
Recombinant FGF21 Attenuates Polychlorinated Biphenyl-Induced NAFLD/NASH by Modulating Hepatic Lipocalin-2 Expression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
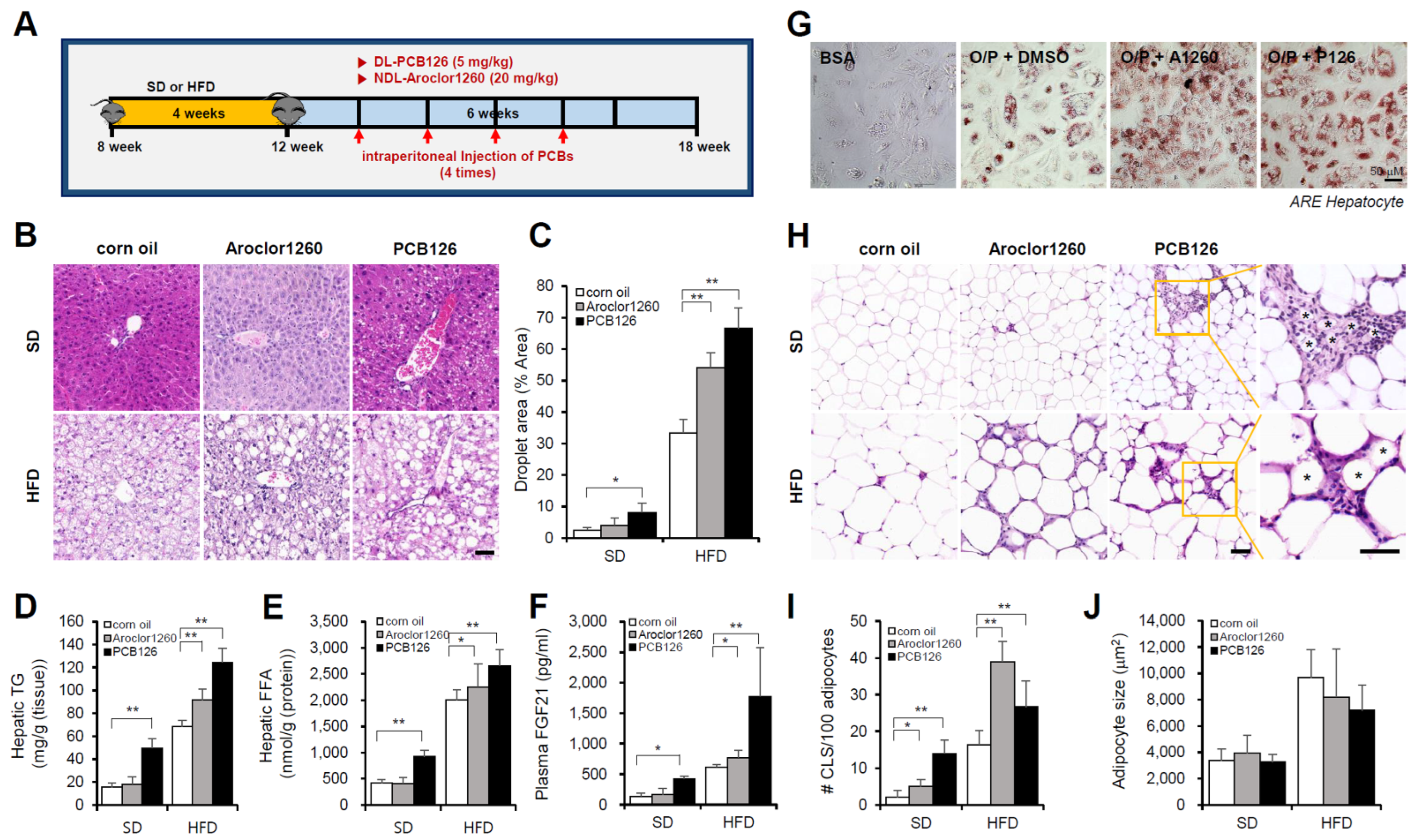
2.1. PCB Exposure Induces NAFLD/NASH in Mice Fed a SD or HFD
2.1.1. Aroclor1260 Exposure Induces NAFLD/NASH in Mice Fed a HFD
2.1.2. PCB126 Exposure Induces NAFLD/NASH in Mice Fed a SD or HFD
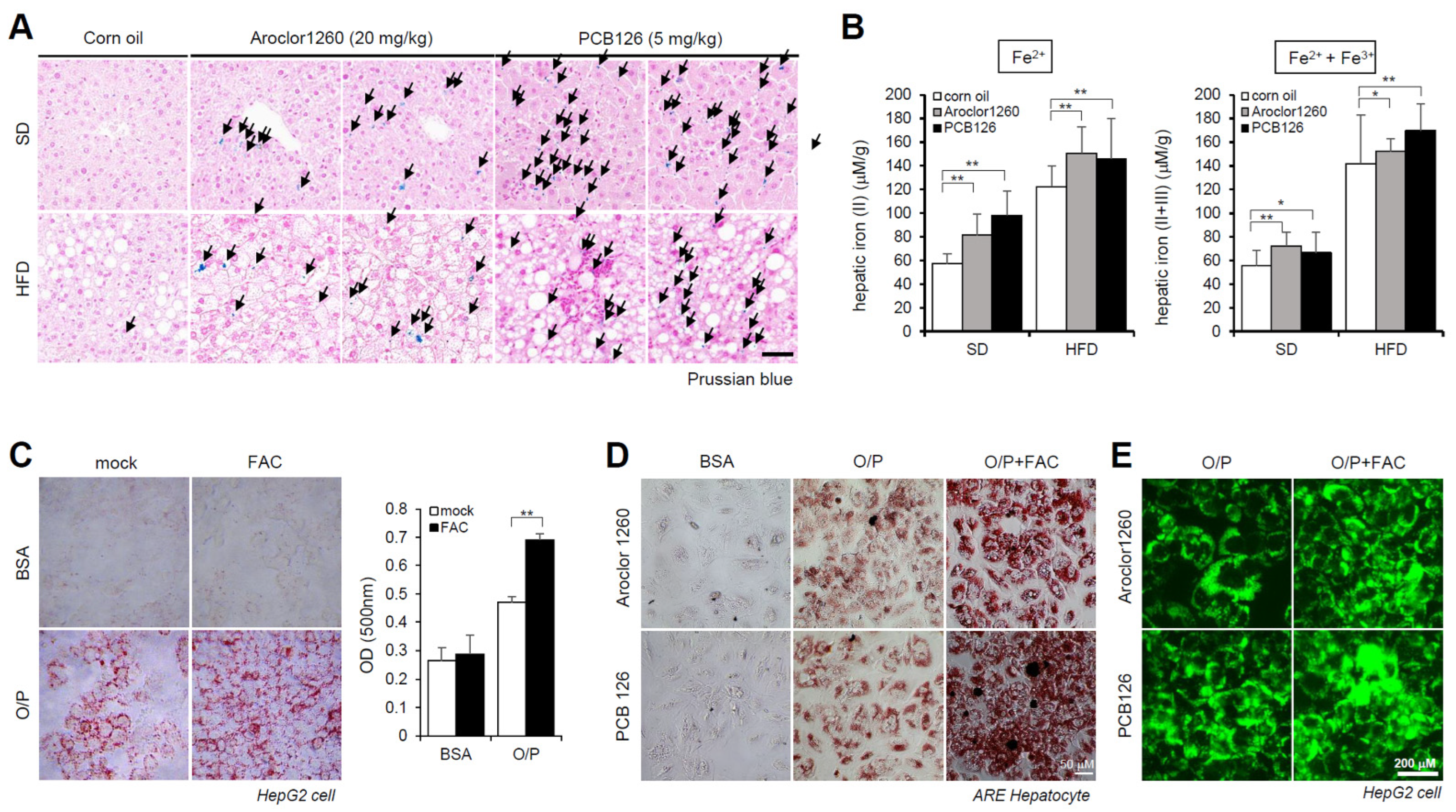
2.2. PCB Exposure Induces Hepatic Iron Overload
2.3. PCB Exposure Accelerates Hepatic Inflammation
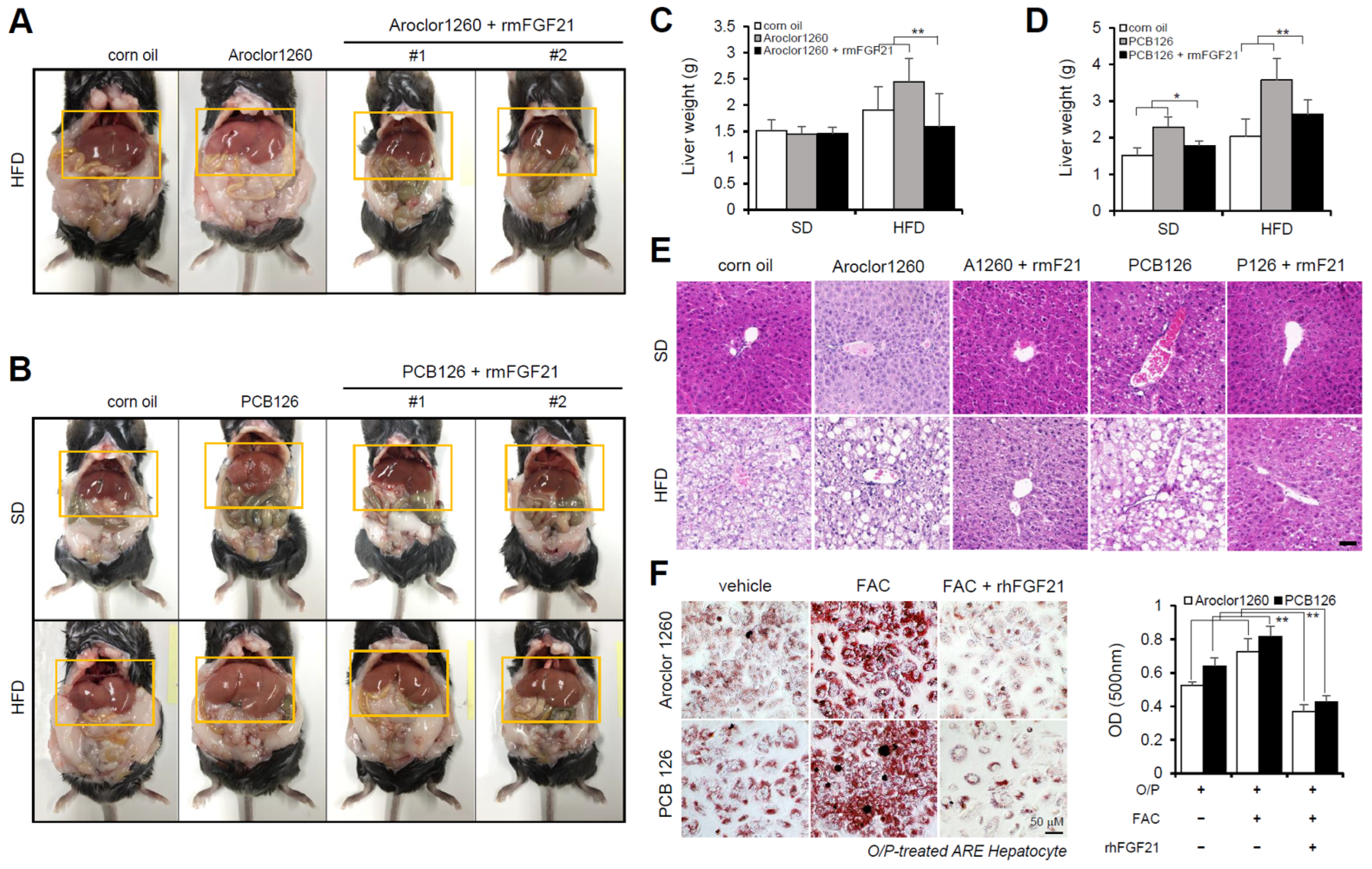
2.4. Recombinant FGF21 Improved Hepatic Steatosis in PCB-Induced NAFLD/NASH Models
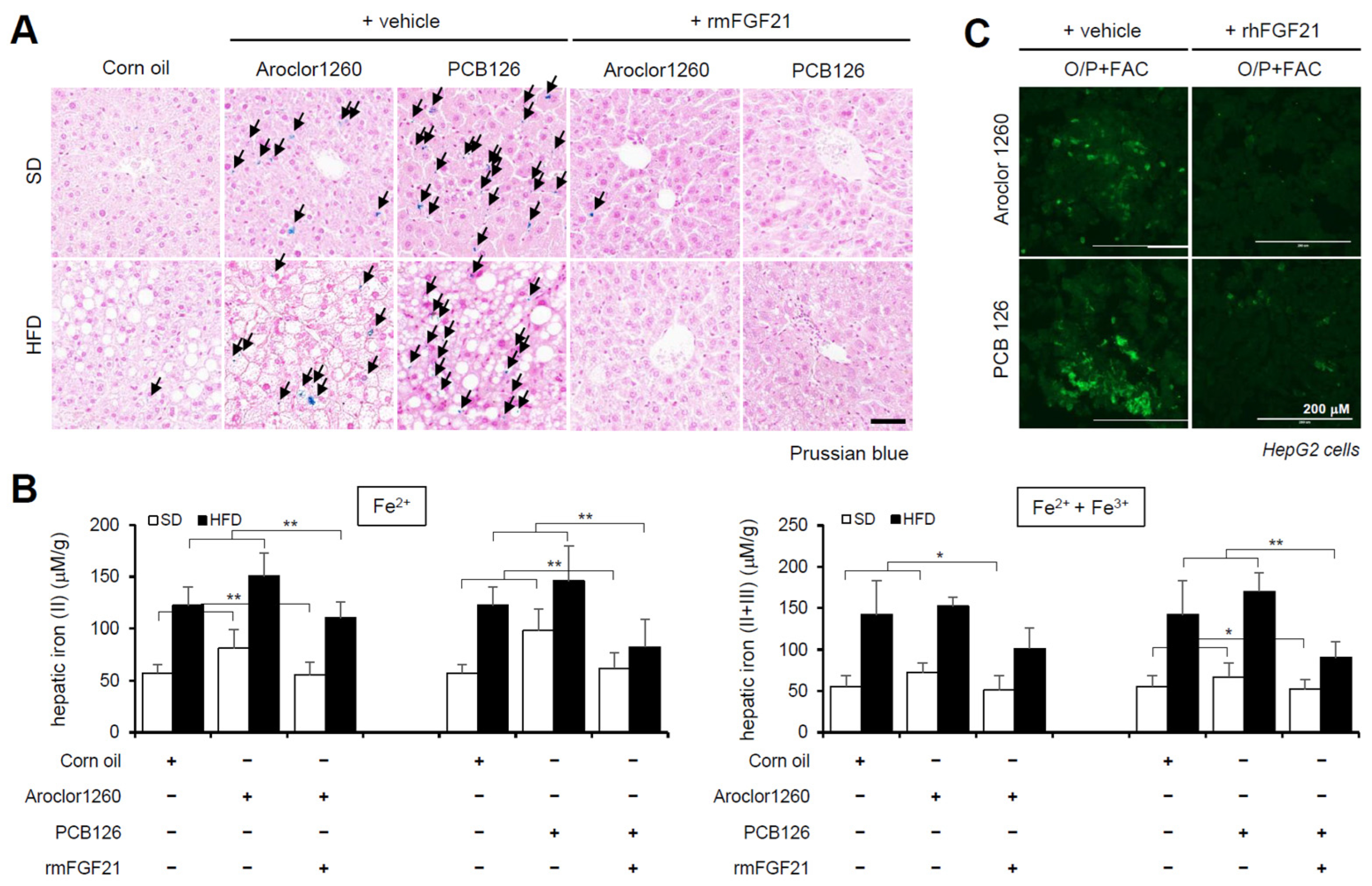
2.5. Recombinant FGF21 Attenuated HIO in PCB-Induced NAFLD/NASH Models
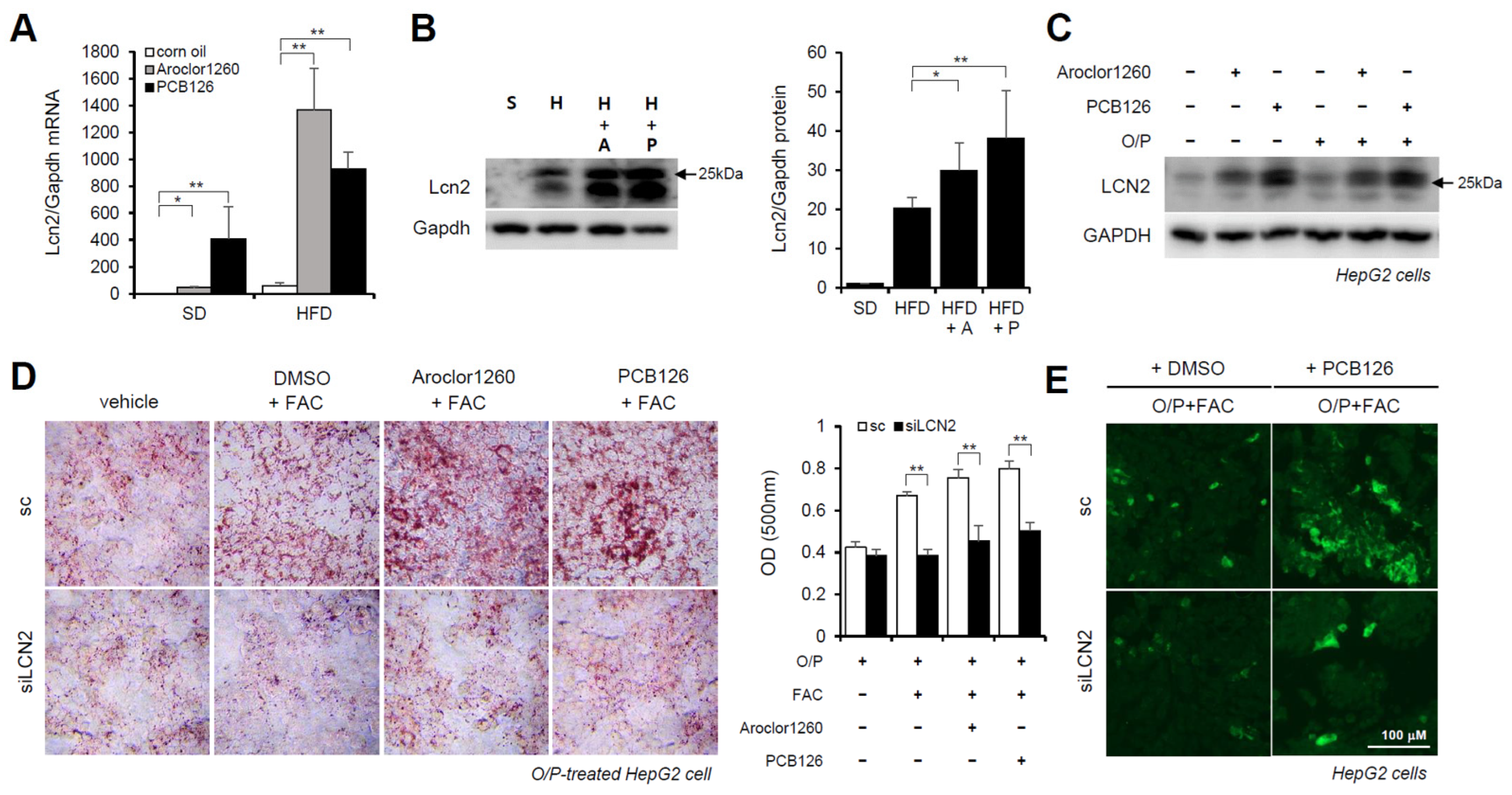
2.6. Hepatic LCN2 Mediates PCB-Induced Hepatic Lipid and Iron Accumulation
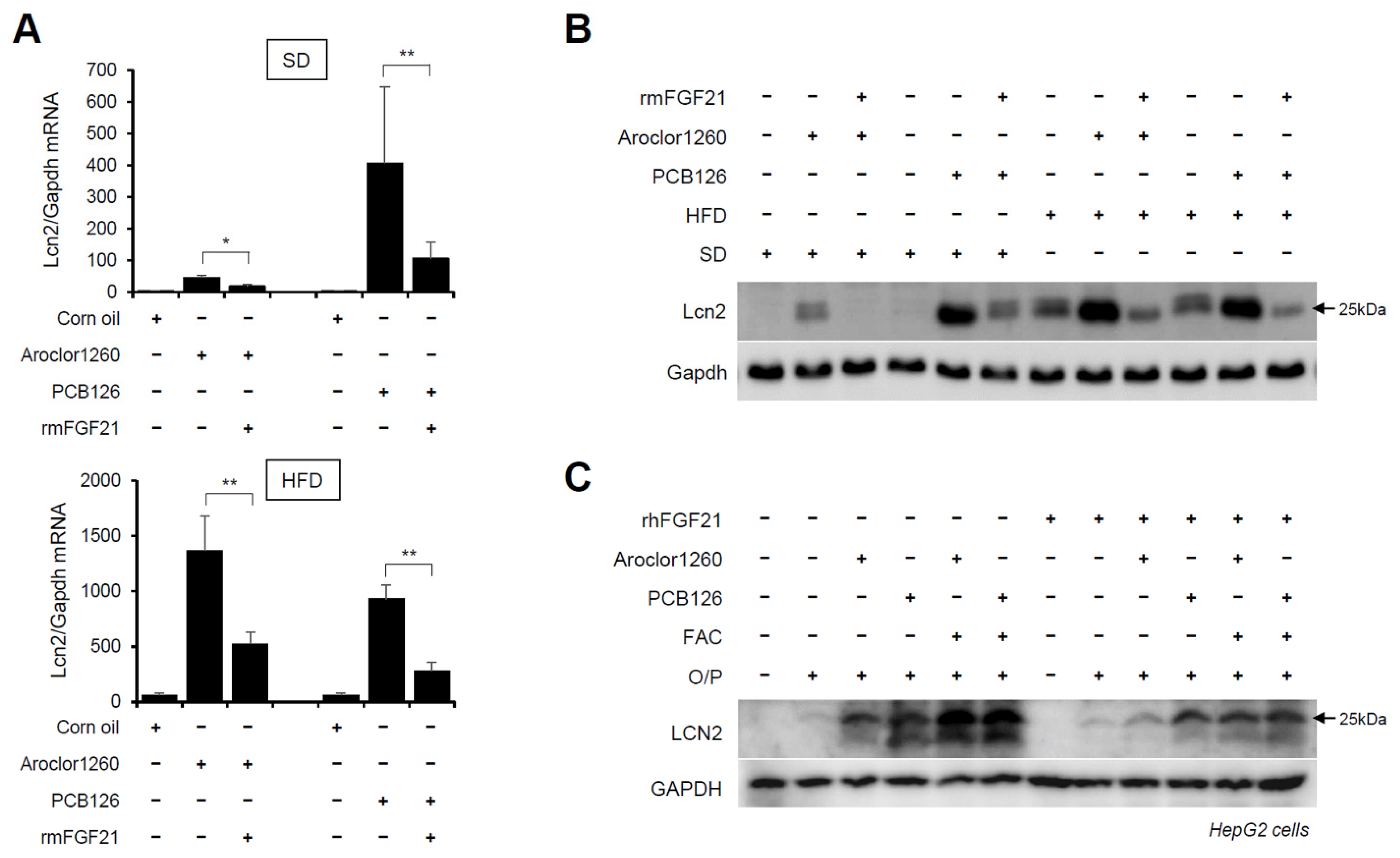
2.7. Recombinant FGF21 Reduces PCB-Induced Overexpression of Hepatic LCN2
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Animals
4.3. Cell Culture and Treatment of Free Fatty Acids
4.4. RNA Interference and Transfection
4.5. Histological Analysis
4.6. Analysis of Metabolites
4.7. Fluorescence Staining and Confocal Microscopy
4.8. Oil Red O Staining
4.9. Western Blot Analysis
4.10. RNA Isolation and Quantitative Real-Time PCR Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [PubMed]
- Pafili, K.; Roden, M. Nonalcoholic Fatty Liver Disease (NAFLD) from Pathogenesis to Treatment Concepts in Humans. Mol. Metab. 2021, 50, 101122. [Google Scholar] [PubMed]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma: A Weighty Connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated Biphenyls, Lead, and Mercury are Associated with Liver Disease in American Adults: NHANES 2003–2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Clair, H.B.; Pinkston, C.M.; Rai, S.N.; Pavuk, M.; Dutton, N.D.; Brock, G.N.; Prough, R.A.; Falkner, K.C.; McClain, C.J.; Cave, M.C. Liver Disease in a Residential Cohort with Elevated Polychlorinated Biphenyl Exposures. Toxicol. Sci. 2018, 164, 39–49. [Google Scholar] [CrossRef]
- Cave, M.; Falkner, K.C.; Ray, M.; Joshi-Barve, S.; Brock, G.; Khan, R.; Bon Homme, M.; McClain, C.J. Toxicant-associated Steatohepatitis in Vinyl Chloride Workers. Hepatology 2010, 51, 474–481. [Google Scholar] [CrossRef]
- Shi, H.; Jan, J.; Hardesty, J.E.; Falkner, K.C.; Prough, R.A.; Balamurugan, A.N.; Mokshagundam, S.P.; Chari, S.T.; Cave, M.C. Polychlorinated Biphenyl Exposures Differentially Regulate Hepatic Metabolism and Pancreatic Function: Implications for Nonalcoholic Steatohepatitis and Diabetes. Toxicol. Appl. Pharmacol. 2019, 363, 22–33. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR Signaling Pathways and Regulatory Functions. Biochim. Open 2018, 7, 1–9. [Google Scholar]
- Boucher, M.; Lefebvre, C.; Chapados, N.A. The Effects of PCB126 on Intra-Hepatic Mechanisms Associated with Non Alcoholic Fatty Liver Disease. J. Diabetes Metab. Disord. 2015, 14, 88. [Google Scholar] [CrossRef]
- Gadupudi, G.S.; Klaren, W.D.; Olivier, A.K.; Klingelhutz, A.J.; Robertson, L.W. PCB126-Induced Disruption in Gluconeogenesis and Fatty Acid Oxidation Precedes Fatty Liver in Male Rats. Toxicol. Sci. 2016, 149, 98–110. [Google Scholar] [CrossRef]
- Wahlang, B.; Prough, R.A.; Falkner, K.C.; Hardesty, J.E.; Song, M.; Clair, H.B.; Clark, B.J.; States, J.C.; Arteel, G.E.; Cave, M.C. Polychlorinated Biphenyl-Xenobiotic Nuclear Receptor Interactions Regulate Energy Metabolism, Behavior, and Inflammation in Non-Alcoholic-Steatohepatitis. Toxicol. Sci. 2016, 149, 396–410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wahlang, B.; Falkner, K.C.; Clair, H.B.; Al-Eryani, L.; Prough, R.A.; States, J.C.; Coslo, D.M.; Omiecinski, C.J.; Cave, M.C. Human Receptor Activation by Aroclor 1260, a Polychlorinated Biphenyl Mixture. Toxicol. Sci. 2014, 140, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Song, M.; Beier, J.I.; Falkner, K.C.; Al-Eryani, L.; Clair, H.B.; Prough, R.A.; Osborne, T.S.; Malarkey, D.E.; States, J.C. Evaluation of Aroclor 1260 Exposure in a Mouse Model of Diet-Induced Obesity and Non-Alcoholic Fatty Liver Disease. Toxicol. Appl. Pharmacol. 2014, 279, 380–390. [Google Scholar] [CrossRef]
- Safe, S.H. Polychlorinated Biphenyls (PCBs): Environmental Impact, Biochemical and Toxic Responses, and Implications for Risk Assessment. Crit. Rev. Toxicol. 1994, 24, 87–149. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R. Metabolism Disrupting Chemicals and Metabolic Disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Papalou, O.; Kandaraki, E.A.; Papadakis, G.; Diamanti-Kandarakis, E. Endocrine Disrupting Chemicals: An Occult Mediator of Metabolic Disease. Front. Endocrinol. 2019, 10, 112. [Google Scholar] [CrossRef]
- Wahlang, B.; Alexander II, N.C.; Li, X.; Rouchka, E.C.; Kirpich, I.A.; Cave, M.C. Polychlorinated Biphenyls Altered Gut Microbiome in CAR and PXR Knockout Mice Exhibiting Toxicant-Associated Steatohepatitis. Toxicol. Rep. 2021, 8, 536–547. [Google Scholar] [CrossRef]
- Britton, L.J.; Subramaniam, V.N.; Crawford, D.H. Iron and Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2016, 22, 8112. [Google Scholar] [CrossRef]
- Datz, C.; Müller, E.; Aigner, E. Iron Overload and Non-Alcoholic Fatty Liver Disease. Minerva Endocrinol. 2016, 42, 173–183. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in Fatty Liver and in the Metabolic Syndrome: A Promising Therapeutic Target. J. Hepatol. 2011, 55, 920–932. [Google Scholar] [CrossRef]
- Wang, C.; Babitt, J.L. Liver Iron Sensing and Body Iron Homeostasis. Blood J. Am. Soc. Hematol. 2019, 133, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Theurl, M.; Lederer, D.; Haufe, H.; Dietze, O.; Strasser, M.; Datz, C.; Weiss, G. Pathways Underlying Iron Accumulation in Human Nonalcoholic Fatty Liver Disease. Am. J. Clin. Nutr. 2008, 87, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Britton, L.; Bridle, K.; Reiling, J.; Santrampurwala, N.; Wockner, L.; Ching, H.; Stuart, K.; Subramaniam, V.N.; Jeffrey, G.; Pierre, T.S.; et al. Hepatic Iron Concentration Correlates with Insulin Sensitivity in Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2018, 2, 644–653. [Google Scholar] [CrossRef] [PubMed]
- George, D.K.; Goldwurm, S.; Macdonald, G.A.; Cowley, L.L.; Walker, N.I.; Ward, P.J.; Jazwinska, E.C.; Powell, L.W. Increased Hepatic Iron Concentration in Nonalcoholic Steatohepatitis is Associated with Increased Fibrosis. Gastroenterology 1998, 114, 311–318. [Google Scholar] [CrossRef]
- Kowdley, K.V. Iron Overload in Patients with Chronic Liver Disease. Gastroenterol. Hepatol. 2016, 12, 695. [Google Scholar]
- Kim, H.Y.; Kwon, W.Y.; Park, J.B.; Lee, M.H.; Oh, Y.J.; Suh, S.; Baek, Y.H.; Jeong, J.S.; Yoo, Y.H. Hepatic STAMP2 Mediates Recombinant FGF21-induced Improvement of Hepatic Iron Overload in Nonalcoholic Fatty Liver Disease. FASEB J. 2020, 34, 12354–12366. [Google Scholar] [CrossRef]
- Kjeldsen, L.; Bainton, D.F.; Sengelov, H.; Borregaard, N. Identification of Neutrophil Gelatinase-Associated Lipocalin as a Novel Matrix Protein of Specific Granules in Human Neutrophils. Blood 1994, 83, 799–807. [Google Scholar] [CrossRef]
- Xiao, X.; Yeoh, B.S.; Vijay-Kumar, M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu. Rev. Nutr. 2017, 37, 103–130. [Google Scholar] [CrossRef]
- Krizanac, M.; Mass Sanchez, P.B.; Weiskirchen, R.; Asimakopoulos, A. A Scoping Review on Lipocalin-2 and its Role in Non-Alcoholic Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 2865. [Google Scholar] [CrossRef]
- Viau, A.; El Karoui, K.; Laouari, D.; Burtin, M.; Nguyen, C.; Mori, K.; Pillebout, E.; Berger, T.; Mak, T.W.; Knebelmann, B.; et al. Lipocalin 2 is Essential for Chronic Kidney Disease Progression in Mice and Humans. J. Clin. Investig. 2010, 120, 4065–4076. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Weiskirchen, S.; Weiskirchen, R. Lipocalin 2 (LCN2) Expression in Hepatic Malfunction and Therapy. Front. Physiol. 2016, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, E.; Andersen, B. FGF19 and FGF21 for the Treatment of NASH—two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 from Mice to Human. Front. Endocrinol. 2020, 11, 601349. [Google Scholar] [CrossRef] [PubMed]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The Effects of LY2405319, an FGF21 Analog, in Obese Human Subjects with Type 2 Diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A. FGF-21 as a Novel Metabolic Regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y. Fibroblast Growth Factor 21 Analogs for Treating Metabolic Disorders. Front. Endocrinol. 2015, 6, 168. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.M.; Guan, Y.; Gao, B. Targeting Adipose Tissue to Tackle NASH: SPARCL1 as an Emerging Player. J. Clin. Investig. 2021, 131, e153640. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological Mechanisms and Therapeutic Targets of Fatty Liver Diseases. Cell. Mol. Immunol. 2021, 18, 73–91. [Google Scholar] [CrossRef]
- Jin, J.; Wahlang, B.; Shi, H.; Hardesty, J.E.; Falkner, K.C.; Head, K.Z.; Srivastava, S.; Merchant, M.L.; Rai, S.N.; Cave, M.C. Dioxin-Like and Non-Dioxin-Like PCBs Differentially Regulate the Hepatic Proteome and Modify Diet-Induced Nonalcoholic Fatty Liver Disease Severity. Med. Chem. Res. 2020, 29, 1247–1263. [Google Scholar] [CrossRef]
- Sun, J.; Fang, R.; Wang, H.; Xu, D.; Yang, J.; Huang, X.; Cozzolino, D.; Fang, M.; Huang, Y. A Review of Environmental Metabolism Disrupting Chemicals and Effect Biomarkers Associating Disease Risks: Where Exposomics Meets Metabolomics. Environ. Int. 2022, 158, 106941. [Google Scholar] [CrossRef]
- Costello, E.; Rock, S.; Stratakis, N.; Eckel, S.P.; Walker, D.I.; Valvi, D.; Cserbik, D.; Jenkins, T.; Xanthakos, S.A.; Kohli, R. Exposure to Per-and Polyfluoroalkyl Substances and Markers of Liver Injury: A Systematic Review and Meta-Analysis. Environ. Health Perspect. 2022, 130, 046001. [Google Scholar] [CrossRef]
- Rantakokko, P.; Männistö, V.; Airaksinen, R.; Koponen, J.; Viluksela, M.; Kiviranta, H.; Pihlajamäki, J. Persistent Organic Pollutants and Non-Alcoholic Fatty Liver Disease in Morbidly Obese Patients: A Cohort Study. Environ. Health 2015, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Deierlein, A.L.; Rock, S.; Park, S. Persistent Endocrine-Disrupting Chemicals and Fatty Liver Disease. Curr. Environ. Health Rep. 2017, 4, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Hardesty, J.E.; Jin, J.; Falkner, K.C.; Cave, M.C. Polychlorinated Biphenyls and Nonalcoholic Fatty Liver Disease. Curr. Opin. Toxicol. 2019, 14, 21–28. [Google Scholar] [CrossRef]
- Ruzzin, J.; Petersen, R.; Meugnier, E.; Madsen, L.; Lock, E.; Lillefosse, H.; Ma, T.; Pesenti, S.; Sonne, S.B.; Marstrand, T.T. Persistent Organic Pollutant Exposure Leads to Insulin Resistance Syndrome. Environ. Health Perspect. 2010, 118, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Boga, S.; Alkim, H.; Alkim, C.; Koksal, A.R.; Bayram, M.; Yilmaz Ozguven, M.B.; Tekin Neijmann, S. The Relationship of Serum Hemojuvelin and Hepcidin Levels with Iron Overload in Nonalcoholic Fatty Liver Disease. J. Gastrointestin Liver Dis. 2015, 24, 293–300. [Google Scholar] [CrossRef]
- Sumida, Y.; Nakashima, T.; Yoh, T.; Furutani, M.; Hirohama, A.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Okanoue, T. Serum Thioredoxin Levels as a Predictor of Steatohepatitis in Patients with Nonalcoholic Fatty Liver Disease. J. Hepatol. 2003, 38, 32–38. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron Depletion by Phlebotomy Improves Insulin Resistance in Patients with Nonalcoholic Fatty Liver Disease and Hyperferritinemia: Evidence from a Case-Control Study. Off. J. Am. Coll. Gastroenterol. ACG 2007, 102, 1251–1258. [Google Scholar] [CrossRef]
- Iwasa, M.; Hara, N.; Iwata, K.; Ishidome, M.; Sugimoto, R.; Tanaka, H.; Fujita, N.; Kobayashi, Y.; Takei, Y. Restriction of Calorie and Iron Intake Results in Reduction of Visceral Fat and Serum Alanine Aminotransferase and Ferritin Levels in Patients with Chronic Liver Disease. Hepatol. Res. 2010, 40, 1188–1194. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Iron Homeostasis and the Inflammatory Response. Annu. Rev. Nutr. 2010, 30, 105–122. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Lin, R.; Li, L.; Zhang, D.; Li, X.; Liu, S. PCB-77 Disturbs Iron Homeostasis through Regulating Hepcidin Gene Expression. Gene 2013, 532, 146–151. [Google Scholar] [CrossRef]
- Wang, X.; Xia, T. New Insights into Disruption of Iron Homeostasis by Environmental Pollutants. J. Environ. Sci. 2015, 34, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Encarnação, T.; Pais, A.A.; Campos, M.G.; Burrows, H.D. Endocrine Disrupting Chemicals: Impact on Human Health, Wildlife and the Environment. Sci. Prog. 2019, 102, 3–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, Y.; Zhong, K.; Wang, C.; Xu, X. The Associations between Endocrine Disrupting Chemicals and Markers of Inflammation and Immune Responses: A Systematic Review and Meta-Analysis. Ecotoxicol. Environ. Saf. 2022, 234, 113382. [Google Scholar] [CrossRef] [PubMed]
- Feng, D. The Alteration of Immune Cells in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Liver Res. 2020, 4, 23–27. [Google Scholar] [CrossRef]
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased Hepatic Myeloperoxidase Activity in Obese Subjects with Nonalcoholic Steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482. [Google Scholar] [CrossRef]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A Front-line Defender Against Phagocytosed Microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.H.; Jeong, S.; Kim, H.; Ahn, K.S.; Cho, J.Y.; Yoon, Y.; Han, H. Increased Urinary Lipocalin-2 Reflects Matrix Metalloproteinase-9 Activity in Chronic Hepatitis C with Hepatic Fibrosis. Tohoku J. Exp. Med. 2010, 222, 319–327. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; van de Leur, E.; Zimmermann, H.W.; Karlmark, K.R.; Tihaa, L.; Haas, U.; Tacke, F.; Berger, T.; Mak, T.W.; Weiskirchen, R. Protective Effects of Lipocalin-2 (LCN2) in Acute Liver Injury Suggest a Novel Function in Liver Homeostasis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 660–673. [Google Scholar] [CrossRef]
- Xiao, X.; Yeoh, B.S.; Saha, P.; Olvera, R.A.; Singh, V.; Vijay-Kumar, M. Lipocalin 2 Alleviates Iron Toxicity by Facilitating Hypoferremia of Inflammation and Limiting Catalytic Iron Generation. Biometals 2016, 29, 451–465. [Google Scholar] [CrossRef]
- Ye, D.; Yang, K.; Zang, S.; Lin, Z.; Chau, H.; Wang, Y.; Zhang, J.; Shi, J.; Xu, A.; Lin, S. Lipocalin-2 Mediates Non-Alcoholic Steatohepatitis by Promoting Neutrophil-Macrophage Crosstalk Via the Induction of CXCR2. J. Hepatol. 2016, 65, 988–997. [Google Scholar] [CrossRef]
- Wieser, V.; Tymoszuk, P.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Pfister, A.; Lichtmanegger, L.; Gerner, R.; Drach, M. Lipocalin 2 Drives Neutrophilic Inflammation in Alcoholic Liver Disease. J. Hepatol. 2016, 64, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Law, I.K.; Xu, A.; Lam, K.S.; Berger, T.; Mak, T.W.; Vanhoutte, P.M.; Liu, J.T.; Sweeney, G.; Zhou, M.; Yang, B. Lipocalin-2 Deficiency Attenuates Insulin Resistance Associated with Aging and Obesity. Diabetes 2010, 59, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, M.; Müller, T.D. A New FGF21 Analog for the Treatment of Fatty Liver Disease. Diabetes 2020, 69, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wu, Y.; Ye, X.; Ma, L.; Qi, J.; Yu, D.; Wei, Y.; Lin, G.; Ren, G.; Li, D. FGF21 Ameliorates Nonalcoholic Fatty Liver Disease by Inducing Autophagy. Mol. Cell. Biochem. 2016, 420, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.; Lindberg, R.A. Fibroblast Growth Factor 21 Reverses Hepatic Steatosis, Increases Energy Expenditure, and Improves Insulin Sensitivity in Diet-Induced Obese Mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Tillman, E.J.; Rolph, T. FGF21: An Emerging Therapeutic Target for Non-Alcoholic Steatohepatitis and Related Metabolic Diseases. Front. Endocrinol. 2020, 11, 601290. [Google Scholar] [CrossRef]
- National Toxicology Program. Toxicology and Carcinogenesis Studies of a Binary Mixture of 3,3′,4,4′,5-Pentachlorobiphenyl (PCB 126) (Cas no. 57465-28-8) and 2,2′,4,4′,5,5′-Hexachlorobiphenyl (PCB 153) (CAS no. 35065-27-1) in Female Harlan Sprague-Dawley Rats (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser. 2006, 530, 1–258. [Google Scholar]
- Kim, H.Y.; Park, S.Y.; Lee, M.H.; Rho, J.H.; Oh, Y.J.; Jung, H.U.; Yoo, S.H.; Jeong, N.Y.; Lee, H.J.; Suh, S. Hepatic STAMP2 Alleviates High Fat Diet-Induced Hepatic Steatosis and Insulin Resistance. J. Hepatol. 2015, 63, 477–485. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.Y.; Yoo, Y.H. Recombinant FGF21 Attenuates Polychlorinated Biphenyl-Induced NAFLD/NASH by Modulating Hepatic Lipocalin-2 Expression. Int. J. Mol. Sci. 2022, 23, 8899. https://doi.org/10.3390/ijms23168899
Kim HY, Yoo YH. Recombinant FGF21 Attenuates Polychlorinated Biphenyl-Induced NAFLD/NASH by Modulating Hepatic Lipocalin-2 Expression. International Journal of Molecular Sciences. 2022; 23(16):8899. https://doi.org/10.3390/ijms23168899
Chicago/Turabian StyleKim, Hye Young, and Young Hyun Yoo. 2022. "Recombinant FGF21 Attenuates Polychlorinated Biphenyl-Induced NAFLD/NASH by Modulating Hepatic Lipocalin-2 Expression" International Journal of Molecular Sciences 23, no. 16: 8899. https://doi.org/10.3390/ijms23168899
APA StyleKim, H. Y., & Yoo, Y. H. (2022). Recombinant FGF21 Attenuates Polychlorinated Biphenyl-Induced NAFLD/NASH by Modulating Hepatic Lipocalin-2 Expression. International Journal of Molecular Sciences, 23(16), 8899. https://doi.org/10.3390/ijms23168899