
Diclofenac-Induced Cytotoxicity in Direct and Indirect Co-Culture of HepG2 Cells with Differentiated THP-1 Cells

Abstract
1. Introduction
2. Results
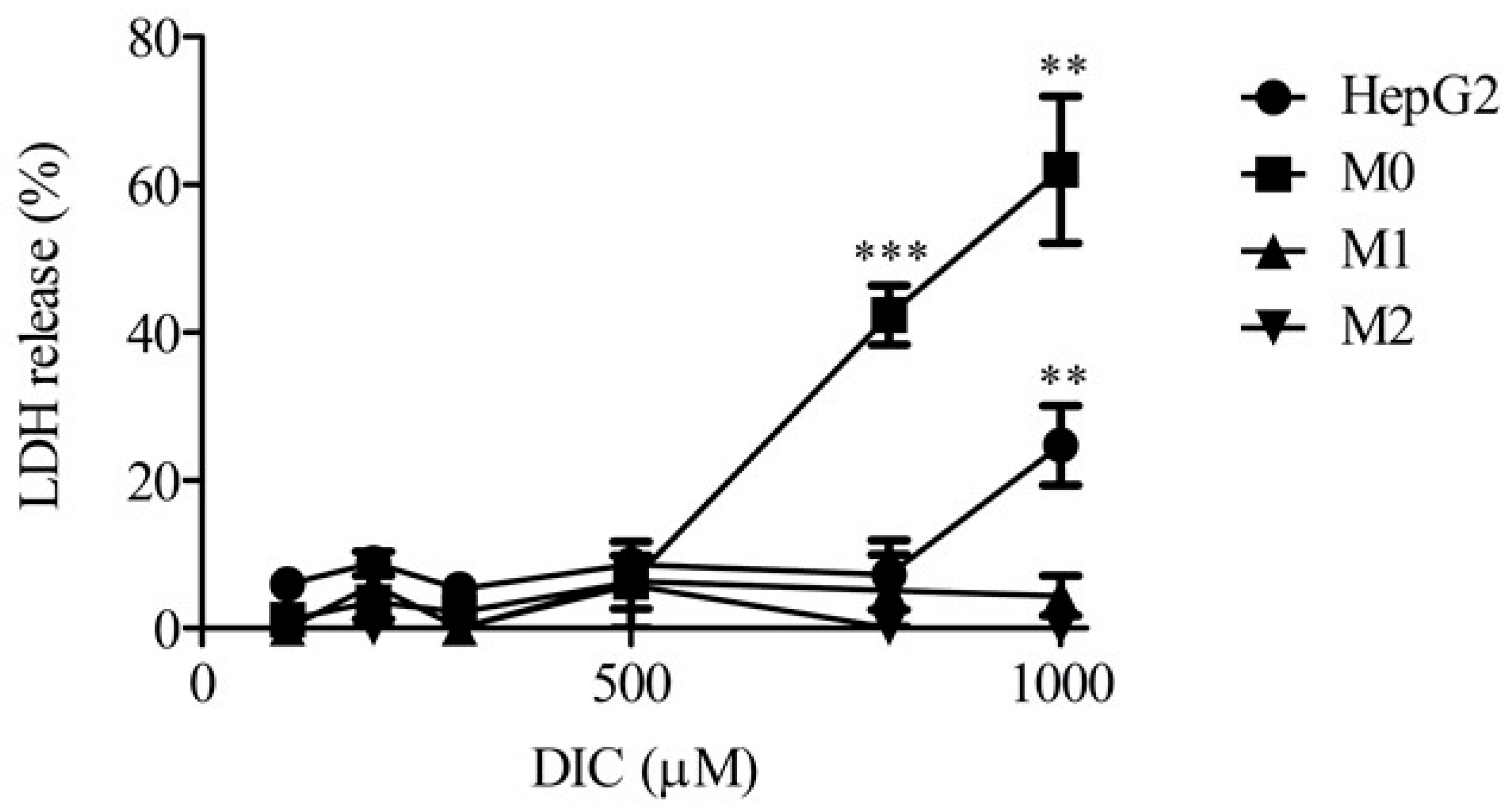
2.1. Lactate Dehydrogenase Release from Monocultures of HepG2 Cells, M0, M1, and M2
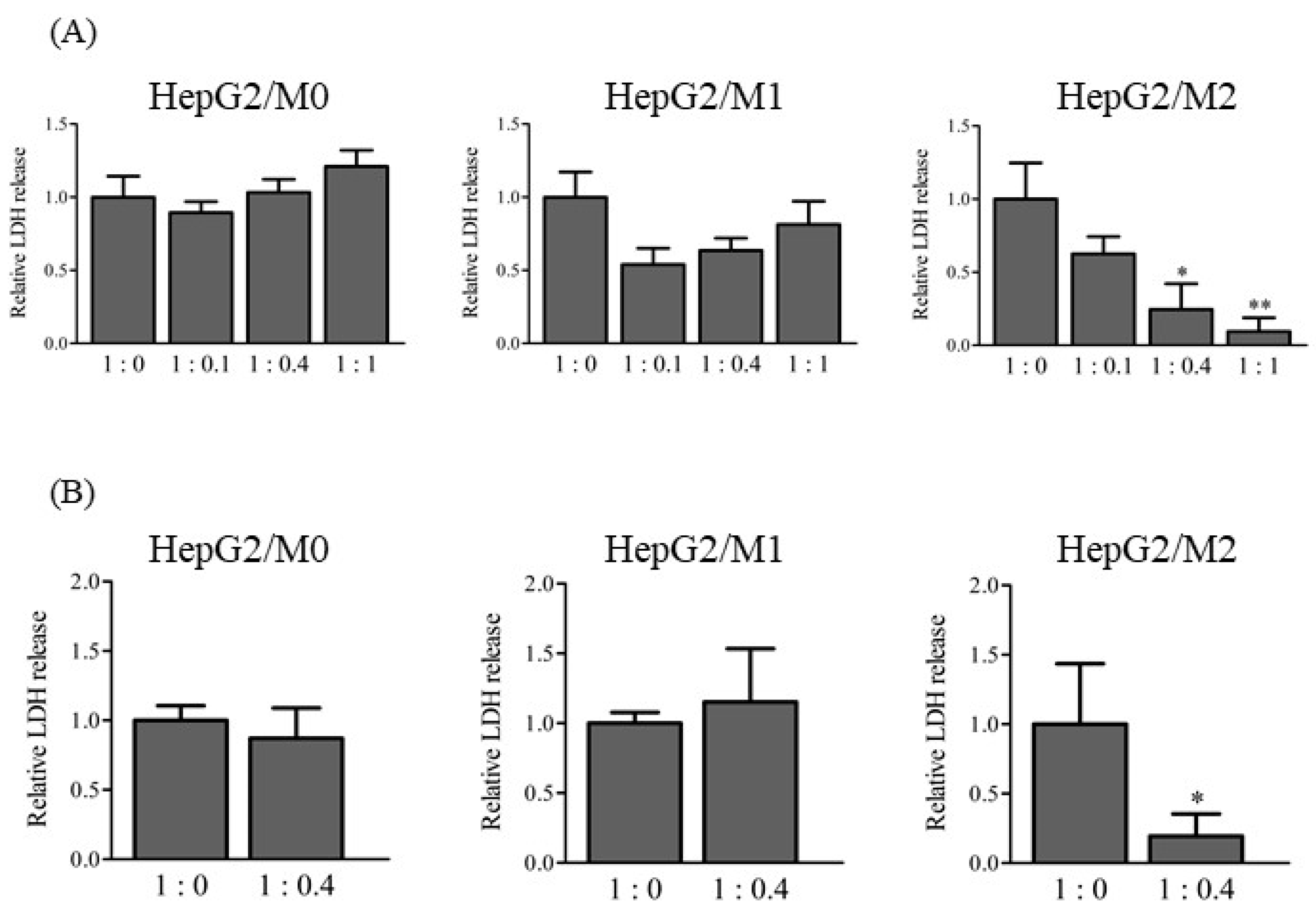
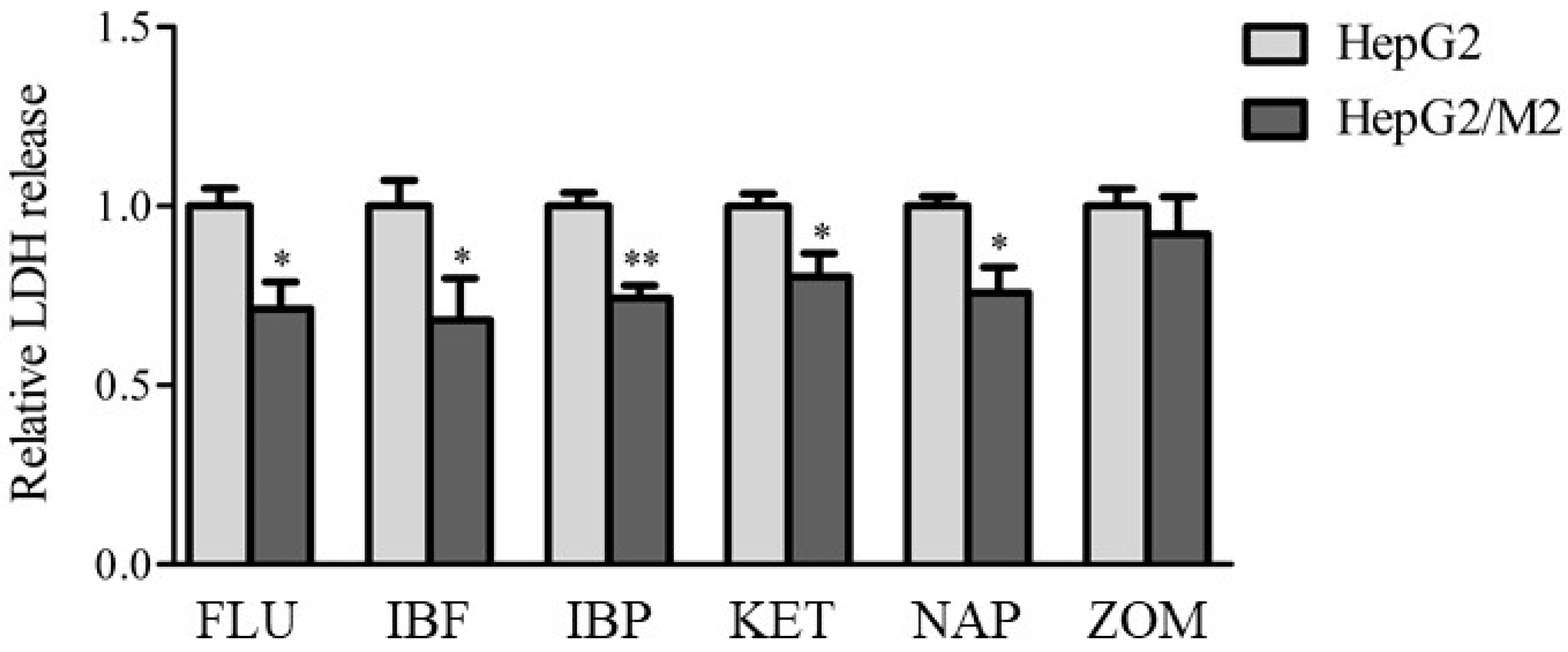
2.2. LDH Release in Direct and Indirect Co-Cultures of HepG2 Cells with M0, M1, and M2
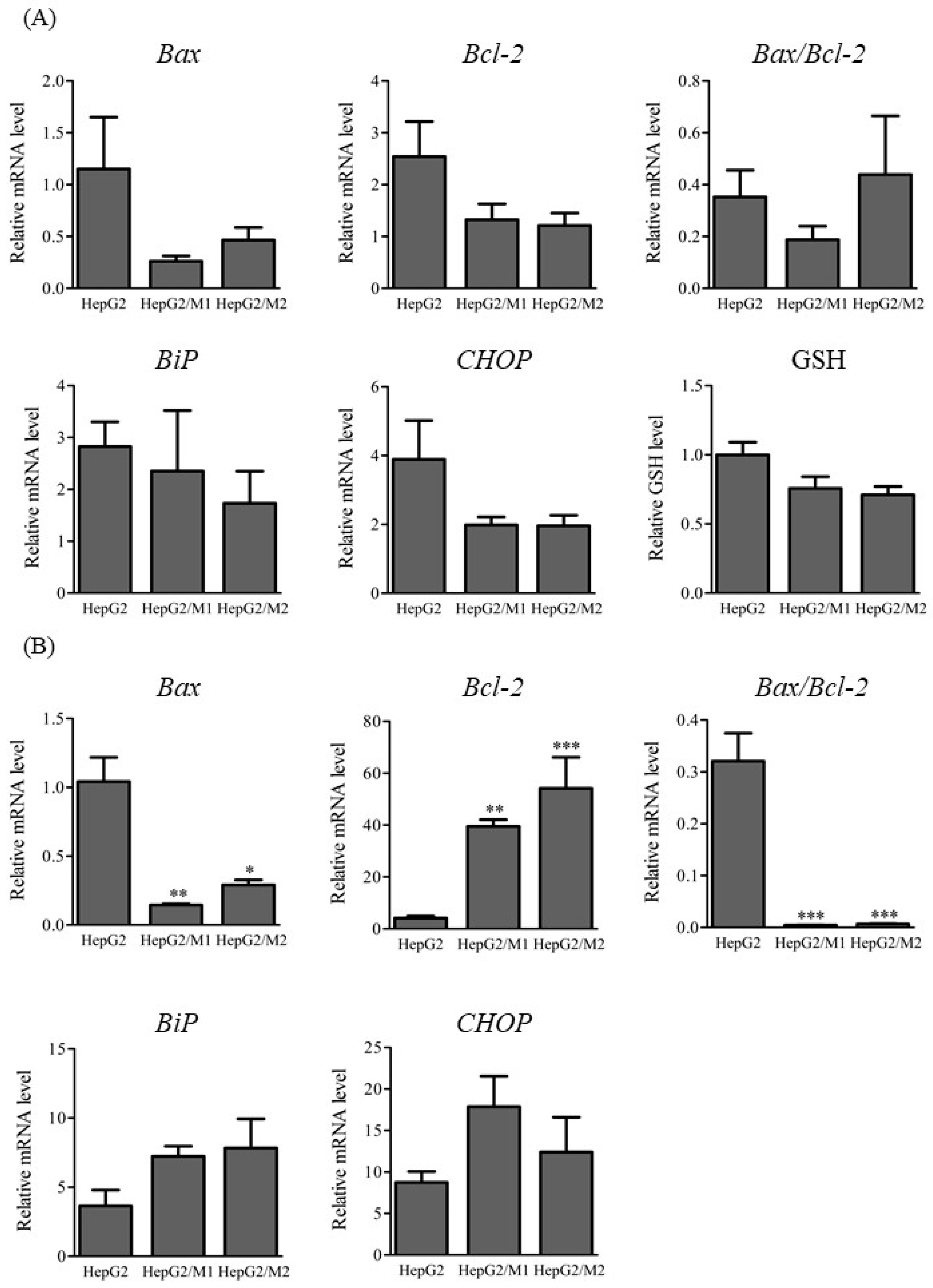
2.3. mRNA Levels of Apoptosis- and ER Stress-Associated Factors and Glutathione Levels
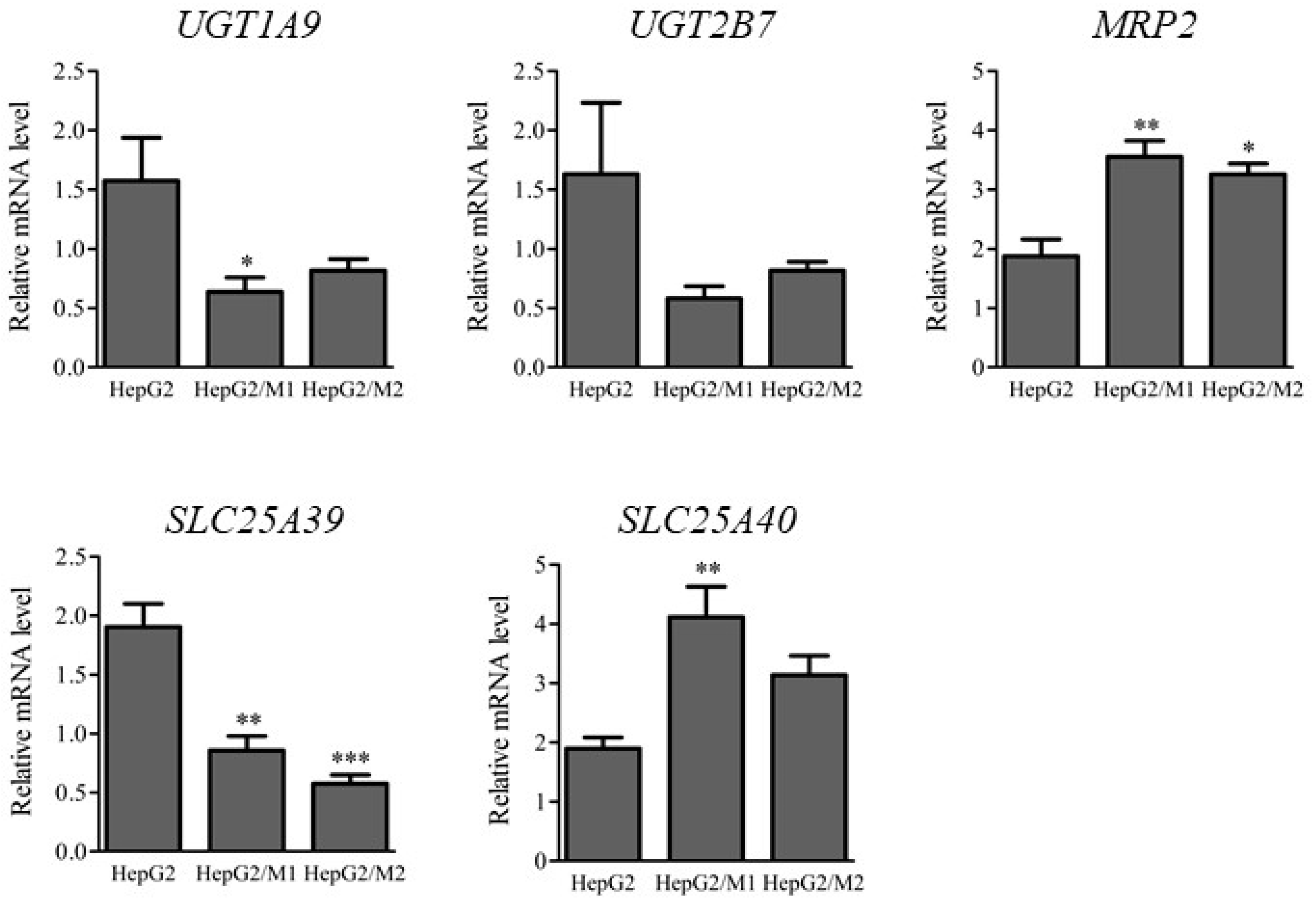
2.4. mRNA Levels of UGTs and Transporters
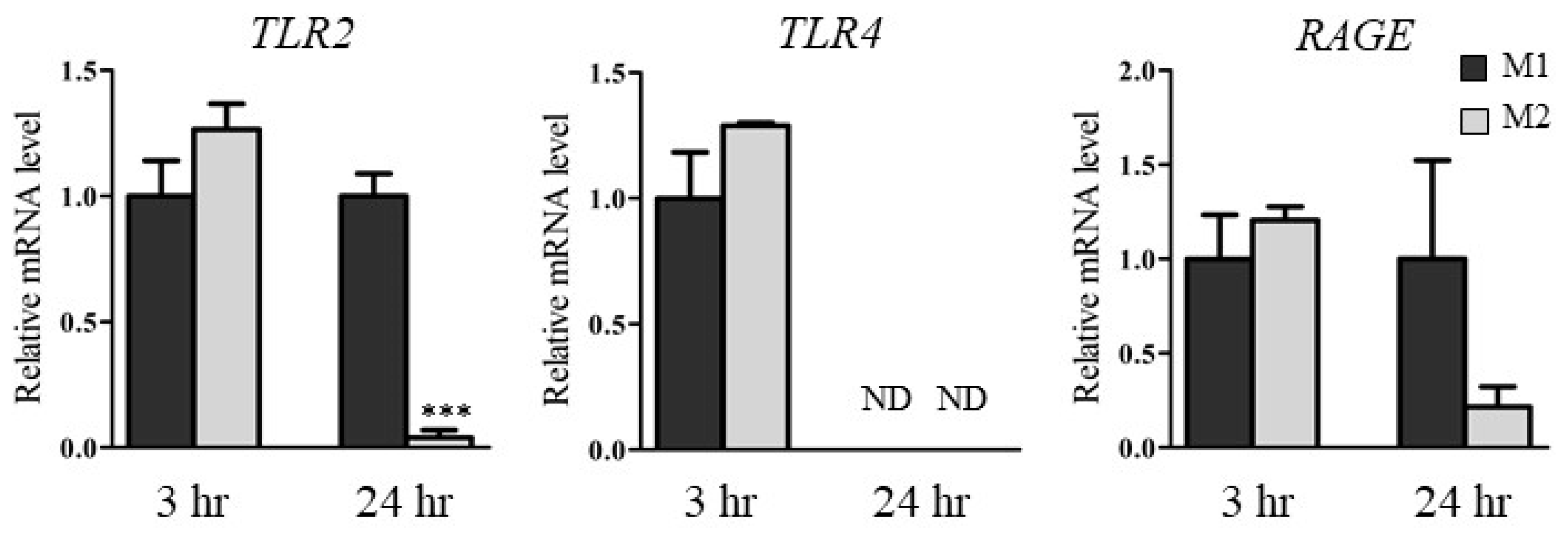
2.5. HMGB1 Concentrations in Medium and mRNA Levels of HMGB1 Receptors in M1 and M2
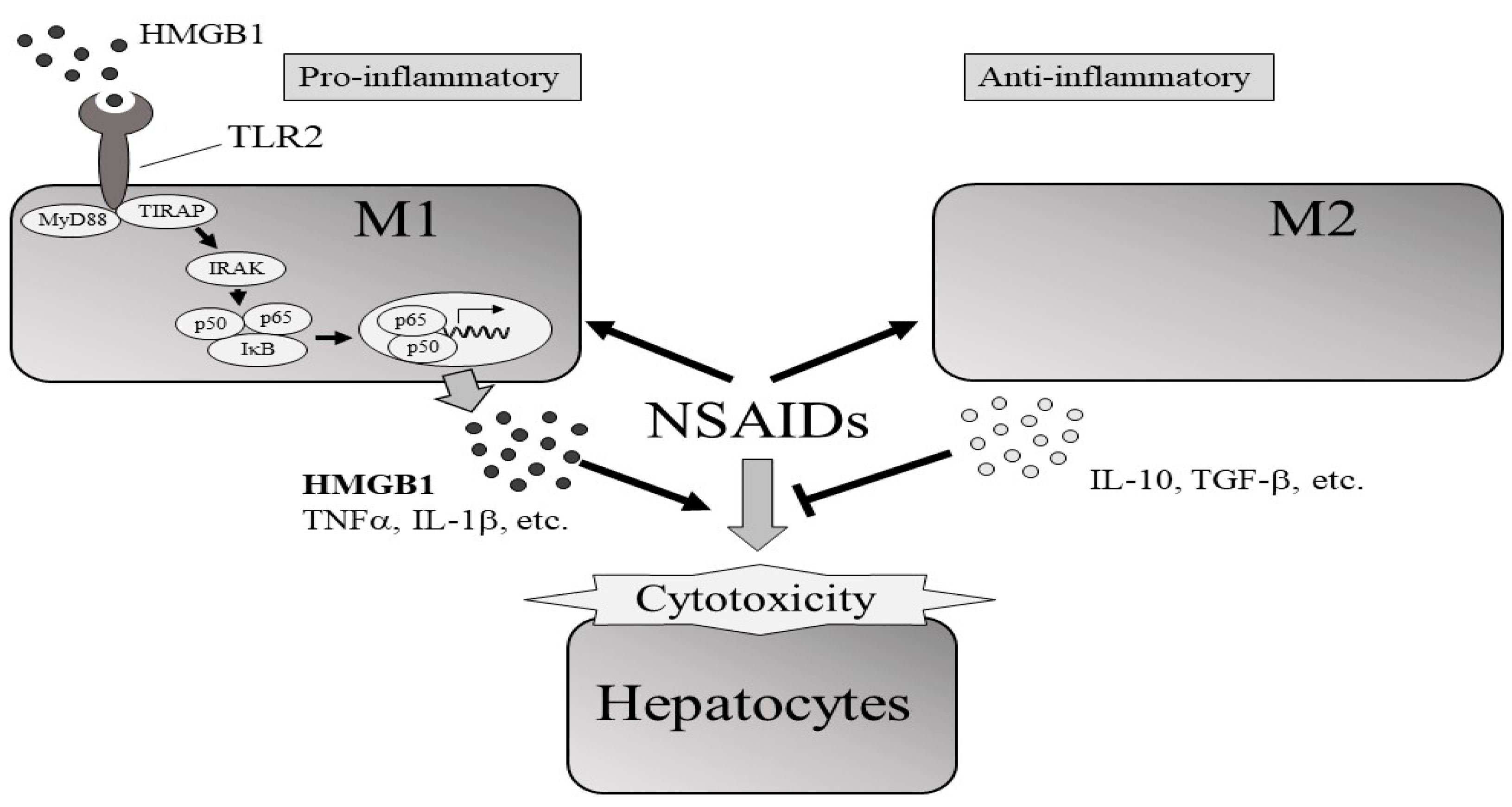
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. LDH Leakage Assay
4.4. mRNA Levels of UGTs, Transporters, HMGB1 Receptors, and ER Stress-Associated Factors
4.5. Intracellular GSH Measurement
4.6. HMGB1 Concentrations in the Medium
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AG | acyl glucuronide |
| Bax | Bcl-2 associated protein X |
| Bcl-2 | B cell lymphoma/leukemia 2 |
| DIC | Diclofenac |
| DILI | drug-induced liver injury |
| DMEM | Dulbecco’s modified Eagle’s medium |
| ER | endoplasmic reticulum |
| FLU | flurbiprofen |
| HMGB1 | high mobility group box 1 |
| IBF | ibufenac |
| IBP | ibuprofen |
| IFN | interferon |
| IL | interleukin |
| KET | ketoprofen |
| LDH | lactate dehydrogenase |
| LPS | lipopolysaccharide |
| MRP | multidrug resistance-associated protein |
| NAP | naproxen |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| PMA | phorbol 12-myristate 13-acetate |
| RAGE | receptor for advanced glycation end products |
| TLR | toll-like receptor |
| UGT | UDP-glucuronosyl transferase |
| ZOM | zomepirac |
References
- Adams, D.H.; Ju, C.; Ramaiah, S.K.; Uetrecht, J.; Jaeschke, H. Mechanisms of immune-mediated liver injury. Toxicol. Sci. 2010, 115, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Boelsterli, U.A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol. In Vitro 2016, 31, 93–102. [Google Scholar] [CrossRef]
- Venkataraman, H.; den Braver, M.W.; Vermeulen, N.P.E.; Commandeur, J.N.M. Cytochrome P450-mediated bioactivation of mefenamic acid to quinoneimine intermediates and inactivation by human glutathione S-transferases. Chem. Res. Toxicol. 2014, 27, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Hashimoto, R.; Shibata, M.; Shimada, H.; Iwaki, M. Involvement of Reactive Metabolites of Diclofenac in Cytotoxicity in Sandwich-Cultured Rat Hepatocytes. Int. J. Toxicol. 2017, 36, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Kaneto, A.; Ishibashi, M.; Kobayashi, A.; Shimada, H.; Iwaki, M. Involvement of diclofenac acyl-β-d-glucuronide in diclofenac-induced cytotoxicity in glutathione-depleted isolated murine hepatocytes co-cultured with peritoneal macrophages. Toxicol. Mech. Methods 2019, 29, 203–210. [Google Scholar] [CrossRef]
- Benesic, A.; Rotter, I.; Dragoi, D.; Weber, S.; Leitl, A.; Buchholtz, M.-L.; Gerbes, A.L. Development and Validation of a Test to Identify Drugs That Cause Idiosyncratic Drug-Induced Liver Injury. Clin. Gastroenterol. Hepatol. 2018, 16, 1488–1494.e5. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. RUCAM in Drug and Herb Induced Liver Injury: The Update. Int. J. Mol. Sci. 2015, 17, 14. [Google Scholar] [CrossRef]
- Banks, A.T.; Zimmerman, H.J.; Ishak, K.G.; Harter, J.G. Diclofenac-associated hepatotoxicity: Analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 1995, 22, 820–827. [Google Scholar] [CrossRef]
- Dunk, A.A.; Walt, R.P.; Jenkins, W.J.; Sherlock, S.S. Diclofenac hepatitis. Br. Med. J. 1982, 284, 1605–1606. [Google Scholar] [CrossRef][Green Version]
- Grillo, M.P.; Knutson, C.G.; Sanders, P.E.; Waldon, D.J.; Hua, F.; Ware, J.A. Studies on the chemical reactivity of diclofenac acyl glucuronide with glutathione: Identification of diclofenac-S-acyl-glutathione in rat bile. Drug Metab. Dispos. 2003, 31, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Tang, W.; Ngui, J.; Tephly, T.; Braun, M. Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol. Sci. 2001, 61, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Li, J.; Huebert, N.; Caldwell, G.W.; Du, Y.; Zhong, H. Detection of a novel reactive metabolite of diclofenac: Evidence for CYP2C9-mediated bioactivation via arene oxides. Drug Metab. Dispos. 2005, 33, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Kenny, J.R.; Maggs, J.L.; Meng, X.; Sinnott, D.; Clarke, S.E.; Park, B.K.; Stachulski, A.V. Syntheses and characterization of the acyl glucuronide and hydroxy metabolites of diclofenac. J. Med. Chem. 2004, 47, 2816–2825. [Google Scholar] [CrossRef] [PubMed]
- Kretz-Rommel, A.; Boelsterli, U.A. Diclofenac covalent protein binding is dependent on acyl glucuronide formation and is inversely related to P450-mediated acute cell injury in cultured rat hepatocytes. Toxicol. Appl. Pharmacol. 1993, 120, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef]
- Sunman, J.A.; Hawke, R.L.; LeCluyse, E.L.; Kashuba, A.D.M. Kupffer cell-mediated IL-2 suppression of CYP3A activity in human hepatocytes. Drug Metab. Dispos. 2004, 32, 359–363. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 91. [Google Scholar] [CrossRef]
- Iwamura, A.; Watanabe, K.; Akai, S.; Nishinosono, T.; Tsuneyama, K.; Oda, S.; Kume, T.; Yokoi, T. Zomepirac Acyl Glucuronide Is Responsible for Zomepirac-Induced Acute Kidney Injury in Mice. Drug Metab. Dispos. 2016, 44, 888–896. [Google Scholar] [CrossRef]
- Yamashita, S.; Oda, S.; Endo, H.; Tsuneyama, K.; Yokoi, T. Neutrophil depletion protects against zomepirac-induced acute kidney injury in mice. Chem. Biol. Interact. 2018, 279, 102–110. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Zhang, C.C.; Gdynia, G.; Ehemann, V.; Roth, W. The HMGB1 protein sensitizes colon carcinoma cells to cell death triggered by pro-apoptotic agents. Int. J. Oncol. 2015, 46, 667–676. [Google Scholar] [CrossRef]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.-Y.; Strassheim, D.; Sohn, J.-W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.-Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Zhang, P.; Yu, Y.; Lu, H.; Liu, Y.; Ni, P.; Su, X.; Wang, D.; Liu, Y.; Wang, J.; et al. HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Phenotype in Experimental Autoimmune Myocarditis Development. Sci. Rep. 2016, 6, 21884. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Eickhoff, A.; Brown, A.C.; Neuman, M.G.; Schulze, J. Diagnostic Biomarkers in Liver Injury by Drugs, Herbs, and Alcohol: Tricky Dilemma after EMA Correctly and Officially Retracted Letter of Support. Int. J. Mol. Sci. 2019, 21, 212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yen, F.S.; Zhu, X.G.; Timson, R.C.; Weber, R.; Xing, C.; Liu, Y.; Allwein, B.; Luo, H.; Yeh, H.-W.; et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 2021, 599, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Westerink, W.M.A.; Schoonen, W.G.E.J. Phase II enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. In Vitro 2007, 21, 1592–1602. [Google Scholar] [CrossRef]
- Kawase, A.; Mukai, H.; Tateishi, S.; Kuroda, S.; Kazaoka, A.; Satoh, R.; Shimada, H.; Sugiura, R.; Iwaki, M. Protein Kinase N Family Negatively Regulates Constitutive Androstane Receptor-Mediated Transcriptional Induction of Cytochrome P450 2b10 in the Livers of Mice. J. Pharmacol. Exp. Ther. 2021, 379, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Nasako, H.; Akizuki, R.; Takashina, Y.; Eguchi, H.; Matsunaga, T.; Yoshino, Y.; Endo, S.; Ikari, A. Elevation of Chemosensitivity of Lung Adenocarcinoma A549 Spheroid Cells by Claudin-2 Knockdown through Activation of Glucose Transport and Inhibition of Nrf2 Signal. Int. J. Mol. Sci. 2021, 22, 6582. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, J.; Naruse, K.; Sado, T.; Uchiyama, T.; Makino, M.; Yamauchi, A.; Ota, H.; Sakuramoto-Tsuchida, S.; Itaya-Hironaka, A.; Takasawa, S.; et al. Involvement of Receptor for Advanced Glycation Endproducts in Hypertensive Disorders of Pregnancy. Int. J. Mol. Sci. 2019, 20, 5462. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 h | 24 h | |
|---|---|---|
| HMGB1 (ng/mL) | ||
| HepG2 | ND | 0.0371 ± 0.0371 |
| HepG2/M1 | 1.78 ± 0.268 *** ### | 0.201 ± 0.184 |
| HepG2/M2 | 0.00719 ± 0.00719 | 0.294 ± 0.170 |
| Gene | Primer Sequence (5′–3′) | Product Size (bp) |
|---|---|---|
| hBiP | Forward: CTGGCTCCTAGAGTACAAGAAAAAG Reverse: ATATTGGTCTACAAGGAGCAGCAAC | 177 |
| hCHOP | Forward: ATTGCCTTTCTCTTCGGACAC Reverse: TTCTTCCTCTTCATTTCCAGGAGG | 127 |
| hBax | Forward: TCATGGGCTGGACATTGGAC Reverse: GCGTCCCAAAGTAGGAGAGG | 145 |
| hBcl-2 | Forward: GAACTGGGGGAGGATTGTGG Reverse: GCCGGTTCAGGTACTCAGTC | 125 |
| hUGT1A9 | Forward: GGGGGCATGAGGTGGTTGTA Reverse: CATTGAGCATGGGCAAAAGCT | 142 |
| hUGT2B7 | Forward: ACAGCAACTGGAAAACAAGCA Reverse: ACACCAGCACCTTTCCACAA | 121 |
| hMRP2 | Forward: AGTCACATGTCCATCCACTGTT Reverse: AGGATGACCTTTCATCCCAACC | 83 |
| hSLC25A39 | Forward: TGCCCTTCTCAGCCCTGTA Reverse: CACAAAGCTCATGCCCACAG | 101 |
| hSLC25A40 | Forward: ACCCACTCCCCAAAGGAAAATG Reverse: GTTTGTTGCCTCCCTCTTCAC | 84 |
| hTLR2 | Forward: GACTCTACCAGATGCCTCCC Reverse: AAGTTATTGCCACCAGCTTCC | 135 |
| hTLR4 | Forward: TGGATCAAGGACCAGAGGCA Reverse: GAGGACCGACACACCAATGA | 141 |
| hRAGE | Forward: CCCTGCTCATTGGGGTCATC Reverse: GTACTACTCTCGCCTGCCTC | 139 |
| hβ-actin | Forward: CACCATTGGCAATGAGCGGTTC Reverse: AGGTCTTTGCGGATGTCCACGT | 135 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawase, A.; Takashima, O.; Tanaka, S.; Shimada, H.; Iwaki, M. Diclofenac-Induced Cytotoxicity in Direct and Indirect Co-Culture of HepG2 Cells with Differentiated THP-1 Cells. Int. J. Mol. Sci. 2022, 23, 8660. https://doi.org/10.3390/ijms23158660
Kawase A, Takashima O, Tanaka S, Shimada H, Iwaki M. Diclofenac-Induced Cytotoxicity in Direct and Indirect Co-Culture of HepG2 Cells with Differentiated THP-1 Cells. International Journal of Molecular Sciences. 2022; 23(15):8660. https://doi.org/10.3390/ijms23158660
Chicago/Turabian StyleKawase, Atsushi, Ouka Takashima, Satsuki Tanaka, Hiroaki Shimada, and Masahiro Iwaki. 2022. "Diclofenac-Induced Cytotoxicity in Direct and Indirect Co-Culture of HepG2 Cells with Differentiated THP-1 Cells" International Journal of Molecular Sciences 23, no. 15: 8660. https://doi.org/10.3390/ijms23158660
APA StyleKawase, A., Takashima, O., Tanaka, S., Shimada, H., & Iwaki, M. (2022). Diclofenac-Induced Cytotoxicity in Direct and Indirect Co-Culture of HepG2 Cells with Differentiated THP-1 Cells. International Journal of Molecular Sciences, 23(15), 8660. https://doi.org/10.3390/ijms23158660