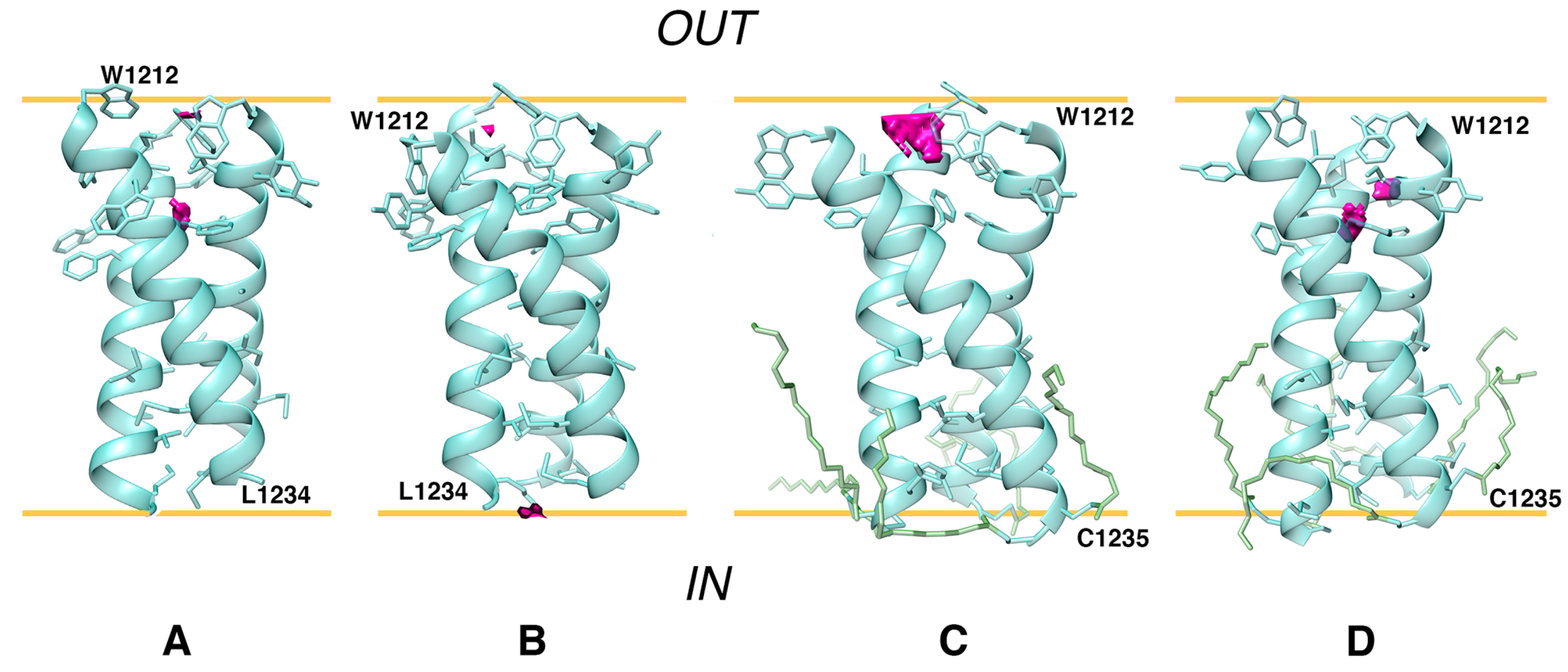
- At the first stage, the boundaries of S-TMD were identified via sequence analysis and further ascertained via Monte Carlo (MC) simulations of a TM monomer in an implicit membrane. Eventually, the fragment chosen to model S-TMD consisted of residues 1212 to 1234.
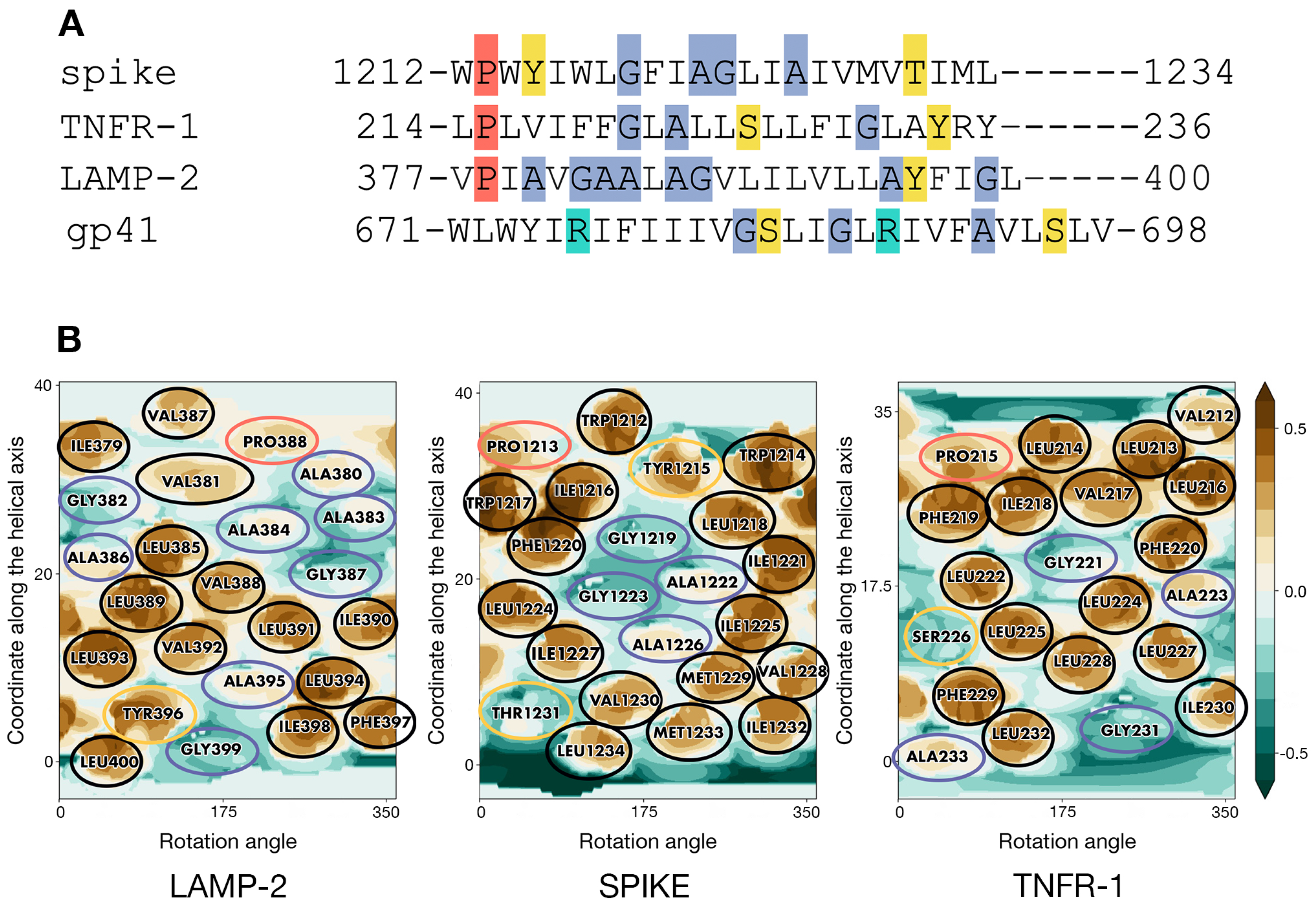
- At the next stage, an optimal structural template was selected among homotrimers of TM helices with known structures. The template had to conform to the following two criteria: (1) the presence of patterns of residues with physicochemical properties in the TM sequence sufficiently similar to those in the fragment of interest in the spike; (2) a good correspondence of the surface geometric and MHP properties between the two TM segments (in the template and spike). In the absence of sequence homology with available potential templates, the main assumption was that TM helices in S-TMD would pack in a similar way as in a template whereof the monomers possess similar sequence/surface motifs. To this end, we scrutinized the TM sequence of the spike and compared it to all TM homotrimers in the PDB database in search of common patterns of charged, polar, hydrophobic, small, and proline residues. For the candidates thus identified, we compared spatial distributions of hydrophobic/hydrophilic properties on their solvent-accessible surface and selected the optimal one(s).
- To enhance the reliability of S-TMD modeling, we used an independent approach. Based on MHP complementarity for TM helices, we predicted dimerization interfaces for the TM segment to identify surfaces likely to be on the helix-helix interfaces and compared them against those in the template(s) to pick the optimum one, with helix–helix contact areas as similar to those predicted for S-TMD as possible.
- One of the candidate templates, the TMD of TNFR-1, was chosen to build a model of S-TMD via homology modeling. To test the stability and viability of this model, MD simulations of this trimer were performed in a model POPC bilayer. POPC was chosen as it mimics, to a sufficient extent, the thickness and composition of the membrane of the endoplasmic reticulum Golgi intermediate compartment (ERGIC), where SARS-CoV-2 virions acquire their envelopes. Additionally, MD simulations of the original template were performed to evaluate its behavior in a POPC bilayer. Based upon MD data, a new, better-packed model trimer (S_OPT) was built via iterative adjustment of the ‘dynamic MHP portraits’ of the interacting helices.
- Finally, palmitoyl tails were added at C1235 and C1236 in the S_OPT model to explore how such modifications could affect the stability of S-TMD and the way it was accommodated by the membrane during MD simulations.
2. Results
2.1. S-TMD Boundaries Identified via Sequence Analysis and Monte Carlo Simulations
2.2. TMD of Tumour Necrosis Factor Receptor-1 as an Optimal Template for S-TMD Modelling
2.3. Preliminary Model of S-TMD Homotrimer Based on Predicted Helix–Helix Interfaces
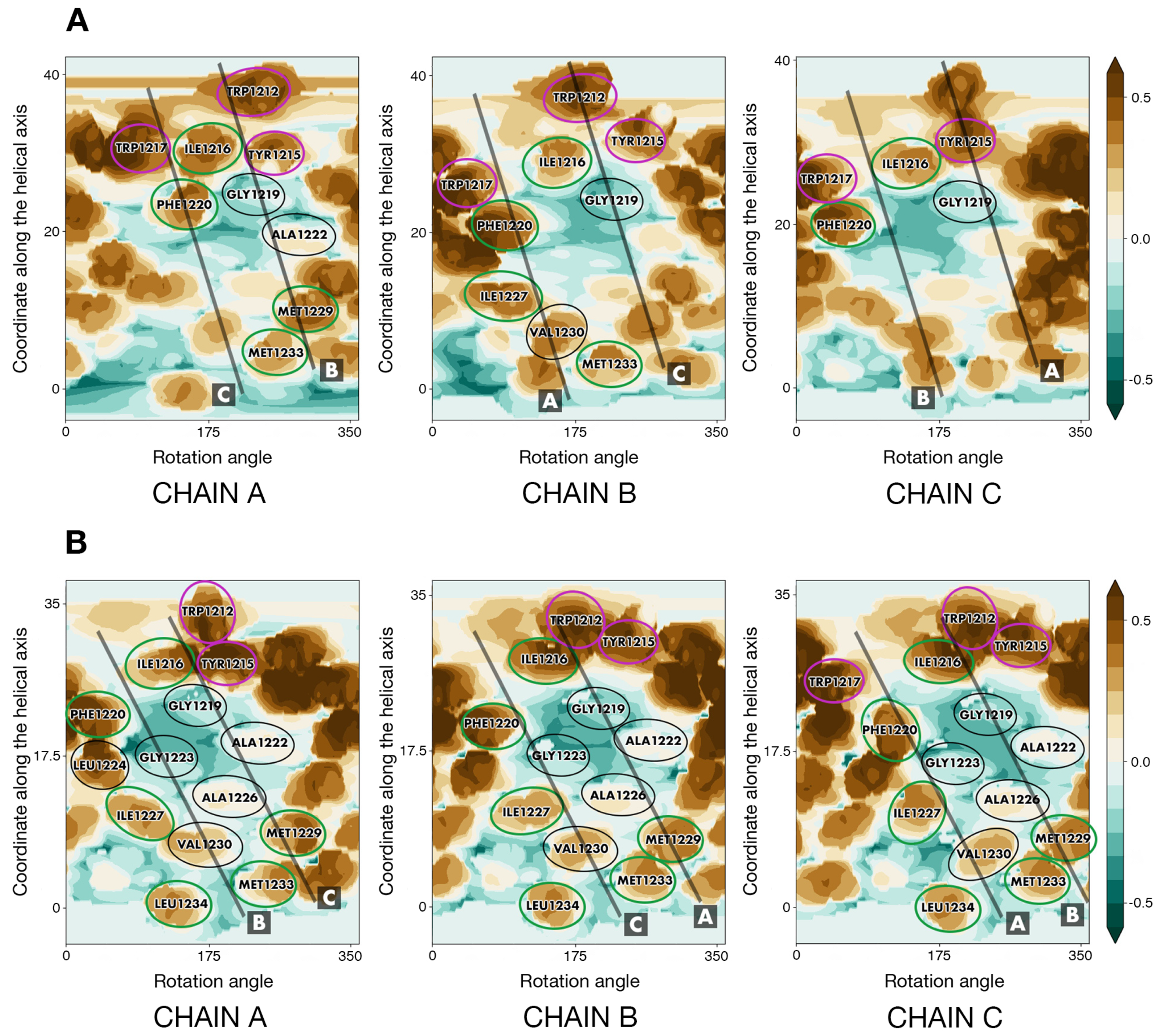
2.4. S_OPT: A Tightly Packed TMD Model Obtained via Iterative Refinement
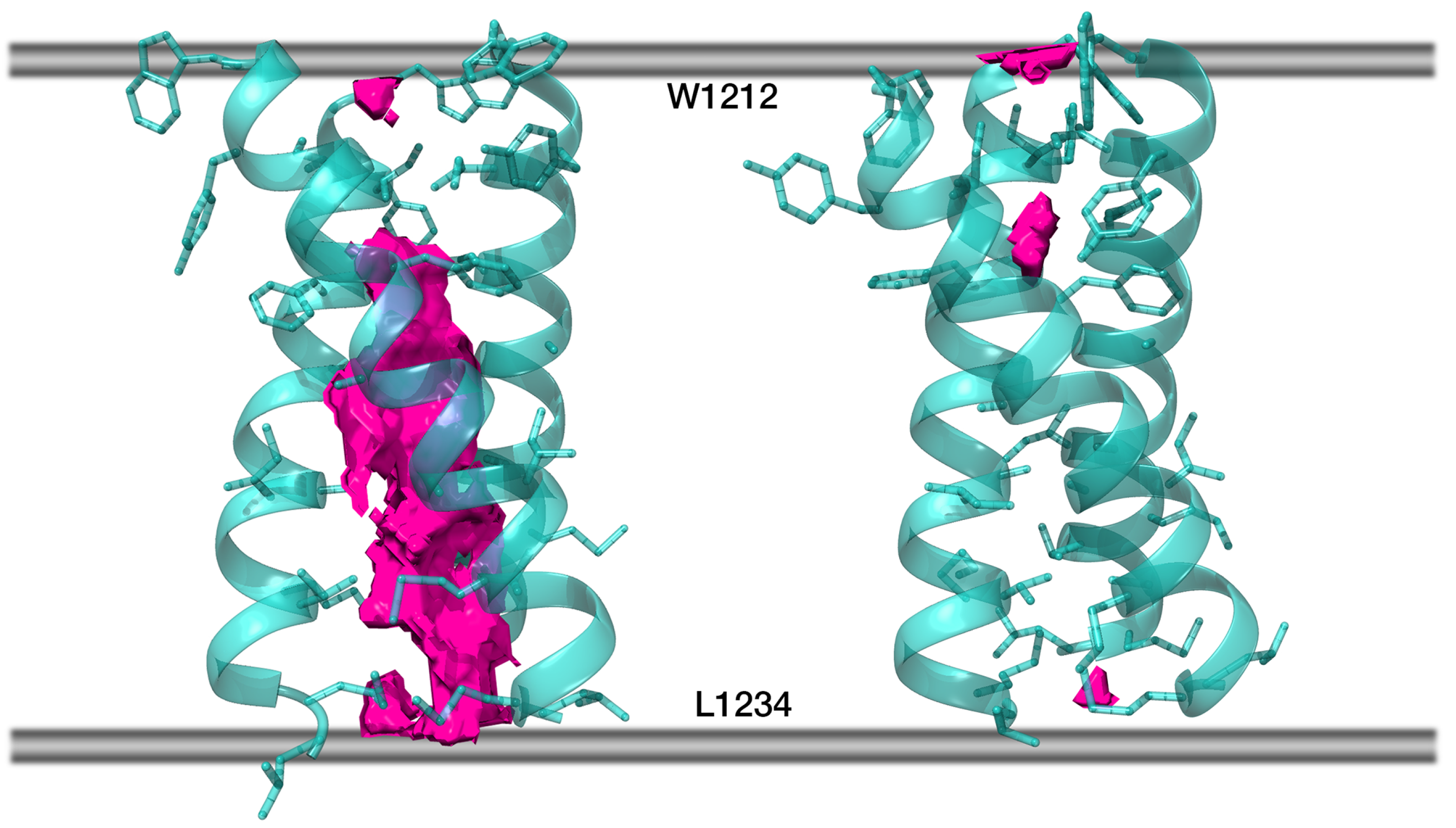
2.5. Model S_OPT Is Highly Stable in a Lipid Bilayer: Results of MD Simulations
2.6. Dynamic Properties of the Template TNRF-1 TMD and Other S-TMD Models
2.6.1. TNFR1_TMD
2.6.2. Other S-TMD Structures
2.7. Palmitoylation of S_OPT Preserves Its Unique Stability in a Model Membrane
3. Discussion
4. Materials and Methods
4.1. Assessment of TMD Boundaries via Sequence Analysis and Monte Carlo Simulations with an Implicit Membrane
4.2. Search for the Template for Homology Modelling of S-TMD
4.2.1. Analysis of TM Sequences in Homotrimers of TMDs with Known Spatial Structure
4.2.2. MHP Mapping and Helix–Helix Interface Analysis
4.3. Modeling of S-TMD
4.4. MD Simulations
4.5. Data Analysis
4.5.1. MD Trajectory Analysis
4.5.2. Free Volume Calculation
4.5.3. Membrane Response Evaluation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TM | transmembrane |
| TMD | transmembrane domain |
| S-TMD | spike transmembrane domain |
| FV | free volume |
| MC | Monte Carlo |
| MHP | molecular hydrophobicity potential. |
References
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Basso, L.G.M.; Zeraik, A.E.; Felizatti, A.P.; Costa-Filho, A.J. Membranotropic and biological activities of the membrane fusion peptides from SARS-CoV spike glycoprotein: The importance of the complete internal fusion peptide domain. Biochim. Biophys. Acta Biomembr. 2021, 1865, 183697. [Google Scholar] [CrossRef]
- Fu, Q.; Chou, J.J. A Trimeric Hydrophobic Zipper Mediates the Intramembrane Assembly of SARS-CoV-2 Spike. J. Am. Chem. Soc. 2021, 143, 8543–8546. [Google Scholar] [CrossRef]
- Mahajan, M.; Bhattacharjya, S. NMR structures and localization of the potential fusion peptides and the pre-transmembrane region of SARS-CoV: Implications in membrane fusion. Biochim. Biophys. Acta 2015, 1848, 721–730. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Sergeeva, O.; Turelli, P.; Qing, E.; Kunz, B.; Raclot, C.; Montoya, J.P.; Abriata, L.A.; Gallagher, T.; et al. S-acylation controls SARS-CoV-2 membrane lipid organization and enhances infectivity. Dev. Cell 2021, 56, 2790–2807. [Google Scholar] [CrossRef]
- Petit, C.M.; Chouljenko, V.N.; Iyer, A.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Rees, D.C.; DeAntonio, L.; Eisenberg, D. Hydrophobic organization of membrane proteins. Science 1989, 245, 510–513. [Google Scholar] [CrossRef]
- Lemmon, M.; Engelman, D.M. Helix-helix interactions inside lipid bilayers. Curr. Opin. Struct. Biol. 1992, 2, 511–518. [Google Scholar] [CrossRef]
- Woo, H.; Park, S.J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- Tan, Z.W.; Tee, W.V.; Samsudin, F.; Guarnera, E.; Bond, P.J.; Berezovsky, I.N. Allosteric perspective on the mutability and druggability of the SARS-CoV-2 Spike protein. Structure 2022, 30, 590–607. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Zhang Lab (University of Michigan) Genome-Wide Structure and Function Modeling of SARS-CoV-2 Virus. Available online: https://zhanglab.ccmb.med.umich.edu/COVID-19 (accessed on 4 March 2021).
- Izvorski, A. Predicted 3D Models of the SARS-CoV-2 Spike Protein Membrane Proximal External Region and Transmembrane Domain. Available online: https://chemrxiv.org/engage/chemrxiv/article-details/60c74f99842e651900db387b (accessed on 17 March 2021).
- Nishima, W.; Kulik, M. Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism. Viruses 2021, 13, 1126. [Google Scholar] [CrossRef]
- Rout, A.K.; Strub, M.P.; Piszczek, G.; Tjandra, N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef]
- Zhao, L.; Fu, Q.; Pan, L.; Piai, A.; Chou, J.J. The Diversity and Similarity of Transmembrane Trimerization of TNF Receptors. Front. Cell Dev. Biol. 2020, 8, 569684. [Google Scholar] [CrossRef]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G.; et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 2016, 353, 172–175. [Google Scholar] [CrossRef]
- Donnelly, D.; Overington, J.P.; Ruffle, S.V.; Nugent, J.H.; Blundell, T.L. Modeling α-helical transmembrane domains: The calculation and use of substitution tables for lipid-facing residues. Protein Sci. 1993, 2, 55–70. [Google Scholar]
- Corver, J.; Broer, R.; van Kasteren, P.; Spaan, W. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: Refinement of the requirements for SARS coronavirus cell entry. Virol. J. 2009, 6, 230. [Google Scholar] [CrossRef] [PubMed]
- Panina, I.; Krylov, N.; Gadalla, M.R.; Aliper, E.; Kordyukova, L.; Veit, M.; Chugunov, A.; Efremov, R. Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity. Int. J. Mol. Sci. 2022, 23, 5091. [Google Scholar] [CrossRef] [PubMed]
- Arbely, E.; Granot, Z.; Kass, I.; Orly, J.; Arkin, I.T. A trimerizing GxxxG motif is uniquely inserted in the severe acute respiratory syndrome (SARS) coronavirus spike protein transmembrane domain. Biochemistry 2006, 45, 11349–11356. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Corver, J.; Broer, R.; van Kasteren, P.; Spaan, W. GxxxG motif of severe acute respiratory syndrome coronavirus spike glycoprotein transmembrane domain is not involved in trimerization and is not important for entry. J. Virol. 2007, 81, 8352–8355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Polyansky, A.A.; Chugunov, A.O.; Volynsky, P.E.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PREDDIMER: A web server for prediction of transmembrane helical dimers. Bioinformatics 2014, 30, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Ng, M.C.K.; Jusoh, S.A.; Tai, H.K.; Siu, S.W.I. TMDIM: An improved algorithm for the structure prediction of transmembrane domains of bitopic dimers. J. Comput. Aided Mol. Des. 2017, 31, 855–865. [Google Scholar] [CrossRef]
- Kordyukova, L.V.; Serebryakova, M.V.; Polyansky, A.A.; Kropotkina, E.A.; Alexeevski, A.V.; Veit, M.; Efremov, R.G.; Filippova, I.Y.; Baratova, L.A. Linker and/or transmembrane regions of influenza A/Group-1, A/Group-2, and type B virus hemagglutinins are packed differently within trimers. Biochim. Biophys. Acta 2011, 1808, 1843–1854. [Google Scholar] [CrossRef]
- Sharma, S.; Juffer, A.H. An atomistic model for assembly of transmembrane domain of T cell receptor complex. J. Am. Chem. Soc. 2013, 135, 2188–2197. [Google Scholar] [CrossRef]
- Vilmen, G.; Banerjee, A.; Freed, E.O. Rafting through the palms: S-acylation of SARS-CoV-2 spike protein induces lipid reorganization. Dev. Cell 2021, 56, 2787–2789. [Google Scholar] [CrossRef]
- Polyansky, A.A.; Volynsky, P.E.; Efremov, R.G. Multistate organization of transmembrane helical protein dimers governed by the host membrane. J. Am. Chem. Soc. 2012, 134, 14390–14400. [Google Scholar] [CrossRef]
- Guillén, J.; Moreno, M.R.; Pérez-Berna, A.J.; Bernabeu, A.; Villalaín, J. Interaction of a peptide from the pre-transmembrane domain of the severe acute respiratory syndrome coronavirus spike protein with phospholipid membranes. J. Phys. Chem. B 2007, 111, 13714–13725. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Lesovoy, D.M.; Bocharova, O.V.; Urban, A.S.; Pavlov, K.V.; Volynsky, P.E.; Efremov, R.G.; Arseniev, A.S. Structural basis of the signal transduction via transmembrane domain of the human growth hormone receptor. BBA—Gen. Subj. 2018, 1862, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Crnjar, A.; Comitani, F.; Hester, W.; Molteni, C. Trans-Cis Proline Switches in a Pentameric Ligand-Gated Ion Channel: How They Are Affected by and How They Affect the Biomolecular Environment. J. Phys. Chem. Lett. 2019, 10, 694–700. [Google Scholar] [CrossRef]
- Yu, J.; Qiao, S.; Guo, R.; Wang, X. Cryo-EM structures of HKU2 and SADS-CoV spike glycoproteins provide insights into coronavirus evolution. Nat. Commun. 2020, 11, 3070. [Google Scholar] [CrossRef] [PubMed]
- Chiliveri, S.C.; Louis, J.M.; Ghirlando, R.; Bax, A. Transient lipid-bound states of spike protein heptad repeats provide insights into SARS-CoV-2 membrane fusion. Sci. Adv. 2021, 7, eabk2226. [Google Scholar] [CrossRef] [PubMed]
- Bippes, C.A.; Muller, D.J. High-resolution atomic force microscopy and spectroscopy of native membrane proteins. Rep. Prog. Phys. 2011, 74, 086601. [Google Scholar] [CrossRef]
- Sumbul, F.; Marchesi, A.; Takahashi, H.; Scheuring, S.; Rico, F. High-Speed Force Spectroscopy for Single Protein Unfolding. Methods Mol. Biol. 2018, 1814, 243–264. [Google Scholar]
- Bernhofer, M.; Kloppmann, E.; Reeb, J.; Rost, B. TMSEG: Novel prediction of transmembrane helices. Proteins 2016, 84, 1706–1716. [Google Scholar] [CrossRef]
- Hofmann, K.; Stoffel, W. TMbase-a database of membrane spanning proteins segments. Biol. Chem. Hoppe Seyler 1993, 347, 166. [Google Scholar]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Nolde, D.E.; Volynskii, P.E.; Arseniev, A.S.; Efremov, R.G. Modeling of peptides and proteins in a membrane environment. I. A solvation model mimicking a lipid bilayer. Russ. J. Bioorg. Chem. 2000, 26, 115–124. [Google Scholar] [CrossRef]
- von Freyberg, B.; Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. J. Comput. Chem. 1991, 12, 1065–1076. [Google Scholar] [CrossRef]
- Némethy, G.; Pottle, M.S.; Sheraga, H.A. Energy parameters in polypeptides. 9. Updating of geometrical parameters, nonbonded interactions, and hydrogen bond interactions for the naturally occurring amino acids. J. Phys. Chem. 1983, 87, 1883–1887. [Google Scholar] [CrossRef]
- Efremov, R.G.; Nolde, D.E.; Vergoten, G.; Arseniev, A.S. A solvent model for simulations of peptides in bilayers. I. Membrane-promoting alpha-helix formation. Biophys. J. 1999, 76, 2448–2459. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Efremov, R.G.; Gulyaev, D.I.; Vergoten, G.; Modyanov, N.N. Application of three-dimensional molecular hydrophobicity potential to the analysis of spatial organization of membrane domains in proteins: I. Hydrophobic properties of transmembrane segments of Na+, K(+)-ATPase. J. Protein Chem. 1992, 11, 665–675. [Google Scholar] [CrossRef]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: A web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2020.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ(1) and χ(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Klauda, J.B.; Monje, V.; Kim, T.; Im, W. Improving the CHARMM force field for polyunsaturated fatty acid chains. J. Phys. Chem. B 2012, 116, 9424–9431. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Krylov, N.A.; Efremov, R.G. libxtc: An efficient library for reading XTC-compressed MD trajectory data. BMC Res. Notes 2021, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Kuzmanic, A.; Hlevnjak, M.; Zagrovic, B. On the Contribution of Linear Correlations to Quasi-harmonic Conformational Entropy in Proteins. J. Chem. Theory Comput. 2012, 8, 3820–3829. [Google Scholar] [CrossRef]
- Douliez, J.P.; Ferrarini, A.; Dufourc, E.J. On the relationship between C-C and C-D order parameters and its use for studying the conformation of lipid acyl chains in biomembranes. J. Chem. Phys. 1998, 109, 2513–2518. [Google Scholar] [CrossRef][Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| Created via template-based modelling | |
| S_TNFR1 | 1208-QYIKWPWYIWLGFIAGLIAIVMVTIMLCCMT-1238 |
| The template used to build the TMD model (TNFR-1) | |
| TNFR1_TMD | 209-GTTVLLPLVIFFGLALLSLLFIGLAYRYQR-238 |
| Built iteratively via the adjustment of the MHP portraits in S_TNFR1 | |
| S_OPT | 1208-QYIKWPWYIWLGFIAGLIAIVMVTIMLCCMT-1238 |
| Based on the recently published NMR structure (PDB ID 7LC8) | |
| S_NMR | 1217-WLGFIAGLIAIVLVTILLSST-1237 |
| S_NMR+ | 1214-WYIWLGFIAGLIAIVLVTILLSSTSCC-1240 |
| S_NMR-WT | 1217-WLGFIAGLIAIVMVTIMLCCM-1237 |
| S_OPT with the mutations present in the recent NMR structure (PDB ID 7LC8) | |
| S_OPT-LC | 1208-QYIKWPWYIWLGFIAGLIAIVLVTILLCCMT-1238 |
| S_OPT-LS | 1208-QYIKWPWYIWLGFIAGLIAIVLVTILLSSMT-1238 |
| S_OPT with palmitoyl tails appended at C1235 and C1236 | |
| S_OPT-PLM | 1208-QYIKWPWYIWLGFIAGLIAIVMVTIMLCCMT-1238 |
| System Composition | MD Length (ns) | No. of Replicas |
|---|---|---|
| A POPC bilayer | ||
| POPC450/Water26276 | 1000 | 1 |
| TNFR1_TMD in a lipid bilayer | ||
| Protein/POPC475/Water42057/Cl−6 | 500 | 1 |
| S_TNFR1 in a lipid bilayer | ||
| Protein/POPC483/Water42060/Cl−3 | 500 | 4 |
| S_OPT in a lipid bilayer | ||
| Protein/POPC485/Water42098/Cl−3 | 1000 | 2 |
| Structures based on the experimentally observed trimer (PDB ID 7LC8) in a lipid bilayer | ||
| S_NMR/POPC488/Water42196 | 500 | 1 |
| S_NMR+/POPC485/Water42148 | 500 | 1 |
| S_NMR-WT/POPC486/Water42198 | 500 | 1 |
| S_OPT with the mutations present in the experimentally observed trimer (PDB ID 7LC8) in a lipid bilayer | ||
| S_OPT-LC/POPC483/ Water42092/Cl−3 | 1000 | 1 |
| S_OPT-LS/POPC484/Water42117/Cl−3 | 1000 | 1 |
| S_OPT palmitoylated at C1235 and C1236 | ||
| Protein/POPC480/Water42084/Cl−3 | 1000 | 1 |
| Protein/POPC474/Water42120/Cl−3 | 1000 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliper, E.T.; Krylov, N.A.; Nolde, D.E.; Polyansky, A.A.; Efremov, R.G. A Uniquely Stable Trimeric Model of SARS-CoV-2 Spike Transmembrane Domain. Int. J. Mol. Sci. 2022, 23, 9221. https://doi.org/10.3390/ijms23169221
Aliper ET, Krylov NA, Nolde DE, Polyansky AA, Efremov RG. A Uniquely Stable Trimeric Model of SARS-CoV-2 Spike Transmembrane Domain. International Journal of Molecular Sciences. 2022; 23(16):9221. https://doi.org/10.3390/ijms23169221
Chicago/Turabian StyleAliper, Elena T., Nikolay A. Krylov, Dmitry E. Nolde, Anton A. Polyansky, and Roman G. Efremov. 2022. "A Uniquely Stable Trimeric Model of SARS-CoV-2 Spike Transmembrane Domain" International Journal of Molecular Sciences 23, no. 16: 9221. https://doi.org/10.3390/ijms23169221
APA StyleAliper, E. T., Krylov, N. A., Nolde, D. E., Polyansky, A. A., & Efremov, R. G. (2022). A Uniquely Stable Trimeric Model of SARS-CoV-2 Spike Transmembrane Domain. International Journal of Molecular Sciences, 23(16), 9221. https://doi.org/10.3390/ijms23169221