
The Effect of Isosaponarin Derived from Wasabi Leaves on Glutamate Release in Rat Synaptosomes and Its Underlying Mechanism

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Isosaponarin Reduces 4-AP-Triggered Ca2+-Dependent Vesicular Glutamate Release from Synaptosomes
2.2. Suppression of N-Type and P/Q-Type Ca2+ Channels Mediates Isosaponarin-Induced Inhibition of Vesicular Glutamate Release
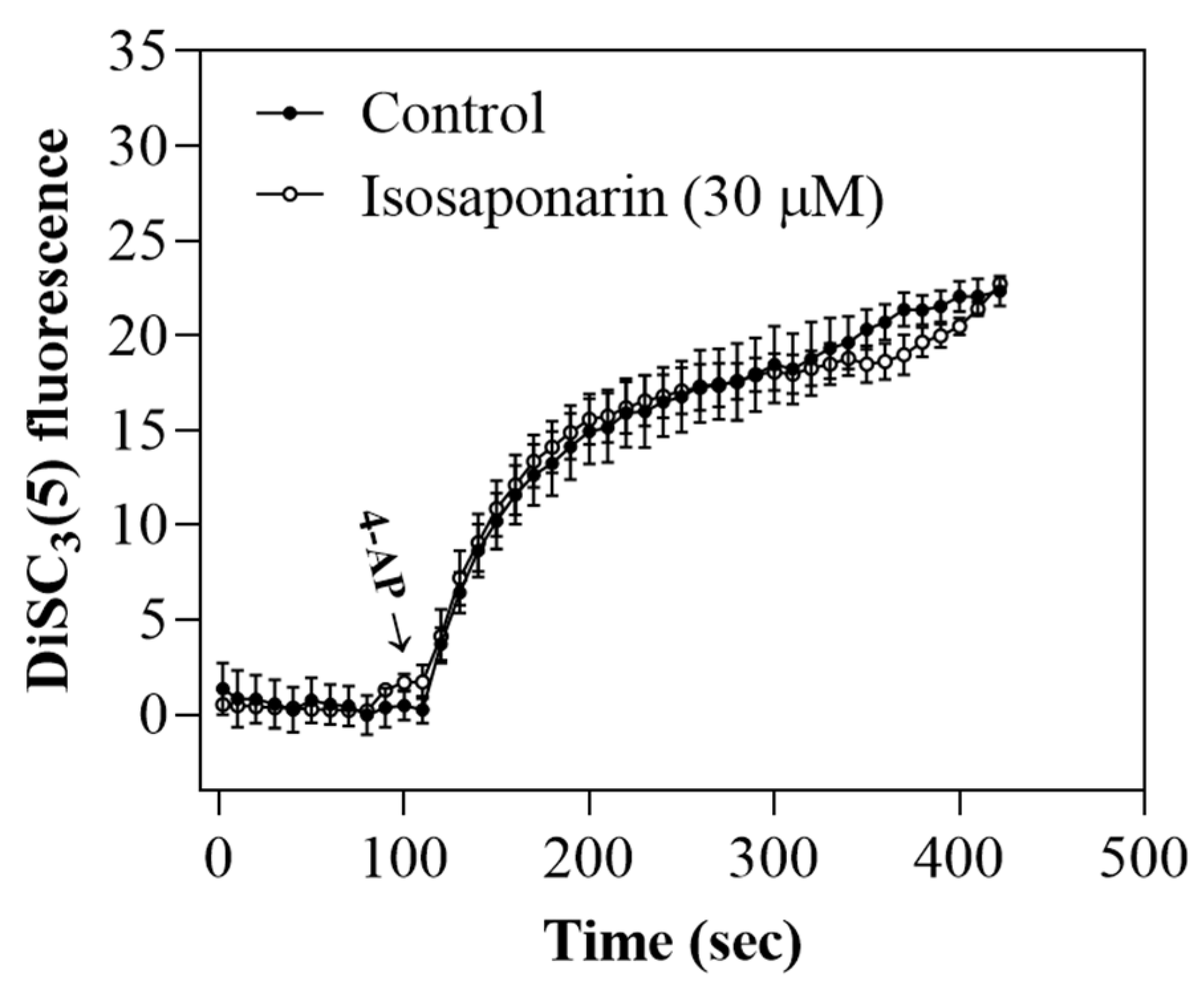
2.3. Isosaponarin Does Not Change the Synaptosomal Membrane Potential
2.4. Isosaponarin-Induced Inhibition of Vesicular Glutamate Release Is Prevented by Ca2+-Dependent PKC Inhibition
2.5. Isosaponarin Decreases SNAP-25 and MARCKS Phosphorylation Evoked by 4-AP in Synaptosomes
2.6. Isosaponarin Decreases the Release of FM1-43 from Synaptosomes
2.7. Isosaponarin Inhibits the 4-AP-Induced Decrease in the Number of Synaptic Vesicles in Synaptosomes
3. Discussion
3.1. Isosaponarin Decreases 4-AP-Evoked Vesicular Glutamate Release by Blocking N- and P/Q-Type Ca2+ Channels
3.2. Suppression of the Ca2+-Dependent PKC/SNAP-25 and MARCKS Pathways and the Consequent Reduction in Available Synaptic Vesicles May Account for Isosaponarin-Induced Inhibition of Vesicular Glutamate Release
4. Materials and Methods
4.1. Animals
4.2. Isolation of Nerve Terminals (Synaptosomes) from the Cortex Regions of the Rat Brains
4.3. Glutamate Release Assay
4.4. Cytosolic Ca2+ Measurements Using Fura-2
4.5. Membrane Potential Measurement Using 3,3,3-Diethylthiacarbocyanine Iodide (DiSC3(5))
4.6. Western Blot
4.7. FM1-43 Release and Image Assay
4.8. The Synaptosomes TEM
4.9. Statistical Analysis
4.10. Chemicals
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Beal, M.F. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992, 6, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Mantas, I.; Flais, I.; Svenningsson, P. Fluoxetine Suppresses Glutamate- and GABA-Mediated Neurotransmission by Altering SNARE Complex. Int. J. Mol. Sci. 2019, 20, 4247. [Google Scholar] [CrossRef]
- Sitges, M.; Sanchez-Tafolla, B.M.; Chiu, L.M.; Aldana, B.I.; Guarneros, A. Vinpocetine inhibits glutamate release induced by the convulsive agent 4-aminopyridine more potently than several antiepileptic drugs. Epilepsy Res. 2011, 96, 257–266. [Google Scholar] [CrossRef]
- Parvez, M.K. Natural or Plant Products for the Treatment of Neurological Disorders: Current Knowledge. Curr. Drug Metab. 2018, 19, 424–428. [Google Scholar] [CrossRef]
- Rehman, M.U.; Wali, A.F.; Ahmad, A.; Shakeel, S.; Rasool, S.; Ali, R.; Rashid, S.M.; Madkhali, H.; Ganaie, M.A.; Khan, R. Neuroprotective Strategies for Neurological Disorders by Natural Products: An update. Curr. Neuropharmacol. 2019, 17, 247–267. [Google Scholar] [CrossRef]
- Hsieh, T.Y.; Chang, Y.; Wang, S.J. Piperine-mediated suppression of voltage-dependent Ca2+ influx and glutamate release in rat hippocampal nerve terminals involves 5HT1A receptors and G protein βγ activation. Food Funct. 2019, 10, 2720–2728. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Wang, S.J.; Huang, S.K. Asiatic acid, an active substance of Centella asiatica, presynaptically depresses glutamate release in the rat hippocampus. Eur. J. Pharmacol. 2019, 865, 172781. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Lu, C.-W.; Hsieh, P.-W.; Chiu, K.-M.; Lee, M.-Y.; Wang, S.-J. Natural Product Isoliquiritigenin Activates GABAB Receptors to Decrease Voltage-Gate Ca2+ Channels and Glutamate Release in Rat Cerebrocortical Nerve Terminals. Biomolecules 2021, 11, 1537. [Google Scholar] [CrossRef]
- Hosoya, T.; Yun, Y.S.; Kunugi, A. Five novel flavonoids from Wasabia japonica. Tetrahedron 2005, 61, 7037–7044. [Google Scholar] [CrossRef]
- Fuke, Y.; Haga, Y.; Ono, H.; Nomura, T.; Ryoyama, K. Anti-carcinogenic activity of 6-methylsulfinylhexyl isothiocyanate-, an active anti-proliferative principal of wasabi (Eutrema wasabi Maxim.). Cytotechnology 1997, 25, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kurata, T.; Misawa, N.; Hosoya, T.; Yamada-Kato, T.; Okunishi, I.; Kumazawa, S. Isolation and Identification of Components from Wasabi (Wasabia japonica Matsumura) Flowers and Investigation of Their Antioxidant and Anti-inflammatory Activities. Food Sci. Technol. Res. 2019, 25, 449–457. [Google Scholar] [CrossRef]
- Yamasaki, M.; Ogawa, T.; Wang, L.; Katsube, T.; Yamasaki, Y.; Sun, X.; Shiwaku, K. Anti-obesity effects of hot water extract from Wasabi (Wasabia japonica Matsum.) leaves in mice fed high-fat diets. Nutr. Res. Pract. 2013, 7, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lee, T.H.; Ham, S.L.; Subedi, L.; Hong, S.M.; Kim, S.Y.; Choi, S.U.; Kim, C.S.; Lee, K.R. Anticancer and Anti-Neuroinflammatory Constituents Isolated from the Roots of Wasabia japonica. Antioxidants 2022, 11, 482. [Google Scholar] [CrossRef]
- Subedi, L.; Venkatesan, R.; Kim, S.Y. Neuroprotective and Anti-Inflammatory Activities of Allyl Isothiocyanate through Attenuation of JNK/NF-κB/TNF-α Signaling. Int. J. Mol. Sci. 2017, 18, 1423. [Google Scholar] [CrossRef]
- Hosoya, T.; Yun, Y.S.; Kunugi, A. Antioxidant phenylpropanoid glycosides from the leaves of Wasabia japonica. Phytochemistry 2008, 69, 827–832. [Google Scholar] [CrossRef]
- Yoshida, S.; Hosoya, T.; Inui, S.; Masuda, H.; Kumazawa, S. Component Analysis of Wasabi Leaves and an Evaluation of their Anti-inflammatory Activity. Food Sci. Technol. Res. 2015, 21, 247–253. [Google Scholar] [CrossRef]
- Chang, C.Y.; Lin, T.Y.; Lu, C.W.; Huang, S.K.; Wang, Y.C.; Chou, S.S.P.; Wang, S.J. Hesperidin inhibits glutamate release and exerts neuroprotection against excitotoxicity induced by kainic acid in the hippocampus of rats. NeuroToxicology 2015, 50, 157–169. [Google Scholar] [CrossRef]
- Chang, Y.; Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Baicalein, a Constituent of Scutellaria baicalensis, Reduces Glutamate Release and Protects Neuronal Cell Against Kainic Acid-Induced Excitotoxicity in Rats. Am. J. Chin. Med. 2016, 44, 943–962. [Google Scholar] [CrossRef]
- Lu, C.-W.; Lin, T.-Y.; Chiu, K.-M.; Lee, M.-Y.; Huang, J.-H.; Wang, S.-J. Silymarin Inhibits Glutamate Release and Prevents against Kainic Acid-Induced Excitotoxic Injury in Rats. Biomedicines 2020, 8, 486. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Akita, K.; Yamada, K.; Okunishi, I. The effect of isosaponarin isolated from wasabi leaf on collagen synthesis in human fibroblasts and its underlying mechanism. J. Nat. Med. 2010, 64, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The glutamatergic nerve terminal. Eur. J. Biochem. 1993, 212, 613–631. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Nicholls, D.G. Transmitter Glutamate Release from Isolated Nerve Terminals: Evidence for Biphasic Release and Triggering by Localized Ca2+. J. Neurochem. 1991, 56, 86–94. [Google Scholar] [CrossRef]
- Poëa-Guyon, S.; Ammar, M.R.; Erard, M.; Amar, M.; Moreau, A.W.; Fossier, P.; Gleize, V.; Vitale, N.; Morel, N. The V-ATPase membrane domain is a sensor of granular pH that controls the exocytotic machinery. J. Cell Biol. 2013, 203, 283–298. [Google Scholar] [CrossRef]
- Millán, C.; Sánchez-Prieto, J. Differential coupling of N- and P/Q-type calcium channels to glutamate exocytosis in the rat cerebral cortex. Neurosci. Lett. 2002, 330, 29–32. [Google Scholar] [CrossRef]
- Wu, L.-G.; Saggau, P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997, 20, 204–212. [Google Scholar] [CrossRef]
- Åkerman, K.E.O.; Scott, I.G.; Heikkila, J.E.; Heinonen, E. Ionic Dependence of Membrane Potential and Glutamate Receptor-Linked Responses in Synaptoneurosomes as Measured with a Cyanine Dye, DiS-C2-(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef]
- Vaughan, P.F.T.; Walker, J.H.; Peers, C. The regulation of neurotransmitter secretion by protein kinase C. Mol. Neurobiol. 1998, 18, 125–155. [Google Scholar] [CrossRef]
- Shearman, M.S.; Shinomura, T.; Oda, T.; Nishizuka, Y. Synaptosomal Protein Kinase C Subspecies: A. Dynamic Changes in the Hippocampus and Cerebellar Cortex Concomitant with Synaptogenesis. J. Neurochem. 1991, 56, 1255–1262. [Google Scholar] [CrossRef]
- Burgoyne, R.; Morgan, A.; Barclay, J.E.; Craig, T.J.; Prescott, G.R.; Ciufo, L.F.; Evans, G.J.; Graham, M.E. Regulation of exocytosis by protein kinase C. Biochem. Soc. Trans. 2005, 33, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.N. Optical detection of synaptic vesicle exocytosis and endocytosis. Curr. Opin. Neurobiol. 1999, 9, 314–320. [Google Scholar] [CrossRef]
- Baldwin, M.L.; Rostas, J.A.; Sim, A.T. Two modes of exocytosis from synaptosomes are differentially regulated by protein phosphatase types 2A and 2B. J. Neurochem. 2003, 85, 1190–1199. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. Calcium-Dependent and-Independent Release of Glutamate from Synaptosomes Monitored by Continuous Fluorometry. J. Neurochem. 1987, 49, 50–57. [Google Scholar] [CrossRef]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef]
- Catterall, W.A.; Few, A.P. Calcium Channel Regulation and Presynaptic Plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, E.; Sánchez-Prieto, J. Presynaptic Modulation of Glutamate Release Targets Different Calcium Channels in Rat Cerebrocortical Nerve Terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef]
- Stevens, C.F.; Sullivan, J.M. Regulation of the Readily Releasable Vesicle Pool by Protein Kinase C. Neuron 1998, 21, 885–893. [Google Scholar] [CrossRef]
- Wu, X.-S.; Wu, L.-G. Protein Kinase C Increases the Apparent Affinity of the Release Machinery to Ca2+ by Enhancing the Release Machinery Downstream of the Ca2+ Sensor. J. Neurosci. 2001, 21, 7928–7936. [Google Scholar] [CrossRef]
- Nagy, G.; Matti, U.; Nehring, R.B.; Binz, T.; Rettig, J.; Neher, E.; Sørensen, J.B. Protein Kinase C-Dependent Phosphorylation of Synaptosome-Associated Protein of 25 kDa at Ser187Potentiates Vesicle Recruitment. J. Neurosci. 2002, 22, 9278–9286. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Nishiki, T.-I.; Omori, A.; Sekiguchi, M.; Kamata, Y.; Kozaki, S.; Takahashi, M. Phosphorylation of 25-kDa Synaptosome-associated Protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J. Biol. Chem. 1996, 271, 14548–14553. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T.; Sihra, T.S.; Nicholls, D.G.; Pocock, J.M. Phosphorylation of synapsin I and MARCKS in nerve terminals is mediated by Ca2+ entry via an Aga-GI sensitive Ca2+ channel which is coupled to glutamate exocytosis. FEBS Lett. 1994, 353, 264–268. [Google Scholar] [CrossRef]
- Chang, Y.; Wang, S.-J. Ginsenoside Rg1 and Rb1 enhance glutamate exocytosis from rat cortical nerve terminals by affecting vesicle mobilization through the activation of protein kinase C. Eur. J. Pharmacol. 2008, 590, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Echinacoside Inhibits Glutamate Release by Suppressing Voltage-Dependent Ca2+ Entry and Protein Kinase C in Rat Cerebrocortical Nerve Terminals. Int. J. Mol. Sci. 2016, 17, 1006. [Google Scholar] [CrossRef]
- Quigley, P.A.; Msghina, M.; Govind, C.K.; Atwood, H.L. Visible Evidence for Differences in Synaptic Effectiveness with Activity-Dependent Vesicular Uptake and Release of FM1-43. J. Neurophysiol. 1999, 81, 356–370. [Google Scholar] [CrossRef][Green Version]
- Barclay, J.W.; Craig, T.J.; Fisher, R.J.; Ciufo, L.F.; Evans, G.J.; Morgan, A.; Burgoyne, R.D. Phosphorylation of Munc18 by Protein Kinase C Regulates the Kinetics of Exocytosis. J. Biol. Chem. 2003, 278, 10538–10545. [Google Scholar] [CrossRef]
- Lou, X.; Korogod, N.; Brose, N.; Schneggenburger, R. Phorbol Esters Modulate Spontaneous and Ca2+-Evoked Transmitter Release via Acting on Both Munc13 and Protein Kinase C. J. Neurosci. 2008, 28, 8257–8267. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Sihra, T.S. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986, 321, 772–773. [Google Scholar] [CrossRef]
- Lu, C.-W.; Lin, C.-J.; Hsieh, P.-W.; Chiu, K.-M.; Lee, M.-Y.; Lin, T.-Y.; Wang, S.-J. An Anthranilate Derivative Inhibits Glutamate Release and Glutamate Excitotoxicity in Rats. Int. J. Mol. Sci. 2022, 23, 2641. [Google Scholar] [CrossRef]
- Hung, Y.-C.; Kuo, Y.-H.; Hsieh, P.-W.; Hsieh, T.-Y.; Kuo, J.-R.; Wang, S.-J. Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism. Int. J. Mol. Sci. 2021, 22, 11447. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Sihra, T.S.; Bogonez, E.; Nicholls, D.G. Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J. Biol. Chem. 1992, 267, 1983–1989. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-W.; Yeh, K.-C.; Chiu, K.-M.; Lee, M.-Y.; Lin, T.-Y.; Wang, S.-J. The Effect of Isosaponarin Derived from Wasabi Leaves on Glutamate Release in Rat Synaptosomes and Its Underlying Mechanism. Int. J. Mol. Sci. 2022, 23, 8752. https://doi.org/10.3390/ijms23158752
Lu C-W, Yeh K-C, Chiu K-M, Lee M-Y, Lin T-Y, Wang S-J. The Effect of Isosaponarin Derived from Wasabi Leaves on Glutamate Release in Rat Synaptosomes and Its Underlying Mechanism. International Journal of Molecular Sciences. 2022; 23(15):8752. https://doi.org/10.3390/ijms23158752
Chicago/Turabian StyleLu, Cheng-Wei, Kun-Chieh Yeh, Kuan-Ming Chiu, Ming-Yi Lee, Tzu-Yu Lin, and Su-Jane Wang. 2022. "The Effect of Isosaponarin Derived from Wasabi Leaves on Glutamate Release in Rat Synaptosomes and Its Underlying Mechanism" International Journal of Molecular Sciences 23, no. 15: 8752. https://doi.org/10.3390/ijms23158752
APA StyleLu, C.-W., Yeh, K.-C., Chiu, K.-M., Lee, M.-Y., Lin, T.-Y., & Wang, S.-J. (2022). The Effect of Isosaponarin Derived from Wasabi Leaves on Glutamate Release in Rat Synaptosomes and Its Underlying Mechanism. International Journal of Molecular Sciences, 23(15), 8752. https://doi.org/10.3390/ijms23158752