
Study of the Experimental and Simulated Vibrational Spectra Together with Conformational Analysis of Thioether Cyanobiphenyl-Based Liquid Crystal Dimers




Abstract
:1. Introduction
2. Results
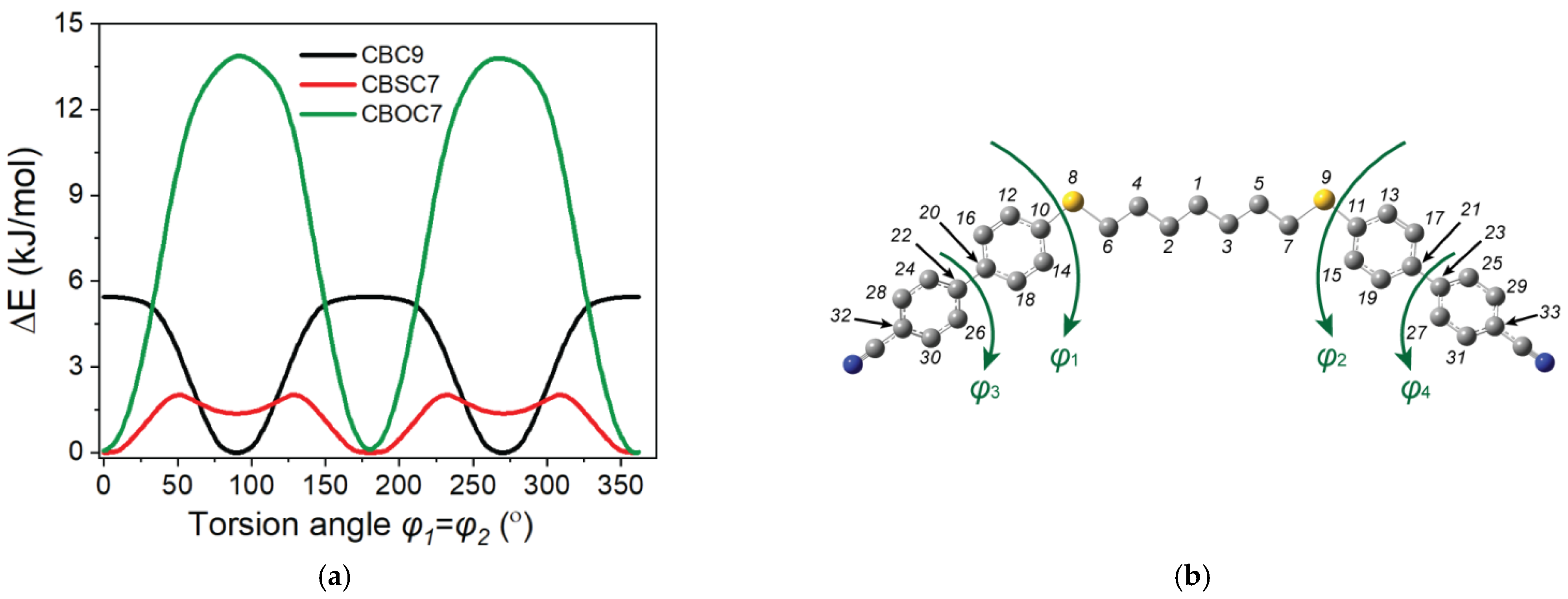
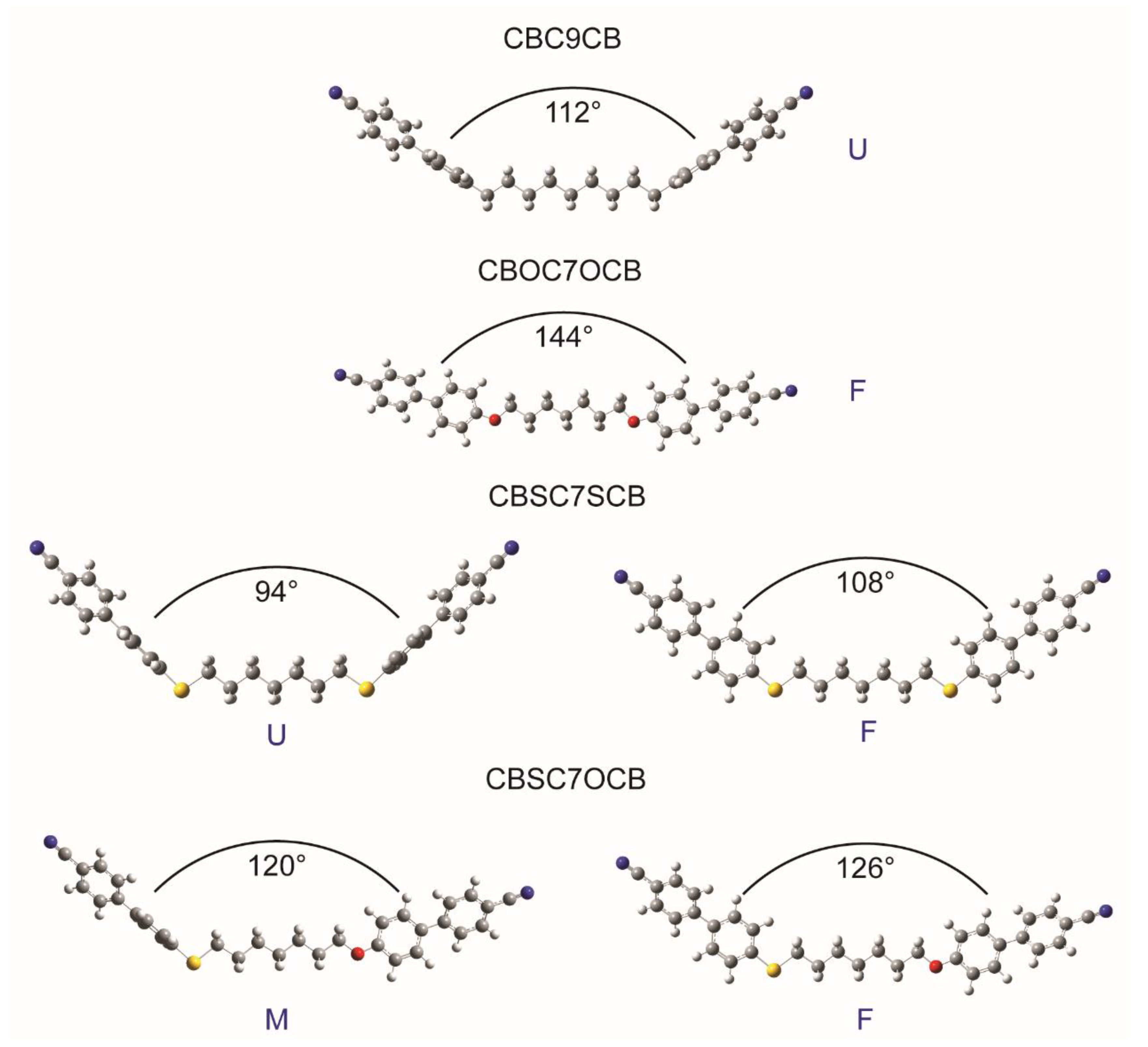
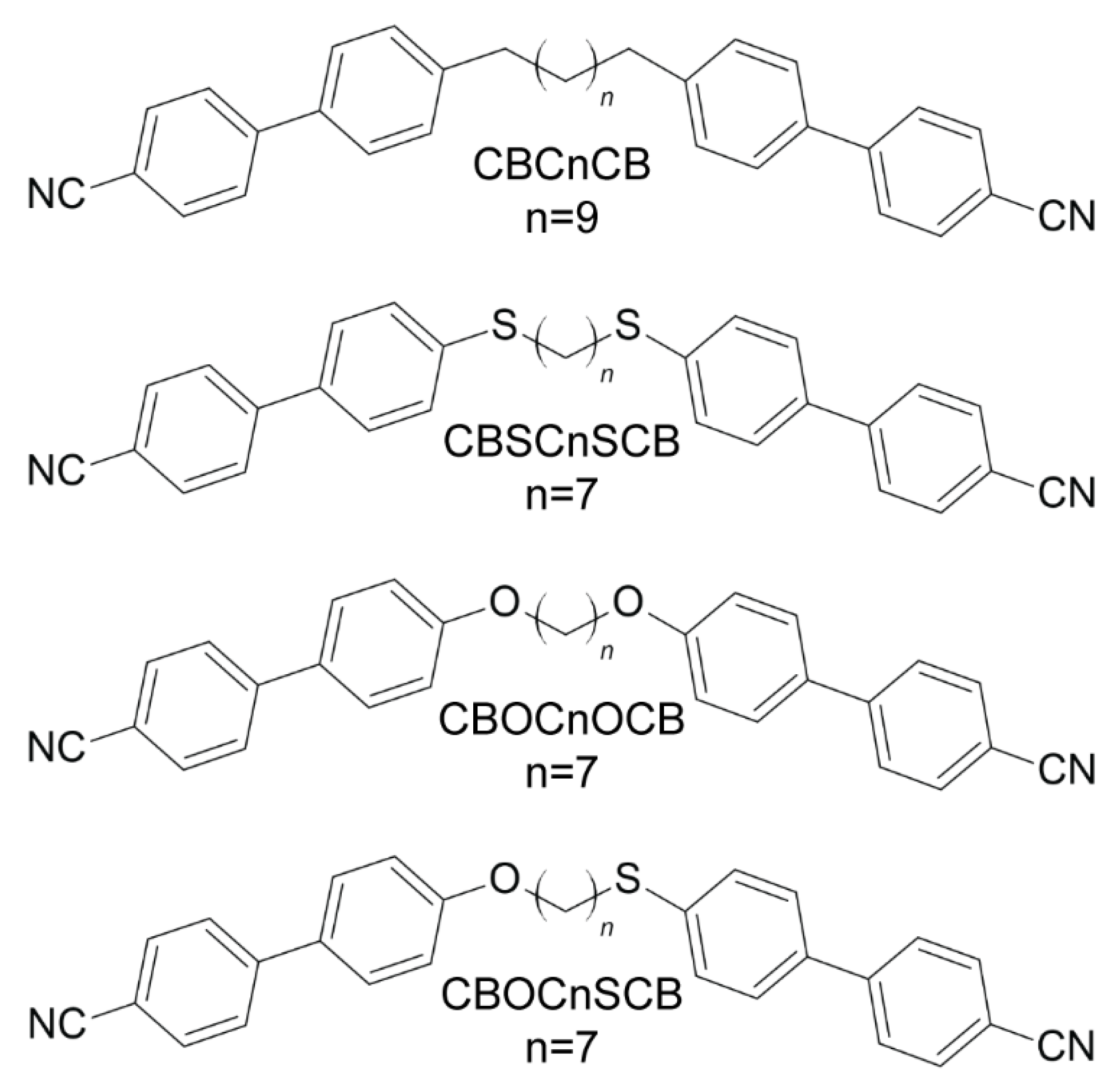
2.1. Molecular Structures
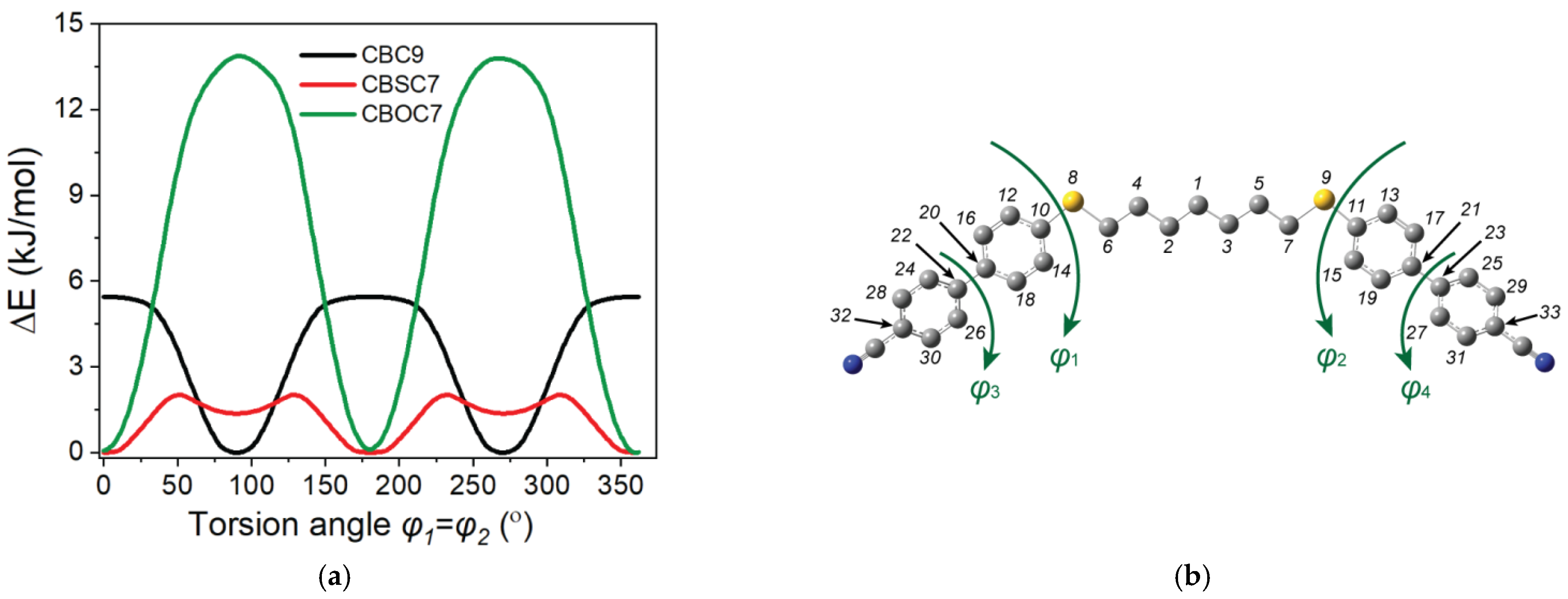
Dihedral Angle Distributions and Conformations
2.2. Molecular Vibrations
2.3. Vibrational Spectra and Their Assignments
- 500–600 cm−1 and 700–900 cm−1 ranges, which cover the deformational vibrations of the carbon atoms (C-C) and hydrogen atoms (C-H) out of the benzene plane;
- 900–1650 cm−1 range, which includes both the characteristic deformation vibrations in the benzene plane as well as the deformation vibrations of the methylene groups of the alkyl chain of the linker in a dimer;
- 2100–2400 cm−1 range, which includes the stretching vibrations of the cyan group (C≡N) that were observed as a sharp and very intense peak in the experimental spectrum;
- 2800–2950 cm−1 range, which includes the C-H stretching vibrations of the methylene groups. In this range, the vibrations were not well reproduced by the theoretical spectra because the calculations did not take into account the anharmonic effect;
- 2900–3100 cm−1 range, which represents the stretching vibrations of the hydrogen atoms (C-H) in the aromatic ring. These vibrations were also not well reproduced by the theoretical frequencies. The bands in this range corresponded to the mixed vibrations, which were strongly overlapping, and the vibrations stretching the C-H hydrogen atoms, significantly disturbed by the Fermi resonance effect.
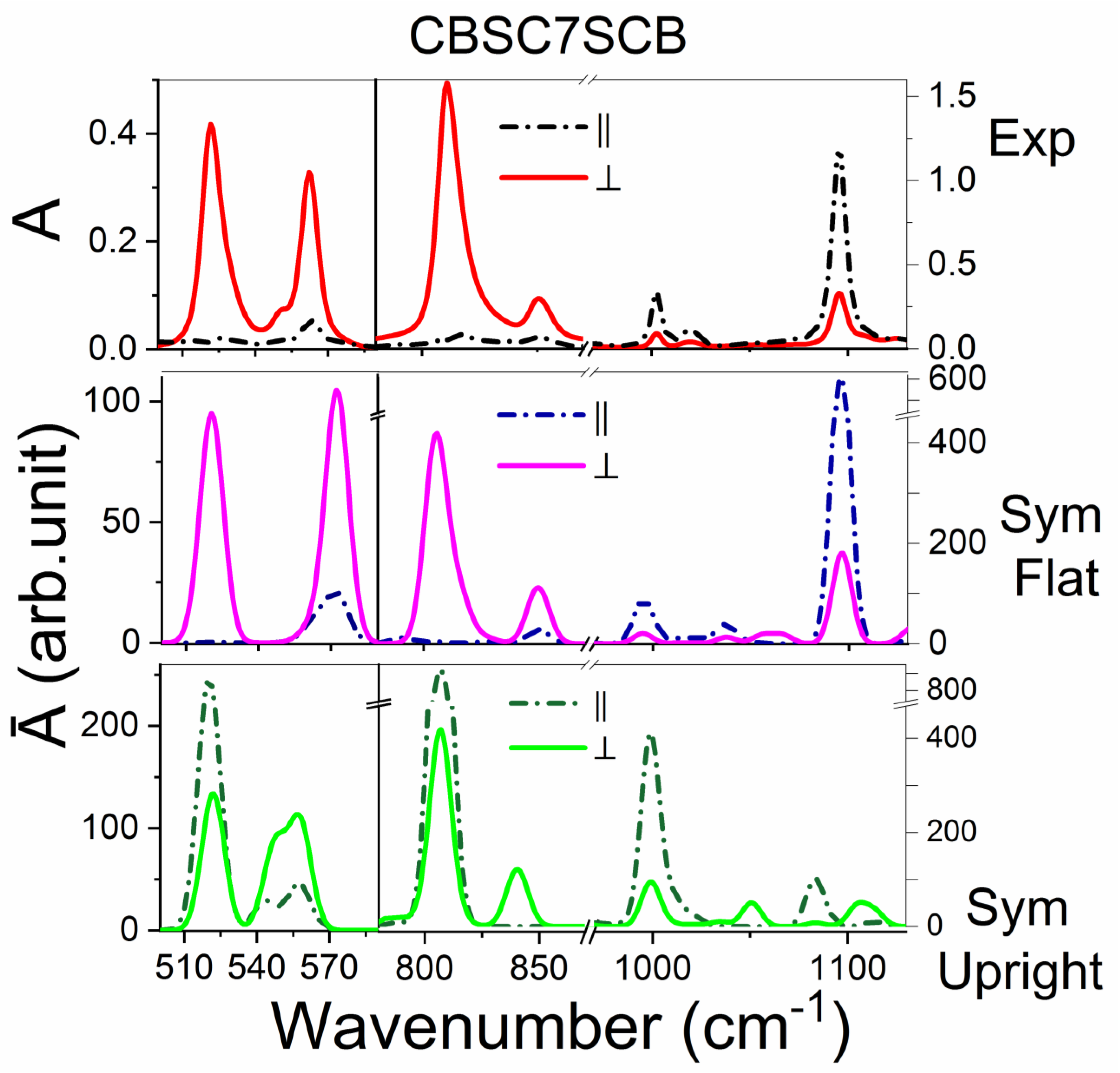
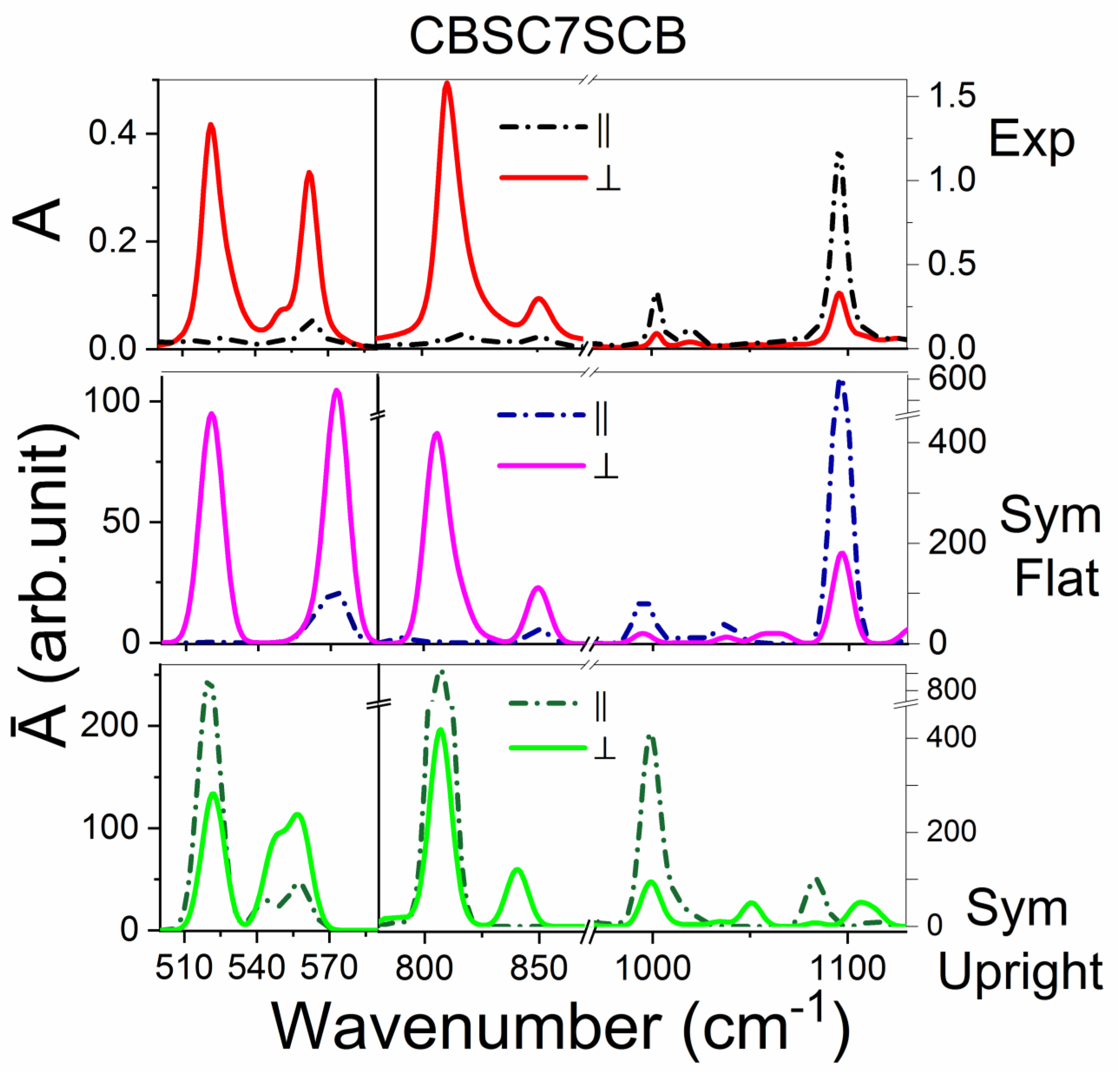
2.3.1. The CBSC7SCB Dimer
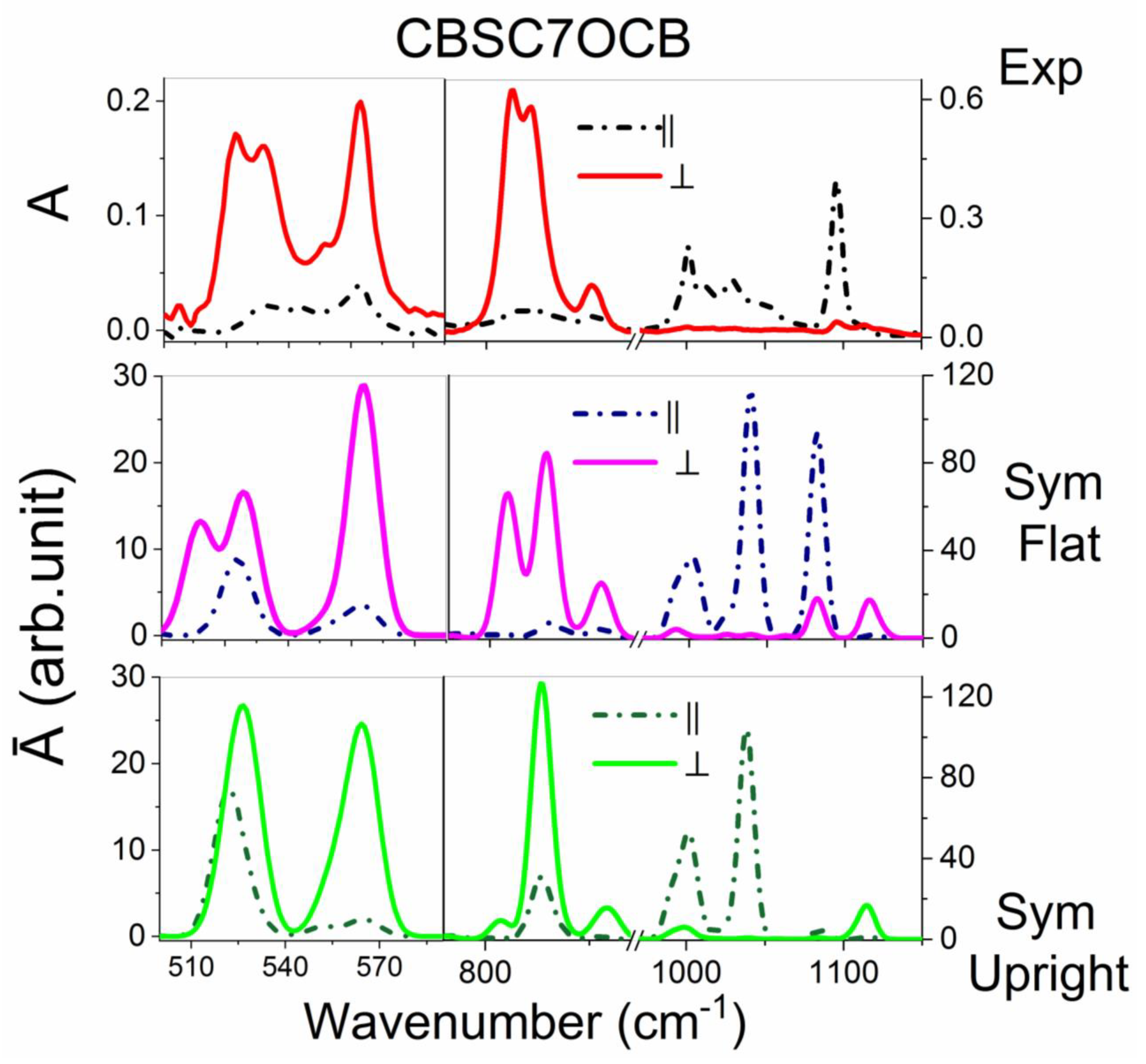
2.3.2. The CBSC7OCB Dimer
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Infrared Spectroscopy
4.3. Density Functional Theory Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Gennes, P.G. Soft Matter. Science 1992, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, S.; Madhusudana, N.V. Liquid Crystals. Annu. Rev. Mater. Sci. 1980, 10, 133. [Google Scholar] [CrossRef]
- Alfutimie, A.; Curtis, R.T.G.J. Tiddy in Handbook of Liquid Crystals, 2nd ed.; Goodby, J.W., Collings, P.J., Kato, T., Tschierske, C., Gleeson, H., Raynes, P., Eds.; Wiley-VCH: Weinheim, Germany, 2014; Volume 6, Chapter 12. [Google Scholar]
- Kirov, N.; Simova, P. Infrared Absorption Spectra of Liquid Crystals. Mol. Cryst. Liq. Cryst. 1975, 30, 59–71. [Google Scholar]
- Fringeli, U.P.; Schadt, M.; Riĥak, P.; Günthard, H.H. Hydrocarbon Chain Ordering in Liquid Crystals Investigated by Means of Infrared Attenuated Total Reflection (IR-ATR) Spectroscopy. Z. Naturforsch. 1976, 31, 1098–1107. [Google Scholar] [CrossRef]
- Kruk, G.; Kocot, A.; Wrzalik, R.; Vij, J.K.; Karthaus, O.; Ringsdorf, H. Infrared absorption study of hexapentyloxytriphenylene a discotic liquid crystal. Liq. Cryst. 1993, 14, 807–819. [Google Scholar] [CrossRef]
- Sakagami, S.; Nonaka, K.; Koga, T.; Takase, A. Infrared Spectroscopic Study of Liquid Crystalline N-[4-(4-n-alkoxybenzoyloxy)-2-hydroxybenzylidene]-3-cyanoanilines. Mol. Cryst. Liq. Cryst. 1998, 312, 23–32. [Google Scholar] [CrossRef]
- Orgasińska, B.; Perova, T.S.; Merkel, K.; Kocot, A.; Vij, J.K. Surface phenomena in discotic liquid crystals investigated using polarized FTIR transmission spectroscopy. Mat. Sci. Eng. C 1999, 8–9, 283–289. [Google Scholar] [CrossRef]
- Jang, W.G.; Park, C.S.; Kim, K.H.; Glaser, M.A.; Clark, N.A. Infrared spectroscopic study of molecular hydrogen bonding in chiral smectic liquid crystals. Phys. Rev. E 2000, 62, 5027–5035. [Google Scholar] [CrossRef]
- Merkel, K.; Wrzalik, R.; Kococt, A. Calculation of vibrational spectra for cyanobiphenyl liquid crystals. J. Mol. Struct. 2001, 563–564, 477–490. [Google Scholar] [CrossRef]
- Wrzalik, R.; Merkel, K.; Kocot, A.; Cieplak, B. Analysis of experimental and simulated vibrational spectra for the antiferroelectric liquid crystal 12OBBB1M6. J. Chem. Phys. 2002, 117, 4889–4895. [Google Scholar] [CrossRef]
- Merkel, K.; Kocot, A.; Vij, J.K.; Korlacki, R.; Mehl, G.H.; Meyer, T. Thermotropic Biaxial Nematic Phase in Liquid Crystalline Organo-Siloxane Tetrapodes. Phys. Rev. Lett. 2004, 93, 237801–237805. [Google Scholar] [CrossRef] [Green Version]
- Merkel, K.; Kocot, A.; Vij, J.K.; Stevenson, P.J.; Panov, A.; Rodriguez, D. Anomalous temperature dependence of layer spacing of de Vries liquid crystals: Compensation model. Appl. Phys. Lett. 2016, 108, 243301–243306. [Google Scholar] [CrossRef]
- Imrie, C.T.; Luckhurst, G.R. Liquid Crystal Dimers and Oligomers in Handbook of Liquid Crystals Vol. 2B: Low Molecular Weight Liquid Crystals; Demus, D., Goodby, J.W., Gray, G.W., Spiess, H.W., Vill, V., Eds.; Wiley-VCH: Weinheim, Germany, 1998; Chapter X; p. 801. [Google Scholar]
- Imrie, C.T.; Henderson, P.A. Liquid crystal dimers and higher oligomers: Between monomers and polymers. Chem. Soc. Rev. 2007, 36, 2096–2124. [Google Scholar] [CrossRef]
- Mandle, R.J. The dependency of twist-bend nematic liquid crystals on molecular structure: A progression from dimers to trimers, oligomers and polymers. Soft Matter 2016, 12, 7883–7901. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.M.; Clarke, M.J.; Blatch, A.E.; Coles, H.J. Structure-flexoelastic properties of bimesogenic liquid crystals. Phys. Rev. E 2007, 75, 041701–041710. [Google Scholar] [CrossRef] [Green Version]
- Shamid, S.M.; Dhakal, S.; Selinger, J.V. Statistical mechanics of bend flexoelectricity and the twist-bend phase in bent-core liquid crystals. Phys. Rev. E 2013, 87, 052503. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Luckhurst, G.R.; Dozov, I. Flexoelectrically driven electroclinic effect in the twist-bend nematic phase of achiral molecules with bent shapes. Phys. Rev. Lett. 2013, 111, 067801–067806. [Google Scholar] [CrossRef]
- Panov, V.P.; Balachandran, R.; Vij, J.K.; Tamba, M.G.; Kohlmeier, A.; Mehl, G.H. Microsecond linear optical response in the unusual nematic phase of achiral bimesogens. Appl. Phys. Lett. 2011, 99, 261903–261906. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Porada, J.H.; Hooper, J.B.; Klittnick, A.; Shen, Y.; Tuchband, M.R.; Korblova, E.; Bedrov, D.; Walba, D.M.; Glaser, M.A.; et al. Chiral heliconical ground state of nanoscale pitch in a nematic liquid crystal of achiral molecular dimers. Proc. Natl. Acad. Sci. USA 2013, 110, 15931–15936. [Google Scholar] [CrossRef] [Green Version]
- Panov, V.P.; Vij, J.K.; Mehl, G.H. Twist-bend nematic phase in cyanobiphenyls and difluoroterphenyls bimesogens. Liq. Cryst. 2017, 44, 147–159. [Google Scholar]
- Meyer, C. Nematic twist-bend phase under external constraints. Liq. Cryst. 2016, 43, 2144–2162. [Google Scholar] [CrossRef]
- Panov, V.P.; Nagaraj, M.; Vij, J.K.; Panarin, Y.P.; Kohlmeier, A.; Tamba, M.G.; Lewis, R.A.; Mehl, G.H. Spontaneous Periodic Deformations in Nonchiral Planar-Aligned Bimesogens with a Nematic-Nematic Transition and a Negative Elastic Constant. Phys. Rev. Lett. 2010, 105, 167801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borshch, V.; Kim, Y.K.; Xiang, J.; Gao, M.; Jákli, A.; Panov, V.P.; Vij, J.K.; Imrie, C.T.; Tamba, M.G.; Mehl, G.H.; et al. Nematic twist-bend phase with nanoscale modulation of molecular orientation. Nat. Commun. 2013, 4, 2635–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsouzi, Z.; Shamid, S.M.; Borshch, V.; Challa, P.K.; Baldwin, A.R.; Tamba, M.G.; Welch, C.; Mehl, G.H.; Gleeson, J.T.; Jakli, O.D.A.; et al. Fluctuation Modes of a Twist-Bend Nematic Liquid Crystal. Phys. Rev. X 2016, 6, 021041-16. [Google Scholar] [CrossRef]
- Chen, D.; Nakata, M.; Shao, R.; Tuchband, M.R.; Shuai, M.; Baumeister, U.; Weissflog, W.; Walba, D.M.; Glaser, M.A.; Maclennan, J.E.; et al. Twist-bend heliconical chiral nematic liquid crystal phase of an achiral rigid bent-core mesogen. Phys. Rev. E 2014, 89, 022506. [Google Scholar] [CrossRef] [Green Version]
- Babakhanova, G.; Parsouzi, Z.; Paladugu, S.; Wang, H.; Nastishin, Y.A.; Shiyanovskii, S.V.; Sprunt, S.; Lavrentovich, O.D. Elastic and viscous properties of the nematic dimer CB7CB. Phys. Rev. E 2017, 96, 062704. [Google Scholar] [CrossRef] [Green Version]
- Connor, P.L.M.; Mandle, R.J. Chemically induced splay nematic phase with micron scale periodicity. Soft Matter 2020, 16, 324–329. [Google Scholar] [CrossRef] [Green Version]
- Merkel, K.; Kocot, A.; Welch, C.; Mehl, G.H. Soft modes of the dielectric response in the twist–bend nematic phase and identification of the transition to a nematic splay bend phase in the CBC7CB dimer. Phys. Chem. Chem. Phys. 2019, 21, 22839–22848. [Google Scholar] [CrossRef]
- Meyer, C.; Blanc, C.; Luckhurst, G.R.; Davidson, P.; Dozov, I. Biaxiality-driven twist-bend to splay-bend nematic phase transition induced by an electric field. Sci. Adv. 2020, 6, eabb8212. [Google Scholar] [CrossRef]
- Zhu, C.; Tuchband, M.R.; Young, A.; Shuai, M.; Scarbrough, A.; Walba, D.M.; Maclennan, J.E.; Wang, C.; Hexemer, A.; Clark, N.A. Resonant carbon k-edge soft x-ray scattering from lattice-free heliconical molecular ordering: Soft dilative elasticity of the twist-bend liquid crystal phase. Phys. Rev. Lett. 2016, 116, 147803–147809. [Google Scholar] [CrossRef] [Green Version]
- Cruickshank, E.; Salamończyk, M.; Pociecha, D.; Strachan, G.J.; Storey, J.M.D.; Wang, C.; Feng, J.; Zhu, C.; Gorecka, E.; Imrie, C.T. Sulfur-linked cyanobiphenyl-based liquid crystal dimers and the twist-bend nematic phase. Liq. Cryst. 2019, 46, 1595–1609. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Feng, J.; Nallapaneni, A.; Arakawa, Y.; Zhao, K.; Zhang, H.; Mehl, G.H.; Zhu, C.; Liu, F. Deciphering helix assembly in the heliconical nematic phase via tender resonant X-ray scattering. J. Mater. Chem. C 2021, 9, 10020–10028. [Google Scholar] [CrossRef]
- Cestari, M.; Diez-Berart, S.; Dunmur, D.A.; Ferrarini, A.; de la Fuente, M.R.; Jackson, D.J.B.; Lopez, D.O.; Luckhurst, G.R.; Perez-Jubindo, M.A.; Richardson, R.M.; et al. Phase behavior and properties of the liquid-crystal dimer 1,7-bis(4-cyanobiphenyl-4-yl) heptane: A twist-bend nematic liquid crystal. Phys. Rev. E 2011, 84, 031704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlem, K.; Čopič, M.; Luckhurst, G.R.; Mertelj, A.; Parri, O.; Richardson, R.M.; Snow, B.D.; Timimi, B.A.; Tuffin, R.P.; Wilkes, D. Chemically induced twist-bend nematic liquid crystals, liquid crystal dimers, and negative elastic constants. Phys. Rev. E 2013, 88, 022503. [Google Scholar] [CrossRef]
- Stevenson, W.D.; Zou, H.; Zeng, X.; Welch, C.; Ungar, G.; Mehl, G.H. Dynamic calorimetry and XRD studies of the nematic and twist-bend nematic phase transitions in a series of dimers with increasing spacer length. Phys. Chem. Chem. Phys. 2018, 20, 25268–25274. [Google Scholar] [CrossRef] [Green Version]
- Salamończyk, M.; Vaupotič, N.; Pociecha, D.; Wang, C.; Zhu, C.; Gorecka, E. Structure of nanoscale-pitch helical phases: Blue phase and twist-bend nematic phase resolved by resonant soft X-ray scattering. Soft Matter 2017, 13, 6694–6699. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, W.; Ahmed, Z.; Zeng, X.; Welch, C.; Ungar, G.; Mehl, G. Molecular organization in the twist–bend nematic phase by resonant X-ray scattering at the Se K-edge and by SAXS, WAXS and GIXRD. Phys. Chem. Chem. Phys. 2017, 19, 13449–13454. [Google Scholar] [CrossRef] [Green Version]
- Tuchband, M.R.; Paterson, D.A.; Salamo ´nczyk, M.; Norman, V.A.; Scarbrough, A.N.; Forsyth, E.; Garcia, E.; Wang, C.; Storey, J.M.; Walba, D.M.; et al. Distinct differences in the nanoscale behaviors of the twist–bend liquid crystal phase of a flexible linear trimer and homologous dimer. Proc. Natl. Acad. Sci. USA 2019, 116, 10698–10704. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.; Feng, C.; Welch, C.; Mehl, G.H.; Feng, J.; Zhu, C.; Gleeson, J.; Sprunt, S.; Jákli, A. The interplay between spatial and heliconical orientational order in twist-bend nematic materials. Phys. Chem. Chem. Phys. 2021, 23, 4055–4063. [Google Scholar] [CrossRef]
- Zhang, Z.; Panov, V.P.; Nagaraj, M.; Mandle, R.J.; Goodby, J.W.; Luckhurst, G.R.; Jones, J.C.; Gleeson, H.F. Raman scattering studies of order parameters in liquid crystalline dimers exhibiting the nematic and twist-bend nematic phases. J. Mater. Chem. C 2015, 3, 10007–10016. [Google Scholar] [CrossRef] [Green Version]
- Knežević, A.; Dokli, I.; Sapunar, M.; Sěgota, S.; Baumeister, U.; Lesac, A. Induced smectic phase in binary mixtures of twist-bend nematogens. Beilstein J. Nanotechnol. 2018, 9, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkel, K.; Loska, B.; Welch, C.; Mehl, G.H.; Kocot, A. Molecular biaxiality determines the helical structure—Infrared measurements of the molecular order in the nematic twist-bend phase of difluoro terphenyl dimer. Phys. Chem. Chem. Phys. 2021, 23, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Merkel, K.; Loska, B.; Welch, C.; Mehl, G.H.; Kocot, A. The role of intermolecular interactions in stabilising the structure of the nematic twist-bend phase. RSC Adv. 2021, 11, 2917–2925. [Google Scholar] [CrossRef] [PubMed]
- Kocot, A.; Loska, B.; Arakawa, Y.; Merkel, K. Structure of the twist-bend nematic phase with respect to the orientational molecular order of the thioether-linked dimers. Phys. Rev. E 2022, 105, 044701. [Google Scholar] [CrossRef]
- Beguin, L.; Emsley, J.W.; Lelli, M.; Lesage, A.; Luckhurst, G.R.; Timimi, B.A.; Zimmermann, H. The chirality of a twist–bend nematic phase identified by NMR spectroscopy. J. Phys. Chem. B 2012, 116, 7940–7951. [Google Scholar] [CrossRef] [PubMed]
- Emsley, W.; Lesot, P.; Luckhurst, G.R.; Meddour, A.; Merlet, D. Chiral solutes can seed the formation of enantiomorphic domains in a twist-bend nematic liquid crystal. Phys. Rev. E 2013, 87, 040501. [Google Scholar] [CrossRef] [PubMed]
- Emsley, J.W.; Lelli, M.; Lesage, A.; Luckhurst, G.R. A comparison of the conformational distributions of the achiral symmetric liquid crystal dimer cb7cb in the achiral nematic and chiral twist-bend nematic phases. J. Phys. Chem. B 2013, 117, 6547–6557. [Google Scholar] [CrossRef]
- Jokisaari, J.P.; Luckhurst, G.R.; Timimi, B.A.; Zhu, J.F.; Zimmermann, H. Twist-bend nematic phase of the liquid crystal dimer cb7cb: Orientational order and conical angle determined by xe-129 and h-2 nmr spectroscopy. Liq. Cryst. 2015, 42, 708–721. [Google Scholar]
- Ivšić, T.; Vinković, M.; Baumeister, U.; Mikleušević, A.; Lesac, A. Towards understanding the NTB phase: A combined experimental, computational and spectroscopic study. RSC Adv. 2016, 6, 5000–5007. [Google Scholar] [CrossRef]
- Forsyth, E.; Paterson, D.A.; Cruickshank, E.; Strachan, G.J.; Gorecka, E.; Walker, R.; Storey, J.M.D.; Imrie, C.T. Liquid crystal dimers and the twist-bend nematic phase: On the role of spacers and terminal alkyl chains. J. Mol. Liq. 2020, 320, 114391. [Google Scholar] [CrossRef]
- Mandle, R.J.; Goodby, J.W. Molecular Flexibility and Bend in Semi-Rigid Liquid Crystals: Implications for the Heliconical Nematic Ground State. Chem. Eur. J. 2019, 25, 14454–14459. [Google Scholar] [CrossRef] [PubMed]
- Archbold, C.T.; Mandle, R.J.; Andrews, J.L.; Cowling, S.J.; Goodby, J.W. Conformational landscapes of bimesogenic compounds and their implications for the formation of modulated nematic phases. Liq. Cryst. 2017, 44, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Pocock, E.E.; Mandle, R.J.; Goodby, J.W. Molecular shape as a means to control the incidence of the nanostructured twist bend phase. Soft Matter 2018, 14, 2508–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pocock, E.E.; Mandle, R.J.; Goodby, J.W. Experimental and Computational Study of a Liquid Crystalline Dimesogen Exhibiting Nematic, Twist-Bend Nematic, Intercalated Smectic, and Soft Crystalline Mesophases. Molecules 2021, 26, 532. [Google Scholar] [CrossRef] [PubMed]
- Longa, L.; Tomczyk, W. Twist−Bend Nematic Phase from the Landau−de Gennes Perspective. J. Phys. Chem. C 2020, 124, 22761–22775. [Google Scholar] [CrossRef]
- Paterson, D.A.; Gao, M.; Kim, Y.-K.; Jamali, A.; Finley, K.L.; Robles-Herna´ndez, B.; Diez-Berart, S.; Salud, J.; de la Fuente, M.R.; Timimi, B.A.; et al. Understanding the twist-bend nematic phase: The characterisation of 1-(4-cyanobiphenyl-40-yloxy)-6-(4-cyanobiphenyl-40-yl)hexane (CB6OCB) and comparison with CB7CB. Soft Matter 2016, 12, 6827–6840. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, Y.; Komatsu, K.; Tsuji, H. Twist-bend nematic liquid crystals based on thioether linkage. New J. Chem. 2019, 43, 6786–6793. [Google Scholar] [CrossRef]
- Kumar, A. Dependency of the twist-bend nematic phase formation on the molecular shape of liquid crystal dimers: A view through the lens of DFT. J. Mol. Liq. 2022, 354, 118858. [Google Scholar] [CrossRef]
- Lesac, A.; Baumeister, U.; Dokli, I.; Hameršak, Z.; Kontrec, I.D.; Viskić, M.; Knežević, A.; Mandle, R.J. Geometric aspects influencing N-N TB transition—Implication of intramolecular torsion. Liq. Cryst. 2018, 45, 1101–1110. [Google Scholar] [CrossRef]
- Memmer, R. Liquid crystal phases of achiral banana-shaped molecules: A computer simulation study. Liq. Cryst. 2002, 29, 483–496. [Google Scholar] [CrossRef]
- Wilson, M.R.; Yu, G.; Potter, T.D.; Walker, M.; Gray, S.J.; Li, J.; Boyd, N.J. Molecular Simulation Approaches to the Study of Thermotropic and Lyotropic Liquid Crystals. Crystals 2022, 12, 685. [Google Scholar] [CrossRef]
- Yu, G.; Wilson, M.R. All-atom simulations of bent liquid crystal dimers: The twist-bend nematic phase and insights into conformational chirality. Soft Matter 2022, 18, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Jansze, S.M.; Martínez-Felipe, A.; Storey, J.M.D.; Marcelis, A.T.M.; Imrie, C.T. A Twist-Bend Nematic Phase Driven by Hydrogen Bonding. Angew. Chem. Int. Ed. 2015, 54, 643–646. [Google Scholar] [CrossRef]
- Martinez-Felipe, A.; Cook, A.G.; Abberley, J.P.; Walker, R.; Storey, J.M.D.; Imrie, C.T. An FT-IR spectroscopic study of the role of hydrogen bonding in the formation of liquid crystallinity for mixtures containing bipyridines and 4-pentoxybenzoic acid. RSC Adv. 2016, 6, 108164–108179. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.; Pociecha, D.; Abberley, J.P.; Martinez-Felipec, A.; Paterson, D.A.; Forsyth, E.; Lawrence, G.B.; Henderson, P.A.; Storey, J.M.D.; Gorecka, E.; et al. Spontaneous chirality through mixing achiral components: A twist-bend nematic phase driven by hydrogen-bonding between unlike components. Chem. Commun. 2018, 54, 3383–3386. [Google Scholar] [CrossRef]
- Alaasar, M.; Tschierske, C. Nematic phases driven by hydrogen-bonding in liquid crystalline nonsymmetric dimers. Liq. Cryst. 2019, 49, 124–130. [Google Scholar] [CrossRef]
- Kima, J.; Koo, J.; Park, J.; Jeong, K.; Lee, J. Enhancement of flexoelastic ratio of nematic liquid crystal doped with hydrogen-bonded bimesogen molecules. J. Mol. Liq. 2019, 277, 541–545. [Google Scholar] [CrossRef]
- Kato, T.; Gupta, M.; Yamaguchi, D.; Gan, K.P.; Nakayama, M. Supramolecular Association and Nanostructure Formation of Liquid Crystals and Polymers for New Functional Materials. Bull. Chem. Soc. Jpn. 2021, 94, 357–376. [Google Scholar] [CrossRef]
- Knežević, A.; Sapunar, M.; Buljan, A.; Dokli, I.; Hameršak, Z.; Kontrec, D.; Lesac, A. Fine-tuning the effect of p–p interactions on the stability of the NTB phase. Soft Matter 2018, 14, 8466–8474. [Google Scholar] [CrossRef]
- Emsley, J.W.; de Luca, G.; Lesage, A.; Merlet, D.; Pileio, G. The structure and conformation of a mesogenic compound between almost zero and almost complete orientational order. Liq Cryst. 2007, 34, 1071–1093. [Google Scholar] [CrossRef]
- Arakawa, Y.; Ishida, Y.; Tsuji, H. Ether- and Thioether-Linked Naphthalene-Based Liquid-Crystal Dimers: Influence of Chalcogen Linkage and Mesogenic-Arm Symmetry on the Incidence and Stability of the Twist–Bend Nematic Phase. Chem. Eur. J. 2020, 26, 3767–3775. [Google Scholar] [CrossRef]
- Merkel, K.; Loska, B.; Arakawa, Y.; Mehl, G.H.; Karcz, J.; Kocota, A. Increased intermolecular interactions and cluster formation at the onset of the twist-bend nematic phase in thioether cyanobiphenyl-based liquid crystal dimers. arXiv 2022, arXiv:2206.07437. [Google Scholar] [CrossRef]
- Barnes, P.J.; Douglass, A.G.; Heeks, S.K.; Luckhurst, G.R. An enhanced odd-even effect of liquid crystal dimers Orientational order in the α,ω-bis(4′-cyanobiphenyl-4-yl)alkanes. Liq. Cryst. 1993, 13, 603–613. [Google Scholar] [CrossRef]
- Carvalho, J.; Cruz, C.; Figueirinhas, J.L.; Tamba, M.G.; Kohlmeier, A.; Mehl, G.H. Proton and deuterium NMR study of the CBC9CB dimer system. J. Phys. Chem. B 2019, 123, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Aluculesei, A.; Cachitas, H.; Carvalho, J.; Chavez, F.V.; Figueirinhas, J.L.; Sebastião, P.J.; Cruz, C.; Tamba, M.G.; Kohlemeir, A.; Mehl, G.H. 1H NMR study of molecular order and dynamics in the liquid crystal CB-C9-CB. Phys. Chem. Chem. Phys. 2019, 21, 4523–4537. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford CT, UK, 2009. [Google Scholar]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Conf. | Potential Energy RMS (kJ/mol) | Torsion Angles, φt (°) | Opening Angle (°) | |||
|---|---|---|---|---|---|---|---|
| φ1 | φ2 | φ3 | φ4 | ||||
| CBC9CB | Upright (U) | −3,843,952.076 0.014 | 92.3 | 86.2 | −38.7 | −38.5 | 112 |
| CBSC7SCB | Upright (U) | −5,521,962.5193 0.0035 | −92.9 | −92.8 | −39.0 | −39.0 | 94 |
| Flat (F) | −5,521,963.9252 0.0024 | 5.8 | 5.8 | −37.2 | −37.2 | 108 | |
| CBSC7OCB | Mixed (M) | −4,673,985.9556 0.0077 | −96.1 | −179.1 | −38.8 | −37.2 | 120 |
| Flat (F) | −4,673,986.6314 0.0037 | −176.3 | −180.0 | −37.4 | −37.6 | 126 | |
| CBOC7OCB | Flat (F) | −3,826,009.436 0.020 | −179.3 | −179.2 | −37.3 | −37.3 | 144 |
| CBC9CB | CBSC7SCB | CBSC7OCB | CBOC7OCB | Assignments | ||||
|---|---|---|---|---|---|---|---|---|
| Ir | Ir | Ir | Ir | |||||
| 520 | vvw | 522 | m | 523 | w | -- | -- | γCC op CB + δCS |
| -- | -- | -- | -- | 532 | w | 532 | w/m | γCC op CB + δCO |
| 554 | w | 562 | w | 562 | w | 562 | w | γCC op CB + δCN |
| 816 | vs | 811 | vs | 813 | vs | -- | γCH op CB | |
| -- | -- | -- | -- | 821 | s, sh | 821 | vs | νsCOC + γCH op CB |
| 836 850 | w | 852 | w | 850 | w | 850 | w | γCH op CB + νCCC sk + δasCH2 rocking |
| 1007 | w | 999 | w | 999 | w | 1000 | w | βCC ip CB, breathable |
| 1026 | vw | 1020 | vw | 1013 | vw | 1013 | vw | νCCC sk + βCH ip CB |
| -- | -- | -- | -- | 1029 1051 | w | 1032 | w | νasCAlO + βCH ip CB |
| -- | -- | 1097 | m | 1095 | m | -- | -- | νasCArS + βCH ip CB |
| 1112 | vw | -- | -- | -- | -- | 1120 | w | βCH ip CB |
| 1185 | m | 1185 | s | 1180 | s | 1178 | s | βCH ip CB |
| -- | -- | -- | -- | 1249 1266 | vs m, sh | 1249 1266 | vs m,sh | νasCArO + βCH ip CB |
| 1284 1315 1360 | vw | 1279 1315 | vw | 1290 1311 | vw | 1290 1313 | vw | γsCH2 wagging γsCH2 twisting |
| 1397 | w | 1395 | w | 1392 | w | 1390 | w | βCH ip CB |
| 1460 | w | 1437 1462 | w | 1435 1472 | w | 1472 | w | βsCH2 scissoring |
| -- 1493 | s | 1484 -- | vs | 1485 1494 | vs | -- 1493 | vs | νCC br |
| -- | -- | -- | -- | 1522 1577 | m vw | 1523 1580 | m w | νCC br + βsCH2 + νasCArO |
| -- 1605 | s | 1594 1604 | vs | -- 1603 | vs | -- 1602 | vs | νCC br |
| 2224 | vs | 2223 | vs | 2223 | vs | 2223 | vs | νCN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocot, A.; Loska, B.; Arakawa, Y.; Mehl, G.H.; Merkel, K. Study of the Experimental and Simulated Vibrational Spectra Together with Conformational Analysis of Thioether Cyanobiphenyl-Based Liquid Crystal Dimers. Int. J. Mol. Sci. 2022, 23, 8005. https://doi.org/10.3390/ijms23148005
Kocot A, Loska B, Arakawa Y, Mehl GH, Merkel K. Study of the Experimental and Simulated Vibrational Spectra Together with Conformational Analysis of Thioether Cyanobiphenyl-Based Liquid Crystal Dimers. International Journal of Molecular Sciences. 2022; 23(14):8005. https://doi.org/10.3390/ijms23148005
Chicago/Turabian StyleKocot, Antoni, Barbara Loska, Yuki Arakawa, Georg H. Mehl, and Katarzyna Merkel. 2022. "Study of the Experimental and Simulated Vibrational Spectra Together with Conformational Analysis of Thioether Cyanobiphenyl-Based Liquid Crystal Dimers" International Journal of Molecular Sciences 23, no. 14: 8005. https://doi.org/10.3390/ijms23148005
APA StyleKocot, A., Loska, B., Arakawa, Y., Mehl, G. H., & Merkel, K. (2022). Study of the Experimental and Simulated Vibrational Spectra Together with Conformational Analysis of Thioether Cyanobiphenyl-Based Liquid Crystal Dimers. International Journal of Molecular Sciences, 23(14), 8005. https://doi.org/10.3390/ijms23148005