
Regulatory T Cell Depletion Using a CRISPR Fc-Optimized CD25 Antibody

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
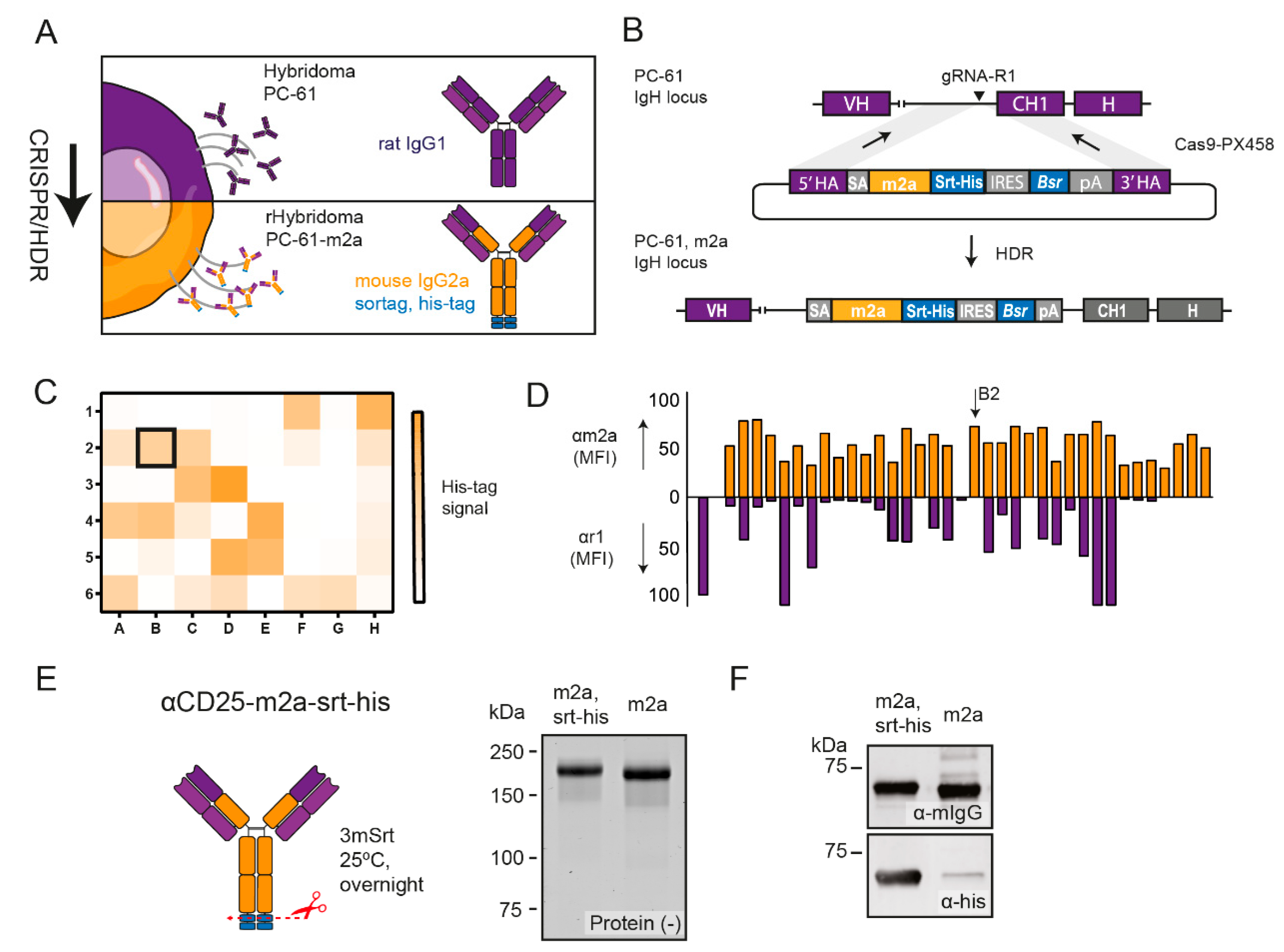
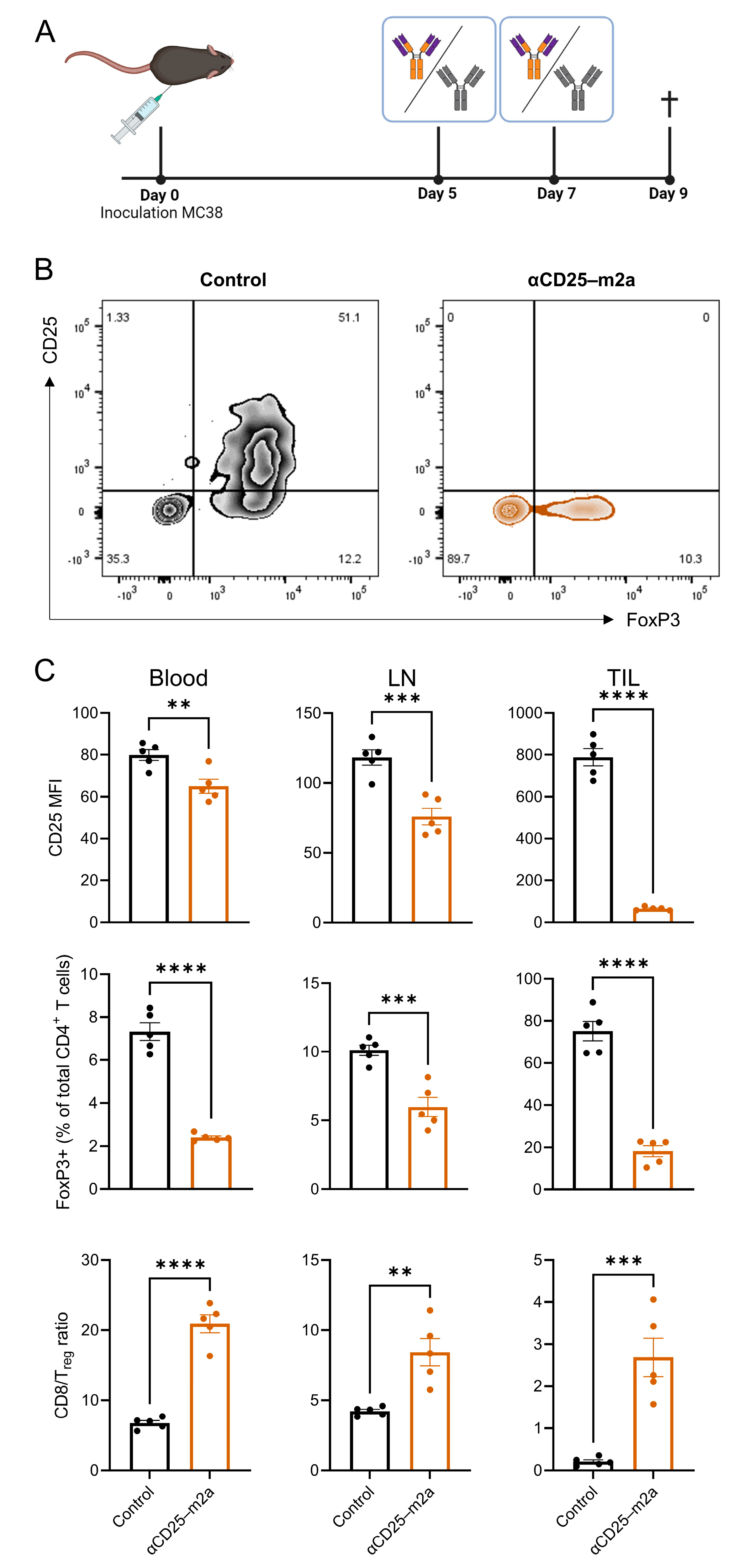
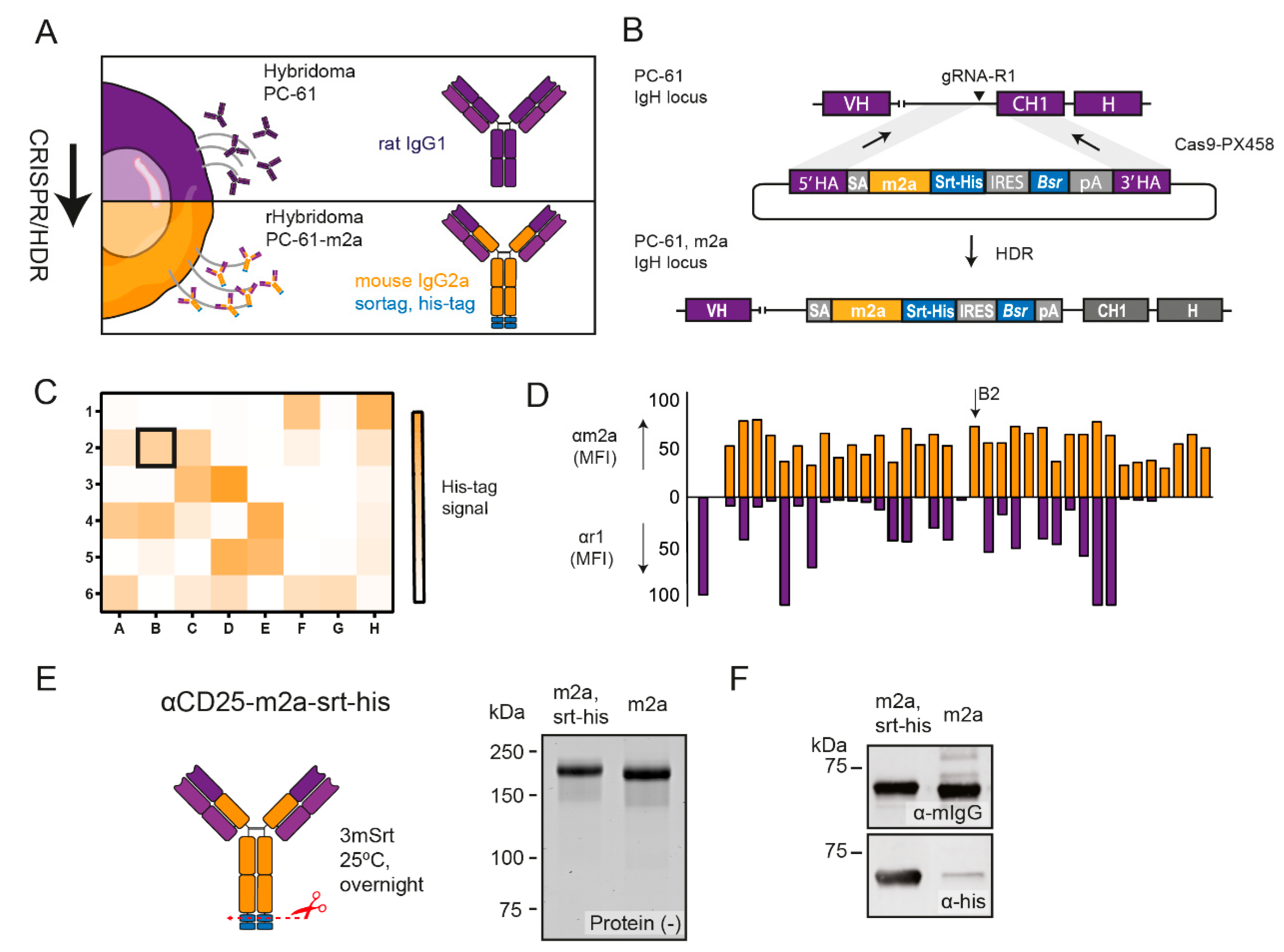
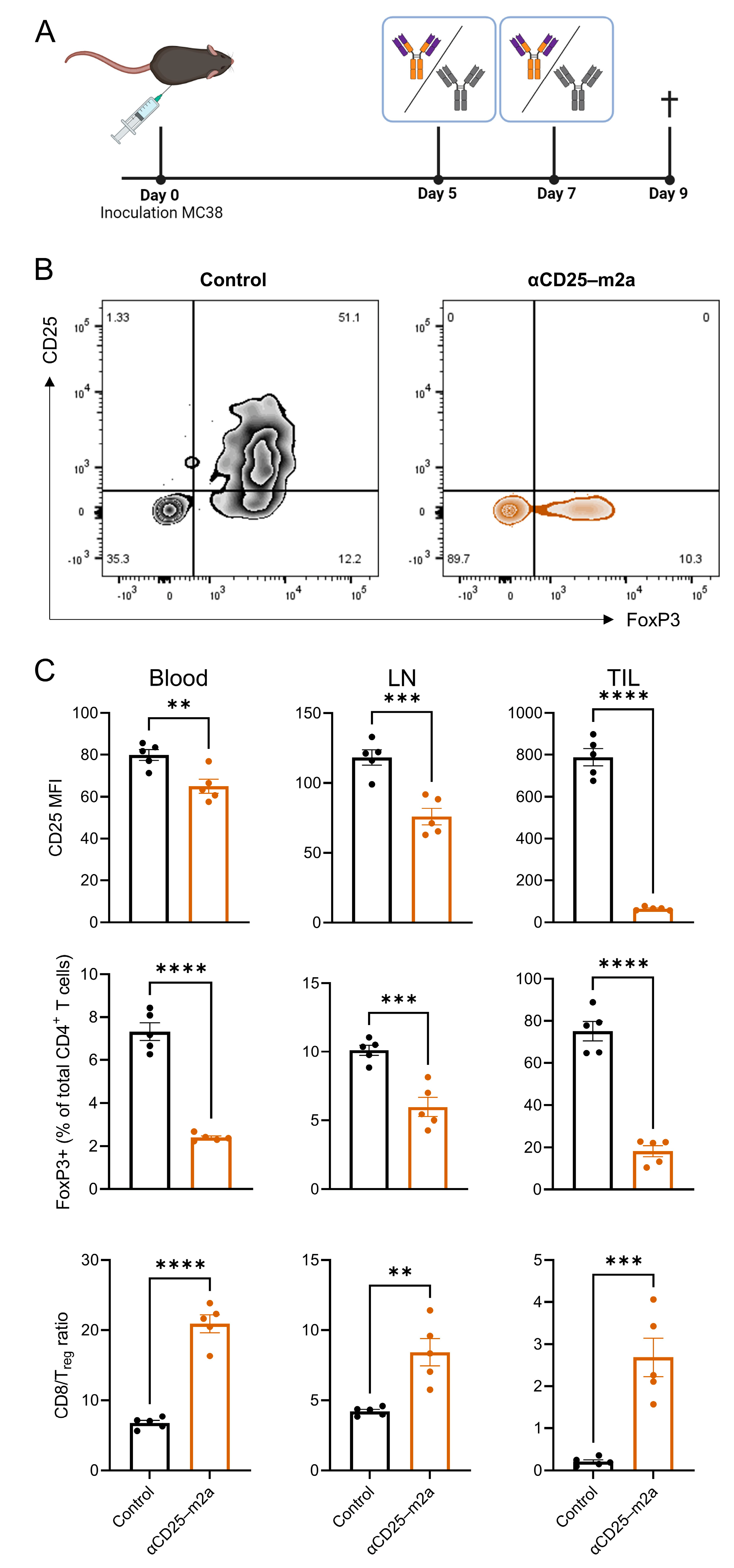
2.1. CRISPR-HDR Can Be Used to Optimize the PC-61 Hybridoma
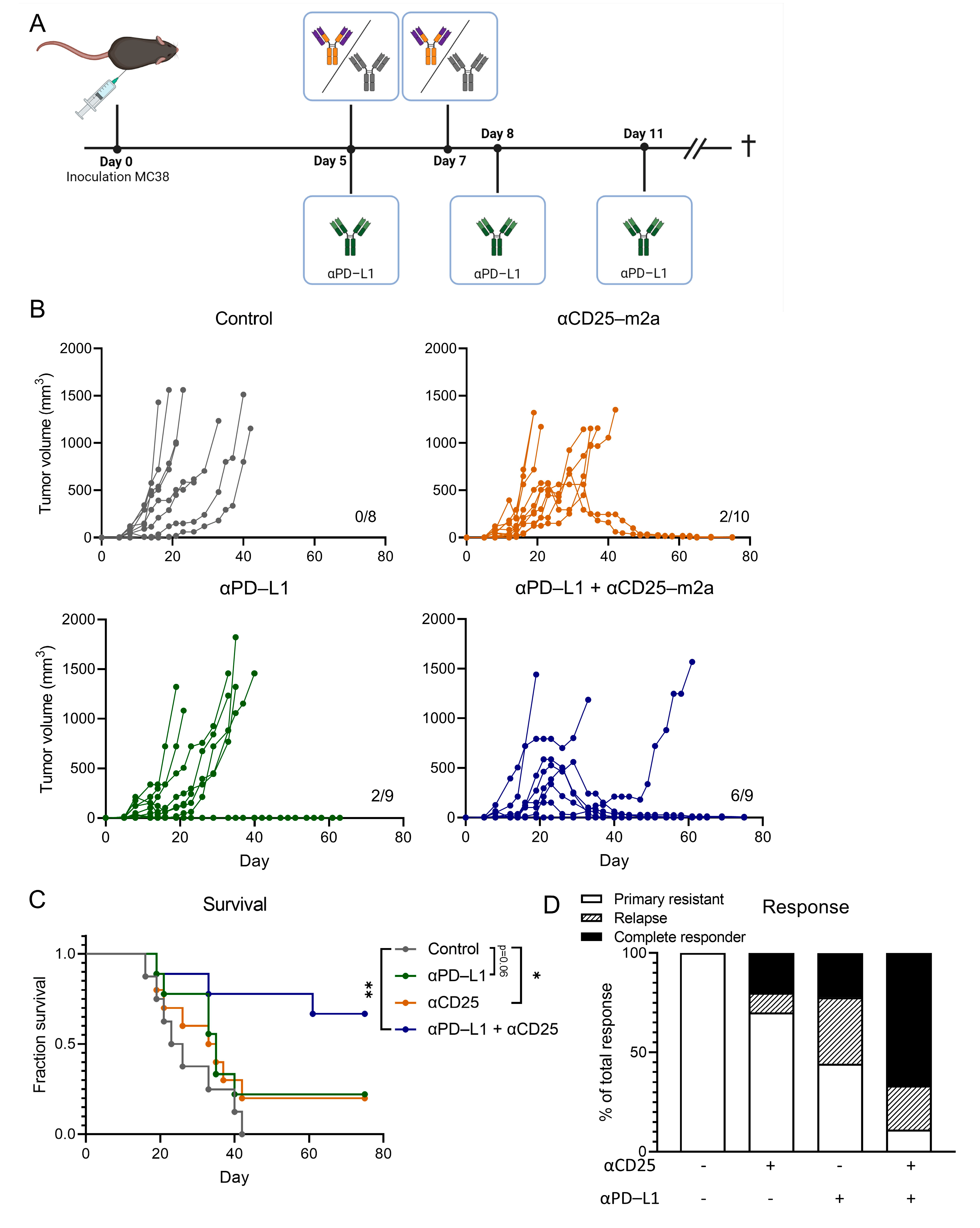
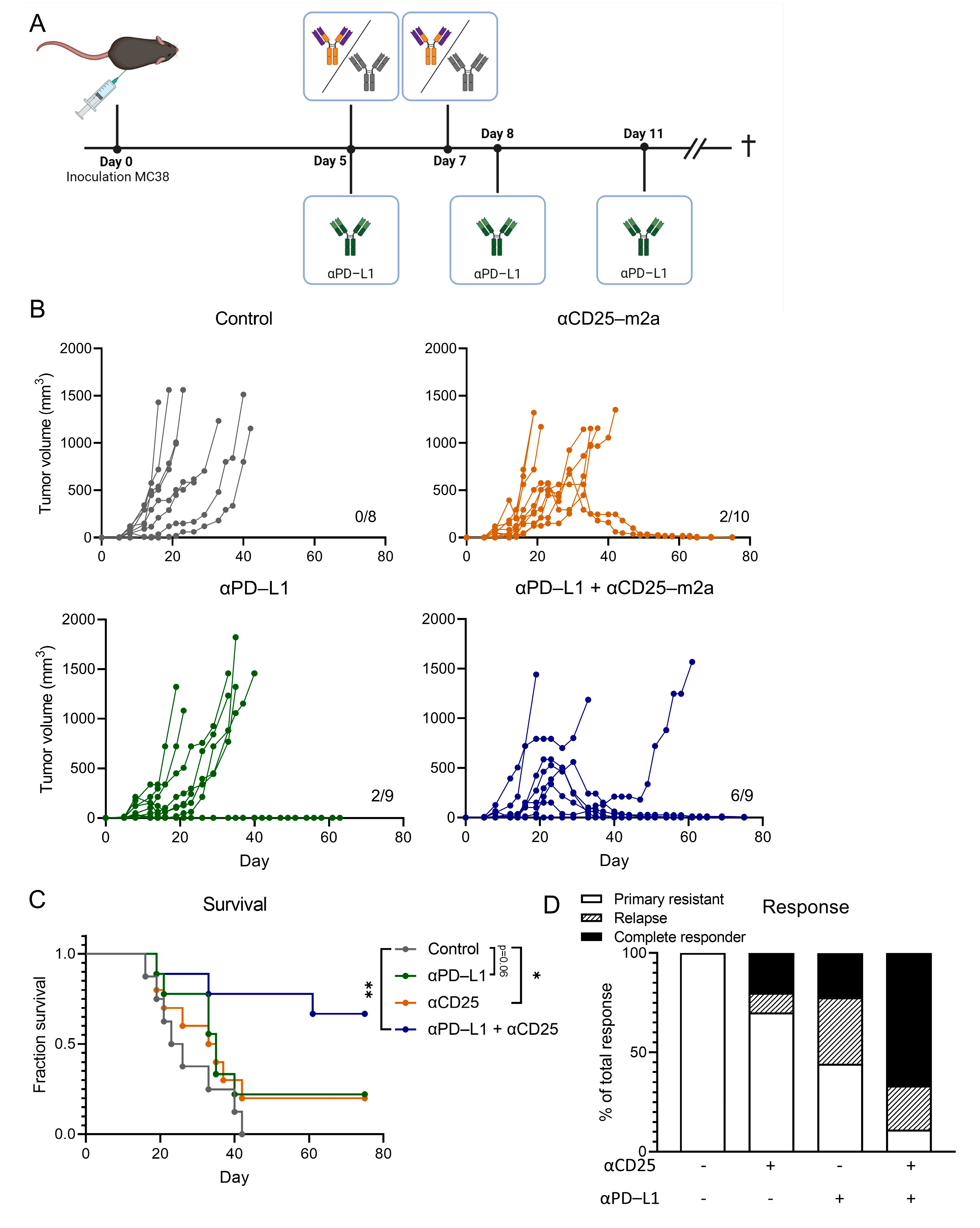
2.2. CD25-m2a Treatment Synergizes with an αPD-L1 Treatment
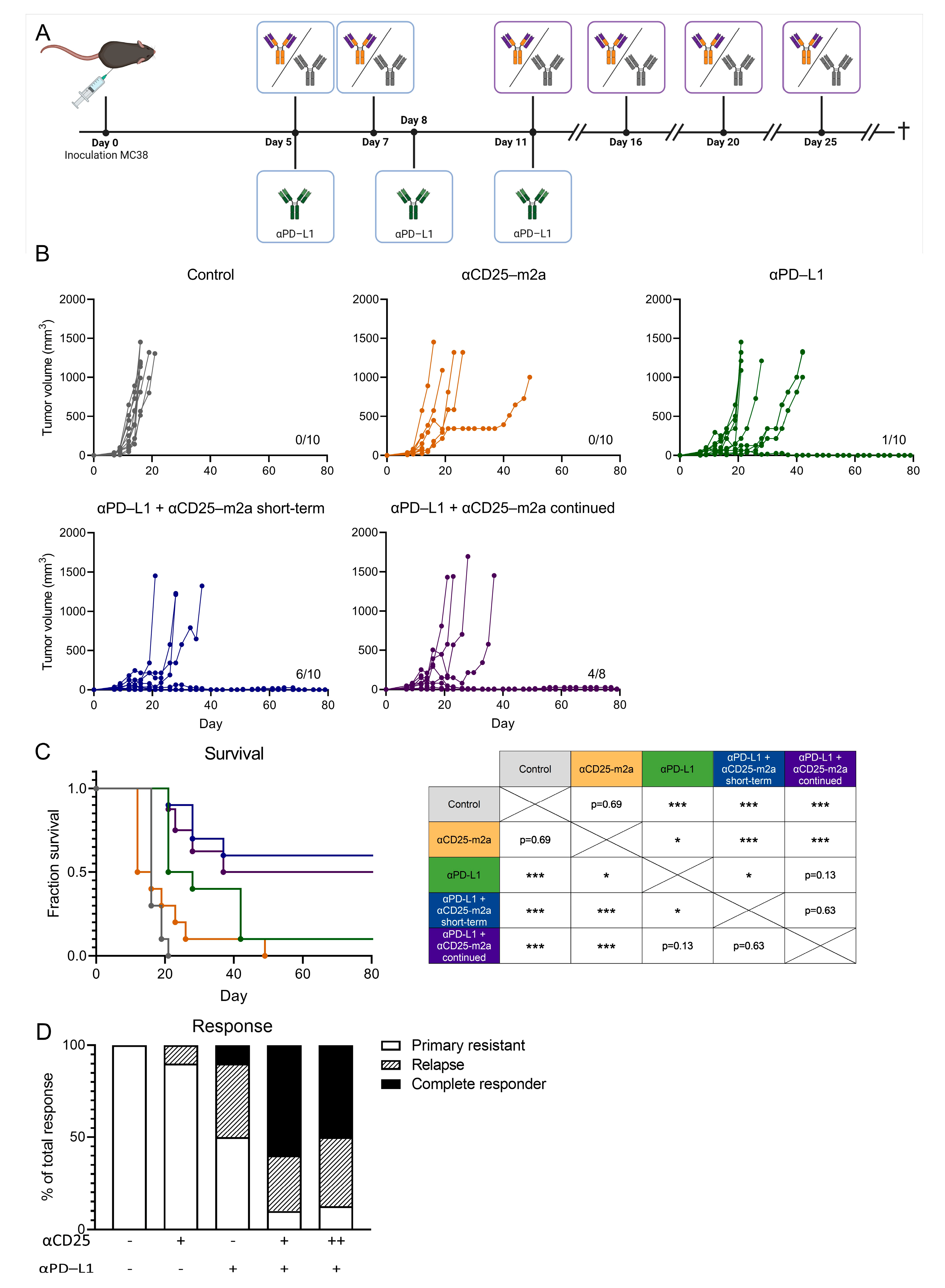
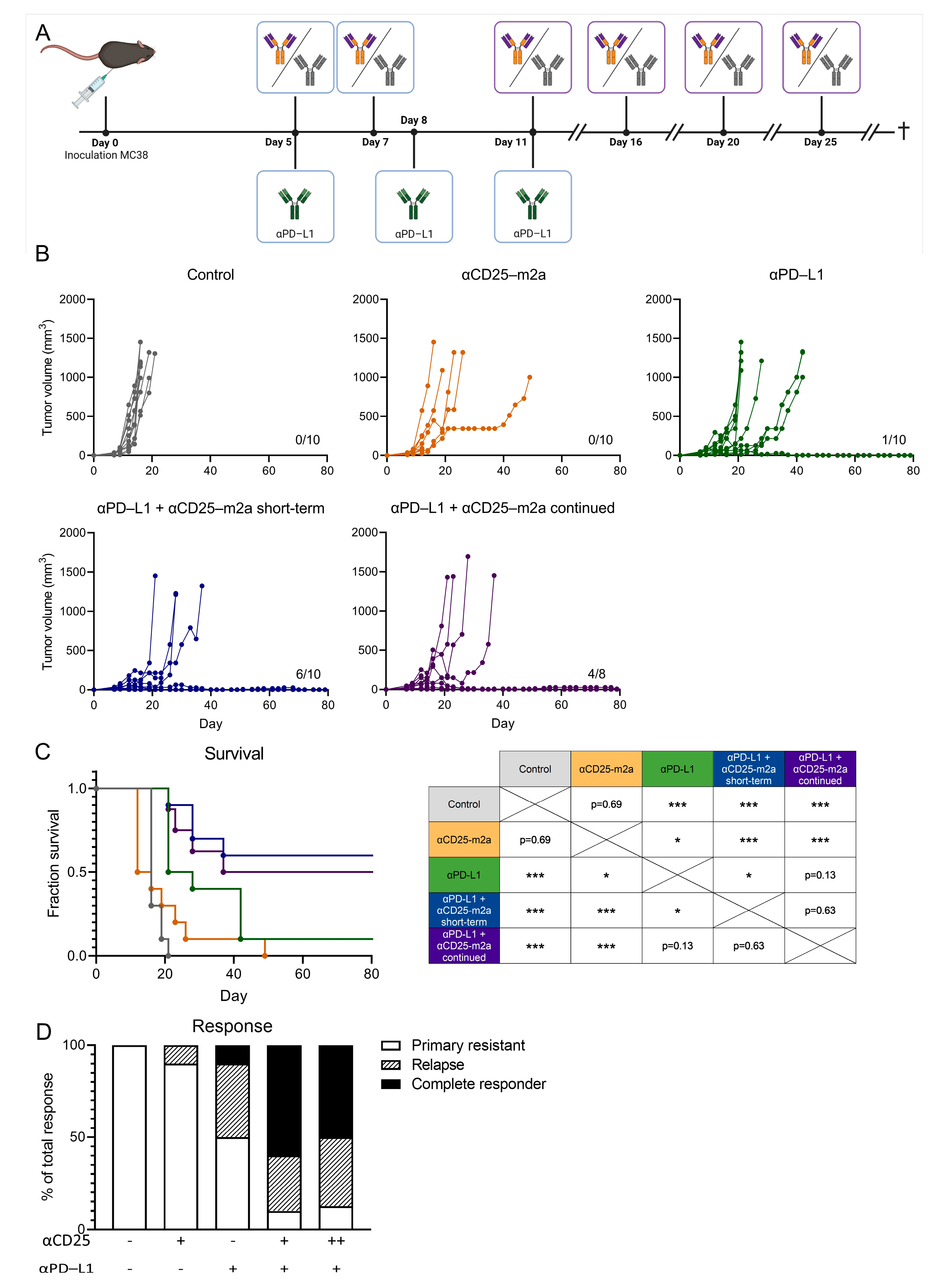
2.3. Continued Treg Depletion Does Not Affect Therapeutic Efficacy
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elpek, K.G.; Lacelle, C.; Singh, N.P.; Yolcu, E.S.; Shirwan, H. CD4+CD25+ T Regulatory Cells Dominate Multiple Immune Evasion Mechanisms in Early but Not Late Phases of Tumor Development in a B Cell Lymphoma Model. J. Immunol. 2007, 178, 6840–6848. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.; Dahm-Vicker, M.; Simon, A.K.; Green, A.; Powrie, F.; Cerundolo, V.; Gallimore, A. Depletion of CD25+ Regulatory Cells Results in Suppression of Melanoma Growth and Induction of Autoreactivity in Mice-PubMed. Cancer Immun. 2002, 2, 1. [Google Scholar]
- Golgher, D.; Jones, E.; Powrie, F.; Elliott, T.; Gallimore, A. Depletion of CD25+ Regulatory Cells Uncovers Immune Responses to Shared Murine Tumor Rejection Antigens-PubMed. Eur. J. Immunol. 2002, 32, 3267–3275. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Butler, M.; Oble, D.A.; Seiden, M.V.; Haluska, F.G.; Kruse, A.; MacRae, S.; Nelson, M.; Canning, C.; Lowy, I.; et al. Immunologic and Clinical Effects of Antibody Blockade of Cytotoxic T Lymphocyte-Associated Antigen 4 in Previously Vaccinated Cancer Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 3005–3010. [Google Scholar] [CrossRef] [Green Version]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 Blockade and GM-CSF Combination Immunotherapy Alters the Intratumor Balance of Effector and Regulatory T Cells. J. Clin. Investg. 2006, 116, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Goding, S.R.; Wilson, K.A.; Xie, Y.; Harris, K.M.; Baxi, A.; Akpinarli, A.; Fulton, A.; Tamada, K.; Strome, S.E.; Antony, P.A. Restoring Immune Function of Tumor-Specific CD4+ T Cells during Recurrence of Melanoma. J. Immunol. 2013, 190, 4899–4909. [Google Scholar] [CrossRef] [Green Version]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient Regulatory T Cell Ablation Deters Oncogene-Driven Breast Cancer and Enhances Radiotherapy. J. Exp. Med. 2013, 210, 2435. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Shen, Y.; Littman, D.R.; Allison, J.P. Limited Tumor Infiltration by Activated T Effector Cells Restricts the Therapeutic Activity of Regulatory T Cell Depletion against Established Melanoma. J. Exp. Med. 2008, 205, 2125. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyi, C.; Postow, M.A. Checkpoint Blocking Antibodies in Cancer Immunotherapy. FEBS Lett. 2014, 588, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heier, I.; Hofgaard, P.O.; Brandtzæg, P.; Jahnsen, F.L.; Karlsson, M. Depletion of CD4+ CD25+ Regulatory T Cells Inhibits Local Tumour Growth in a Mouse Model of B Cell Lymphoma. Clin. Exp. Immunol. 2008, 152, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc γ Receptors Contribute to the Antitumor Activities of Immunoregulatory Receptor-Targeting Antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, F.A.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Setiady, Y.Y.; Coccia, J.A.; Park, P.U. In Vivo Depletion of CD4+FOXP3+ Treg Cells by the PC61 Anti-CD25 Monoclonal Antibody Is Mediated by FcgammaRIII+ Phagocytes. Eur. J. Immunol. 2010, 40, 780–786. [Google Scholar] [CrossRef]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 Blockade Depletes and Selectively Reprograms Regulatory T Cells in Concert with Immunotherapy in Cancer Patients. Sci. Transl. Med. 2012, 4, 134ra62. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.F.M.; Punt, C.J.A.; Lesterhuis, W.J.; Sutmuller, R.P.M.; Brouwer, H.M.L.H.; Scharenborg, N.M.; Klasen, I.S.; Hilbrands, L.B.; Figdor, C.G.; De Vries, I.J.M.; et al. Dendritic Cell Vaccination in Combination with Anti-CD25 Monoclonal Antibody Treatment: A Phase I/II Study in Metastatic Melanoma Patients. Clin. Cancer Res. 2010, 16, 5067–5078. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of Tumor Immunity by Removing CD25+CD4+ T Cells: A Common Basis Between Tumor Immunity and Autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar]
- Dilillo, D.J.; Ravetch Correspondence, J. V Differential Fc-Receptor Engagement Drives an Anti-Tumor Vaccinal Effect ADCC-Mediated Tumor Clearance Requires Engagement of HFcgRIIIA on Macrophages d Selective Engagement of HFcgRIIA on DCs Promotes Long-Term Immunity against Cancer. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 2007 81 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Arce Vargas, F.; Furness, A.J.S.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Miranda Rota, E.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Schoot, J.M.S.; Fennemann, F.L.; Valente, M.; Dolen, Y.; Hagemans, I.M.; Becker, A.M.D.; Le Gall, C.M.; Van Dalen, D.; Cevirgel, A.; Van Bruggen, J.A.C.; et al. Functional Diversification of Hybridoma-Produced Antibodies by CRISPR/HDR Genomic Engineering. Sci. Adv. 2019, 5, eaaw1822. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef]
- Hagemans, I.M.; Wierstra, P.J.; Steuten, K.; Molkenboer-Kuenen, J.D.M.; van Dalen, D.; ter Beest, M.; van der Schoot, J.M.S.; Ilina, O.; Gotthardt, M.; Figdor, C.G.; et al. Multiscale Imaging of Therapeutic Anti-PD-L1 Antibody Localization Using Molecularly Defined Imaging Agents. J. Nanobiotechnol. 2022, 20, 64. [Google Scholar] [CrossRef]
- Möhlmann, S.; Mahlert, C.; Greven, S.; Scholz, P.; Harrenga, A. In Vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem 2011, 12, 1774–1780. [Google Scholar] [CrossRef]
- Matsushita, N.; Pilon-Thomas, S.A.; Martin, L.M.; Riker, A.I. Comparative Methodologies of Regulatory T Cell Depletion in a Murine Melanoma Model. J. Immunol. Methods 2008, 333, 167. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Yun, Z.; Tagawa, T.; Rey-McIntyre, K.; Anraku, M.; De Perrot, M. Tumor Cell Repopulation between Cycles of Chemotherapy Is Inhibited by Regulatory T-Cell Depletion in a Murine Mesothelioma Model. J. Thorac. Oncol. 2011, 6, 1578–1586. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 Can Function as a Negative Regulator of T Cell Activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Ise, W.; Kohyama, M.; Nutsch, K.M.; Lee, H.M.; Suri, A.; Unanue, E.R.; Murphy, T.L.; Murphy, K.M. CTLA-4 Suppresses the Pathogenicity of Self Antigen–Specific T Cells by Cell-Intrinsic and Cell-Extrinsic Mechanisms. Nat. Immunol. 2009, 11, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.M.; Sharpe, A.H. Taming Tissue-Specific T Cells: CTLA-4 Reins in Self-Reactive T Cells. Nat. Immunol. 2010, 11, 109–111. [Google Scholar] [CrossRef]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 Inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcγRs Modulate the Anti-Tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc Receptors Modulate in Vivo Cytoxicity against Tumor Targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Dahan, R.; Barnhart, B.C.; Li, F.; Yamniuk, A.P.; Korman, A.J.; Ravetch, J.V. Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcγR Engagement. Cancer Cell 2016, 29, 820–831. [Google Scholar] [CrossRef] [Green Version]
- J€, F.; Bruhns, P.; Onsson, F.J.; Roopenian, D. Mouse and Human FcR Effector Functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostindie, S.C.; Lazar, G.A.; Schuurman, J.; Parren, P.W.H.I. Avidity in Antibody Effector Functions and Biotherapeutic Drug Design. Nat. Rev. Drug Discov. 2022, 2022, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.; Amann, M.; Goubier, A.; Arce Vargas, F.; Zervas, D.; Qing, C.; Henry, J.Y.; Ghorani, E.; Akarca, A.U.; Marafioti, T.; et al. CD25-Treg-Depleting Antibodies Preserving IL-2 Signaling on Effector T Cells Enhance Effector Activation and Antitumor Immunity. Nat. Cancer 2020, 1, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.; Dorr, B.M.; Liu, D.R. A General Strategy for the Evolution of Bond-Forming Enzymes Using Yeast Display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Elsas, M.J.; van der Schoot, J.M.S.; Bartels, A.; Steuten, K.; van Dalen, D.; Wijfjes, Z.; Figdor, C.G.; van Hall, T.; van der Burg, S.H.; Verdoes, M.; et al. Regulatory T Cell Depletion Using a CRISPR Fc-Optimized CD25 Antibody. Int. J. Mol. Sci. 2022, 23, 8707. https://doi.org/10.3390/ijms23158707
van Elsas MJ, van der Schoot JMS, Bartels A, Steuten K, van Dalen D, Wijfjes Z, Figdor CG, van Hall T, van der Burg SH, Verdoes M, et al. Regulatory T Cell Depletion Using a CRISPR Fc-Optimized CD25 Antibody. International Journal of Molecular Sciences. 2022; 23(15):8707. https://doi.org/10.3390/ijms23158707
Chicago/Turabian Stylevan Elsas, Marit J., Johan M. S. van der Schoot, Alexander Bartels, Kas Steuten, Duco van Dalen, Zacharias Wijfjes, Carl G. Figdor, Thorbald van Hall, Sjoerd H. van der Burg, Martijn Verdoes, and et al. 2022. "Regulatory T Cell Depletion Using a CRISPR Fc-Optimized CD25 Antibody" International Journal of Molecular Sciences 23, no. 15: 8707. https://doi.org/10.3390/ijms23158707
APA Stylevan Elsas, M. J., van der Schoot, J. M. S., Bartels, A., Steuten, K., van Dalen, D., Wijfjes, Z., Figdor, C. G., van Hall, T., van der Burg, S. H., Verdoes, M., & Scheeren, F. A. (2022). Regulatory T Cell Depletion Using a CRISPR Fc-Optimized CD25 Antibody. International Journal of Molecular Sciences, 23(15), 8707. https://doi.org/10.3390/ijms23158707