
The Role of Sex in Acute and Chronic Liver Damage




Abstract
1. Introduction
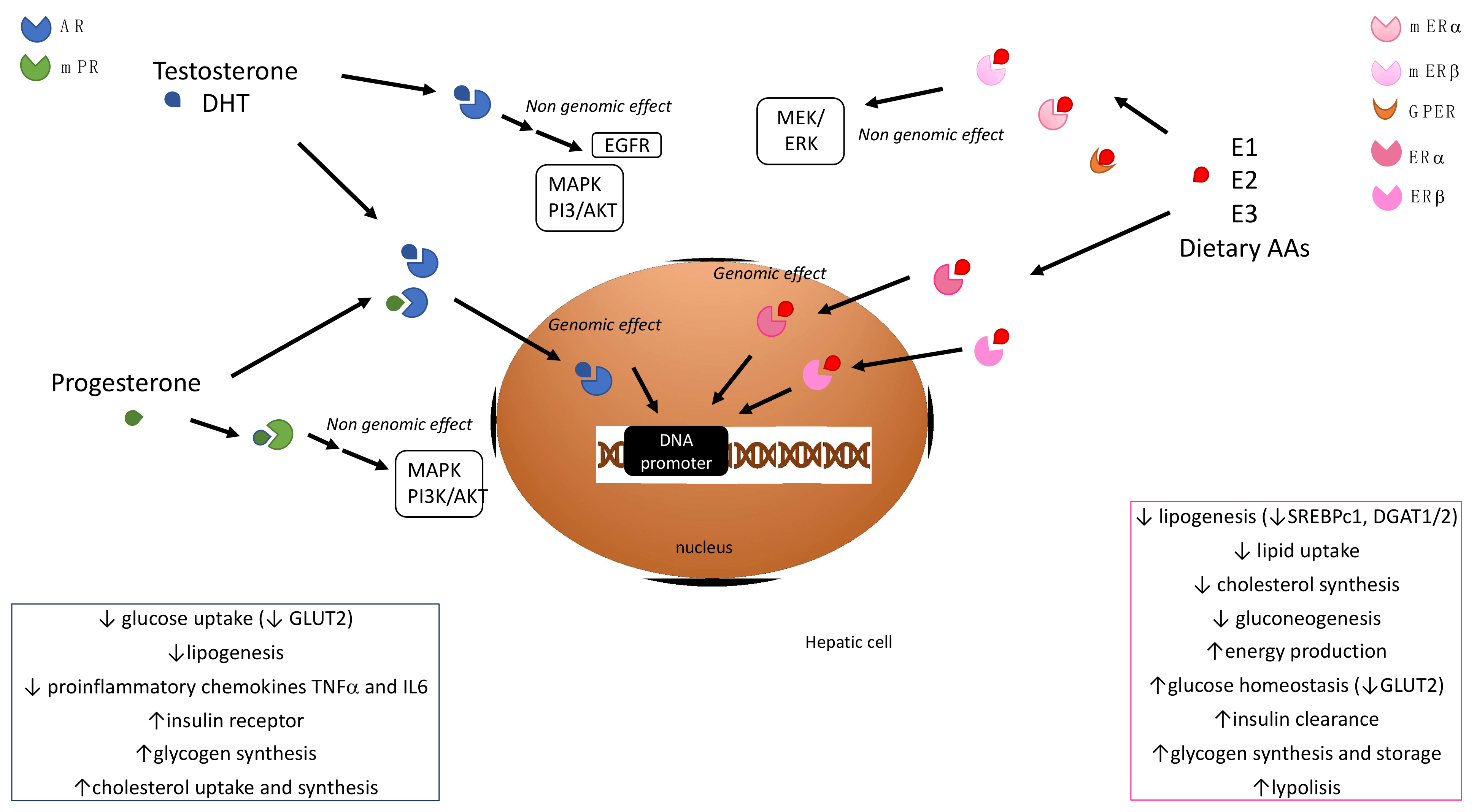
2. Sex Hormone Signaling and Hepatic Metabolism
2.1. Estrogens
2.2. Androgens
2.3. Progestins
3. Acute Liver Injury
3.1. Drug-Induced Liver Injury
3.2. ALF-Associated Comorbidities
3.2.1. Ischemic Hepatitis
3.2.2. Ischemia Reperfusion Injury
3.2.3. Hepatitis A
4. Chronic Liver Injury
4.1. Viral Hepatitis
4.1.1. HBV Hepatitis
4.1.2. HCV Hepatitis
4.2. Alcoholic Liver Disease (ALD)
4.3. Non-Alcoholic Liver Disease (NAFLD)
4.4. Autoimmune Liver Diseases: PBC, PSC, AIH
4.4.1. Primary Biliary Cholangitis
4.4.2. Primary Sclerosing Cholangitis
4.4.3. Autoimmune Hepatitis
4.5. Liver Cancers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, X.; Schadt, E.E.; Wang, S.; Wang, H.; Arnold, A.P.; Ingram-Drake, L.; Drake, T.A.; Lusis, A.J. Tissue-Specific Expression and Regulation of Sexually Dimorphic Genes in Mice. Genome Res. 2006, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular Mechanism of Estrogen-Estrogen Receptor Signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Lampertico, P.; Seletti, C.; Francesca Donato, M.; Ronchi, G.; del Ninno, E.; Colombo, M. The Clinical and Pathogenetic Significance of Estrogen Receptor-Beta Expression in Chronic Liver Diseases and Liver Carcinoma. Cancer 2003, 98, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Miceli, V.; Cocciadiferro, L.; Fregapane, M.; Zarcone, M.; Montalto, G.; Polito, L.M.; Agostara, B.; Granata, O.M.; Carruba, G. Expression of Wild-Type and Variant Estrogen Receptor Alpha in Liver Carcinogenesis and Tumor Progression. Omics A J. Integr. Biol. 2011, 15, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S. Beyond the X Factor: Relevance of Sex Hormones in NAFLD Pathophysiology. Cells 2021, 10, 2502. [Google Scholar] [CrossRef]
- Ribas, V.; Nguyen, M.T.A.; Henstridge, D.C.; Nguyen, A.-K.; Beaven, S.W.; Watt, M.J.; Hevener, A.L. Impaired Oxidative Metabolism and Inflammation Are Associated with Insulin Resistance in ERalpha-Deficient Mice. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E304–E319. [Google Scholar] [CrossRef]
- Shen, M.; Shi, H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, e294278. [Google Scholar] [CrossRef]
- Bryzgalova, G.; Gao, H.; Ahren, B.; Zierath, J.R.; Galuska, D.; Steiler, T.L.; Dahlman-Wright, K.; Nilsson, S.; Gustafsson, J.-A.; Efendic, S.; et al. Evidence That Oestrogen Receptor-Alpha Plays an Important Role in the Regulation of Glucose Homeostasis in Mice: Insulin Sensitivity in the Liver. Diabetologia 2006, 49, 588–597. [Google Scholar] [CrossRef]
- Ahmed-Sorour, H.; Bailey, C.J. Role of Ovarian Hormones in the Long-Term Control of Glucose Homeostasis, Glycogen Formation and Gluconeogenesis. Ann. Nutr. Metab. 1981, 25, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Travison, T.G.; Vesper, H.W.; Orwoll, E.; Wu, F.; Kaufman, J.M.; Wang, Y.; Lapauw, B.; Fiers, T.; Matsumoto, A.M.; Bhasin, S. Harmonized Reference Ranges for Circulating Testosterone Levels in Men of Four Cohort Studies in the United States and Europe. J. Clin. Endocrinol. Metab. 2017, 102, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Courant, F.; Aksglaede, L.; Antignac, J.-P.; Monteau, F.; Sorensen, K.; Andersson, A.-M.; Skakkebaek, N.E.; Juul, A.; Bizec, B.L. Assessment of Circulating Sex Steroid Levels in Prepubertal and Pubertal Boys and Girls by a Novel Ultrasensitive Gas Chromatography-Tandem Mass Spectrometry Method. J. Clin. Endocrinol. Metab. 2010, 95, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. The Roles of Androgen Receptors and Androgen-Binding Proteins in Nongenomic Androgen Actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef]
- Treviño, L.S.; Gorelick, D.A. The Interface of Nuclear and Membrane Steroid Signaling. Endocrinology 2021, 162, bqab107. [Google Scholar] [CrossRef]
- Thomas, P. Membrane Androgen Receptors Unrelated to Nuclear Steroid Receptors. Endocrinology 2019, 160, 772–781. [Google Scholar] [CrossRef]
- Movérare-Skrtic, S.; Venken, K.; Andersson, N.; Lindberg, M.K.; Svensson, J.; Swanson, C.; Vanderschueren, D.; Oscarsson, J.; Gustafsson, J.-A.; Ohlsson, C. Dihydrotestosterone Treatment Results in Obesity and Altered Lipid Metabolism in Orchidectomized Mice. Obesity 2006, 14, 662–672. [Google Scholar] [CrossRef]
- Parthasarathy, C.; Renuka, V.N.; Balasubramanian, K. Sex Steroids Enhance Insulin Receptors and Glucose Oxidation in Chang Liver Cells. Clin. Chim. Acta 2009, 399, 49–53. [Google Scholar] [CrossRef]
- Pal, M.; Gupta, S. Testosterone Supplementation Improves Glucose Homeostasis despite Increasing Hepatic Insulin Resistance in Male Mouse Model of Type 2 Diabetes Mellitus. Nutr. Diabetes 2016, 6, e236. [Google Scholar] [CrossRef]
- Kelly, D.M.; Jones, T.H. Testosterone: A Metabolic Hormone in Health and Disease. J. Endocrinol. 2013, 217, R25–R45. [Google Scholar] [CrossRef]
- Muthusamy, T.; Murugesan, P.; Balasubramanian, K. Sex Steroids Deficiency Impairs Glucose Transporter 4 Expression and Its Translocation through Defective Akt Phosphorylation in Target Tissues of Adult Male Rat. Metabolism 2009, 58, 1581–1592. [Google Scholar] [CrossRef]
- Mohamad, N.-V.; Wong, S.K.; Wan Hasan, W.N.; Jolly, J.J.; Nur-Farhana, M.F.; Ima-Nirwana, S.; Chin, K.-Y. The Relationship between Circulating Testosterone and Inflammatory Cytokines in Men. Aging Male 2019, 22, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Allard, C.; Xu, W.; Mauvais-Jarvis, F. The Role of Androgens in Metabolism, Obesity, and Diabetes in Males and Females. Obesity 2015, 23, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.H.; Dobs, A.S.; Vaidya, D.; Szklo, M.; Gapstur, S.; Kopp, P.; Liu, K.; Ouyang, P. Endogenous Sex Hormones and Glucose Tolerance Status in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2007, 92, 1289–1295. [Google Scholar] [CrossRef]
- Larsson, H.; Ahrén, B. Androgen Activity as a Risk Factor for Impaired Glucose Tolerance in Postmenopausal Women. Diabetes Care 1996, 19, 1399–1403. [Google Scholar] [CrossRef]
- Valadez-Cosmes, P.; Vázquez-Martínez, E.R.; Cerbón, M.; Camacho-Arroyo, I. Membrane Progesterone Receptors in Reproduction and Cancer. Mol. Cell. Endocrinol. 2016, 434, 166–175. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones-Can a Touch of P53 Make a Difference? Front. Endocrinol. 2019, 10, 374. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, X.; Liu, X.; Tian, Z.; Zhang, H.; Qian, X.; Luo, Z.; Wei, D.; Jin, S.; Wang, C.; et al. The Effect of Progesterone and Pregnenolone on Diabetes Status in Chinese Rural Population: A Dose–Response Analysis from Henan Rural Cohort. Eur. J. Endocrinol. 2019, 181, 603–614. [Google Scholar] [CrossRef]
- Weiler, N.; Schlotmann, A.; Schnitzbauer, A.A.; Zeuzem, S.; Welker, M.-W. The Epidemiology of Acute Liver Failure. Dtsch. Arztebl. Int. 2020, 117, 43–50. [Google Scholar] [CrossRef]
- Stravitz, R.T.; Lee, W.M. Acute Liver Failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef]
- Vento, S.; Cainelli, F. Acute Liver Failure. Lancet 2020, 395, 1833. [Google Scholar] [CrossRef]
- Lee, W.M.; Squires, R.H.; Nyberg, S.L.; Doo, E.; Hoofnagle, J.H. Acute Liver Failure: Summary of a Workshop. Hepatology 2008, 47, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Peters, M.G. Liver Disease in Women: The Influence of Gender on Epidemiology, Natural History, and Patient Outcomes. Gastroenterol. Hepatol. 2013, 9, 633–639. [Google Scholar]
- Amacher, D.E. Female Gender as a Susceptibility Factor for Drug-Induced Liver Injury. Hum. Exp. Toxicol. 2014, 33, 928–939. [Google Scholar] [CrossRef]
- Reuben, A.; Koch, D.G.; Lee, W.M. Drug-Induced Acute Liver Failure: Results of a U.S. Multicenter, Prospective Study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef]
- Bizzaro, D.; Crescenzi, M.; Di Liddo, R.; Arcidiacono, D.; Cappon, A.; Bertalot, T.; Amodio, V.; Tasso, A.; Stefani, A.; Bertazzo, V.; et al. Sex-Dependent Differences in Inflammatory Responses during Liver Regeneration in a Murine Model of Acute Liver Injury. Clin. Sci. 2018, 132, 255–272. [Google Scholar] [CrossRef]
- Para, O.; Crispino, P.; Barone, N.; Macis, S.; Airasca, L.; Gnerre, P.; Politi, C. Sex Differences in Adverse Drug Reaction and Liver Disease. Ital. J. Med. 2018, 12, 15–22. [Google Scholar] [CrossRef]
- Soldin, O.P.; Mattison, D.R. Sex Differences in Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2009, 48, 143–157. [Google Scholar] [CrossRef]
- Ruggieri, A.; Gagliardi, M.C.; Anticoli, S. Sex-Dependent Outcome of Hepatitis B and C Viruses Infections: Synergy of Sex Hormones and Immune Responses? Front. Immunol. 2018, 9, 2302. [Google Scholar] [CrossRef]
- Palatini, P.; Martin, S.; Pegoraro, P.; Orlando, R. Enzyme Inhibition and Induction in Liver Disease. Curr. Clin. Pharmacol. 2008, 3, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Parikh, P.M.; Gerussi, A.; Tsochatzis, E. Gender Differences in Liver Disease and the Drug-Dose Gender Gap. Pharmacol. Res. 2017, 120, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-Induced Liver Injury: Interactions between Drug Properties and Host Factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, P.; Aithal, G.P.; Bjornsson, E.S.; Andrade, R.J.; Sawle, A.; Arrese, M.; Barnhart, H.X.; Bondon-Guitton, E.; Hayashi, P.H.; Bessone, F.; et al. Association of Liver Injury From Specific Drugs, or Groups of Drugs, With Polymorphisms in HLA and Other Genes in a Genome-Wide Association Study. Gastroenterology 2017, 152, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Kullak-Ublick, G.A.; Andrade, R.J.; Merz, M.; End, P.; Benesic, A.; Gerbes, A.L.; Aithal, G.P. Drug-Induced Liver Injury: Recent Advances in Diagnosis and Risk Assessment. Gut 2017, 66, 1154–1164. [Google Scholar] [CrossRef]
- Grove, J.I.; Aithal, G.P. Human Leukocyte Antigen Genetic Risk Factors of Drug-Induced Liver Toxicology. Expert Opin. Drug Metab. Toxicol. 2015, 11, 395–409. [Google Scholar] [CrossRef]
- Jeong, H. Altered Drug Metabolism during Pregnancy: Hormonal Regulation of Drug-Metabolizing Enzymes. Expert Opin. Drug Metab. Toxicol. 2010, 6, 689–699. [Google Scholar] [CrossRef]
- Palatini, P.; Orlando, R.; De Martin, S. The Effect of Liver Disease on Inhibitory and Plasma Protein-Binding Displacement Interactions: An Update. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1215–1230. [Google Scholar] [CrossRef]
- Khatri, R.; Fallon, J.K.; Sykes, C.; Kulick, N.; Rementer, R.J.B.; Miner, T.A.; Schauer, A.P.; Kashuba, A.D.M.; Boggess, K.A.; Brouwer, K.L.R.; et al. Pregnancy-Related Hormones Increase UGT1A1-Mediated Labetalol Metabolism in Human Hepatocytes. Front. Pharmacol. 2021, 12, 655320. [Google Scholar] [CrossRef]
- Roelfsema, F.; Veldhuis, J.D. Growth Hormone Dynamics in Healthy Adults Are Related to Age and Sex and Strongly Dependent on Body Mass Index. Neuroendocrinology 2016, 103, 335–344. [Google Scholar] [CrossRef]
- Cho, J.; Kim, L.; Li, Z.; Rose, N.R.; Talor, M.V.; Njoku, D.B. Sex Bias in Experimental Immune-Mediated, Drug-Induced Liver Injury in BALB/c Mice: Suggested Roles for Tregs, Estrogen, and IL-6. PLoS ONE 2013, 8, e61186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Jiang, Z. T-Helper Cell-Mediated Factors in Drug-Induced Liver Injury. J. Appl. Toxicol. 2015, 35, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Spengler, E.; Fontana, R.J. Chapter 2—Acute Liver Failure. In Handbook of Liver Disease, 4th ed.; Friedman, L.S., Martin, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 18–33. ISBN 978-0-323-47874-8. [Google Scholar]
- Maruyama, H.; Shiina, S. Antioxidant Therapy on Ischemic Hepatitis: Here We Are and Where Do We Go? Hepatol. Int. 2020, 14, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Tapper, E.B.; Sengupta, N.; Bonder, A. The Incidence and Outcomes of Ischemic Hepatitis: A Systematic Review with Meta-Analysis. Am. J. Med. 2015, 128, 1314–1321. [Google Scholar] [CrossRef]
- Stewart, D.; Shores, D.; Alaish, S.M. Organ System Response to Cardiac Function—Splanchnic. In Critical Heart Disease in Infants and Children, 3rd ed.; Ungerleider, R.M., Meliones, J.N., Nelson McMillan, K., Cooper, D.S., Jacobs, J.P., Eds.; Elsevier: Philadelphia, PA, USA, 2019; pp. 150–159.e4. ISBN 978-1-4557-0760-7. [Google Scholar]
- Waseem, N.; Chen, P.-H. Hypoxic Hepatitis: A Review and Clinical Update. J. Clin. Transl. Hepatol. 2016, 4, 263–268. [Google Scholar] [CrossRef]
- Ostapowicz, G.; Fontana, R.J.; Schiødt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.B.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a Prospective Study of Acute Liver Failure at 17 Tertiary Care Centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Taylor, R.M.; Tujios, S.; Jinjuvadia, K.; Davern, T.; Shaikh, O.S.; Han, S.; Chung, R.T.; Lee, W.M.; Fontana, R.J. Short and Long-Term Outcomes in Patients with Acute Liver Failure Due to Ischemic Hepatitis. Dig. Dis. Sci. 2012, 57, 777–785. [Google Scholar] [CrossRef]
- Lee, S.H.; Culberson, C.; Korneszczuk, K.; Clemens, M.G. Differential Mechanisms of Hepatic Vascular Dysregulation with Mild vs. Moderate Ischemia-Reperfusion. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1219–G1226. [Google Scholar] [CrossRef]
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia Reperfusion Injury in Liver Transplantation: Cellular and Molecular Mechanisms. Liver Int. 2019, 39, 788–801. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, J.; Wei, Q.; Saeb-Parsy, K.; Xu, X. The Role of Ischemia/Reperfusion Injury in Early Hepatic Allograft Dysfunction. Liver Transpl. 2020, 26, 1034–1048. [Google Scholar] [CrossRef]
- Sanketh, R.; Daqing, M. Hepatic Ischemia-Reperfusion Injury in Liver Transplant Setting: Mechanisms and Protective Strategies. J. Biomed. Res. 2019, 33, 221–234. [Google Scholar] [CrossRef]
- Berrevoet, F.; Hesse, U.J.; de Laere, S.; Jacobs, B.; Pattyn, P.; de Hemptinne, B. Impact of Donor and Recipient Gender on Liver Transplantation. Transpl. Proc. 1997, 29, 3431–3432. [Google Scholar] [CrossRef]
- Jarrar, D.; Wang, P.; Cioffi, W.G.; Bland, K.I.; Chaudry, I.H. The Female Reproductive Cycle Is an Important Variable in the Response to Trauma-Hemorrhage. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1015–H1021. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Pavlick, K.P.; Hines, I.N.; Hoffman, J.M.; Bharwani, S.; Gray, L.; Wolf, R.E.; Grisham, M.B. Selected Contribution: Effects of Gender on Reduced-Size Liver Ischemia and Reperfusion Injury. J. Appl. Physiol. 2001, 91, 2816–2822. [Google Scholar] [CrossRef]
- Diniz-Santos, D.R.; de Melo, M.C.N.; Melo, R.F.; Silva, L.R. Acute Liver Failure Complicating Viral Hepatitis A. Braz. J. Infect. Dis. 2004, 8, 180–183. [Google Scholar] [CrossRef]
- Chang, M.-L.; Liaw, Y.-F. Gender Impacts on the Disease Severity of Overt Acute Hepatitis A: Different from Overt Acute Hepatitis B. Dig. Dis. Sci. 2019, 64, 570–575. [Google Scholar] [CrossRef]
- Manka, P.; Verheyen, J.; Gerken, G.; Canbay, A. Liver Failure Due to Acute Viral Hepatitis (A–E). Visc. Med. 2016, 32, 80–85. [Google Scholar] [CrossRef]
- Giefing-Kröll, C.; Berger, P.; Lepperdinger, G.; Grubeck-Loebenstein, B. How Sex and Age Affect Immune Responses, Susceptibility to Infections, and Response to Vaccination. Aging Cell 2015, 14, 309–321. [Google Scholar] [CrossRef]
- Ruggieri, A.; Anticoli, S.; D’Ambrosio, A.; Giordani, L.; Viora, M. The Influence of Sex and Gender on Immunity, Infection and Vaccination. Ann. Ist. Super. Sanita 2016, 52, 198–204. [Google Scholar] [CrossRef]
- Durazzo, M.; Belci, P.; Collo, A.; Prandi, V.; Pistone, E.; Martorana, M.; Gambino, R.; Bo, S. Gender Specific Medicine in Liver Diseases: A Point of View. World J. Gastroenterol. 2014, 20, 2127–2135. [Google Scholar] [CrossRef]
- Wang, S.-H.; Yeh, S.-H.; Lin, W.-H.; Wang, H.-Y.; Chen, D.-S.; Chen, P.-J. Identification of Androgen Response Elements in the Enhancer I of Hepatitis B Virus: A Mechanism for Sex Disparity in Chronic Hepatitis B. Hepatology 2009, 50, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Chen, P.-J.; Yeh, S.-H. Gender Disparity in Chronic Hepatitis B: Mechanisms of Sex Hormones. J. Gastroenterol. Hepatol 2015, 30, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Yeh, S.-H.; Lin, W.-H.; Yeh, K.-H.; Yuan, Q.; Xia, N.-S.; Chen, D.-S.; Chen, P.-J. Estrogen Receptor α Represses Transcription of HBV Genes via Interaction with Hepatocyte Nuclear Factor 4α. Gastroenterology 2012, 142, 989–998.e4. [Google Scholar] [CrossRef]
- Tan, J.; Surti, B.; Saab, S. Pregnancy and Cirrhosis. Liver Transpl. 2008, 14, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, A.A.M.; Myers, R.P. The Outcomes of Pregnancy in Patients with Cirrhosis: A Population-Based Study. Liver Int. 2010, 30, 275–283. [Google Scholar] [CrossRef]
- Hagström, H.; Höijer, J.; Marschall, H.-U.; Williamson, C.; Heneghan, M.A.; Westbrook, R.H.; Ludvigsson, J.F.; Stephansson, O. Outcomes of Pregnancy in Mothers With Cirrhosis: A National Population-Based Cohort Study of 1.3 Million Pregnancies. Hepatol. Commun. 2018, 2, 1299–1305. [Google Scholar] [CrossRef]
- Lee, C.M.; Lu, S.N.; Changchien, C.S.; Yeh, C.T.; Hsu, T.T.; Tang, J.H.; Wang, J.H.; Lin, D.Y.; Chen, C.L.; Chen, W.J. Age, Gender, and Local Geographic Variations of Viral Etiology of Hepatocellular Carcinoma in a Hyperendemic Area for Hepatitis B Virus Infection. Cancer 1999, 86, 1143–1150. [Google Scholar] [CrossRef]
- Yu, M.-W.; Yeh, S.-H.; Chen, P.-J.; Liaw, Y.-F.; Lin, C.-L.; Liu, C.-J.; Shih, W.-L.; Kao, J.-H.; Chen, D.-S.; Chen, C.-J. Hepatitis B Virus Genotype and DNA Level and Hepatocellular Carcinoma: A Prospective Study in Men. J. Natl. Cancer Inst. 2005, 97, 265–272. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL Recommendations on Treatment of Hepatitis C: Final Update of the Series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef]
- Micallef, J.M.; Kaldor, J.M.; Dore, G.J. Spontaneous Viral Clearance Following Acute Hepatitis C Infection: A Systematic Review of Longitudinal Studies. J. Viral. Hepat. 2006, 13, 34–41. [Google Scholar] [CrossRef]
- Van den Berg, C.H.B.S.; Grady, B.P.X.; Schinkel, J.; van de Laar, T.; Molenkamp, R.; van Houdt, R.; Coutinho, R.A.; van Baarle, D.; Prins, M. Female Sex and IL28B, a Synergism for Spontaneous Viral Clearance in Hepatitis C Virus (HCV) Seroconverters from a Community-Based Cohort. PLoS ONE 2011, 6, e27555. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, A.; Patel, D.; Yarlagadda, S.; Strickland, F.M.; Richardson, B.C. Stronger Inflammatory/Cytotoxic T-Cell Response in Women Identified by Microarray Analysis. Genes Immun. 2009, 10, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Sekhon, H.K.; Kaur, G. Sex Hormones and Immune Dimorphism. Sci. World J. 2014, 2014, 159150. [Google Scholar] [CrossRef] [PubMed]
- Seillet, C.; Laffont, S.; Trémollières, F.; Rouquié, N.; Ribot, C.; Arnal, J.-F.; Douin-Echinard, V.; Gourdy, P.; Guéry, J.-C. The TLR-Mediated Response of Plasmacytoid Dendritic Cells Is Positively Regulated by Estradiol in Vivo through Cell-Intrinsic Estrogen Receptor α Signaling. Blood 2012, 119, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2015, 6, 635. [Google Scholar] [CrossRef]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG Clinical Guideline: Alcoholic Liver Disease. Off. J. Am. Coll. Gastroenterol. ACG 2018, 113, 175–194. [Google Scholar] [CrossRef]
- White, A. Gender Differences in the Epidemiology of Alcohol Use and Related Harms in the United States. ARCR 2020, 40, 1. [Google Scholar] [CrossRef]
- Becker, U.; Deis, A.; Sørensen, T.I.; Grønbaek, M.; Borch-Johnsen, K.; Müller, C.F.; Schnohr, P.; Jensen, G. Prediction of Risk of Liver Disease by Alcohol Intake, Sex, and Age: A Prospective Population Study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef]
- Thomasson, H.R. Gender Differences in Alcohol Metabolism. Physiological Responses to Ethanol. Recent Dev. Alcohol. 1995, 12, 163–179. [Google Scholar] [CrossRef]
- Thurman, R.G. II. Alcoholic Liver Injury Involves Activation of Kupffer Cells by Endotoxin. Am. J. Physiol. 1998, 275, G605–G611. [Google Scholar] [CrossRef]
- Parés, A.; Caballería, J.; Bruguera, M.; Torres, M.; Rodés, J. Histological Course of Alcoholic Hepatitis. Influence of Abstinence, Sex and Extent of Hepatic Damage. J. Hepatol. 1986, 2, 33–42. [Google Scholar] [CrossRef]
- Eagon, P.K.; Lechner, P.S. Effect of Alcohol on Growth Hormone-Related Liver Function and Sex Hormone Homeostasis. In Alcohol and Hormones; Watson, R.R., Ed.; Humana Press: Totowa, NJ, USA, 1995; pp. 325–336. ISBN 978-1-4612-6678-5. [Google Scholar]
- Eagon, P.K. Alcoholic Liver Injury: Influence of Gender and Hormones. World J. Gastroenterol. 2010, 16, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Järveläinen, H.A.; Lukkari, T.A.; Heinaro, S.; Sippel, H.; Lindros, K.O. The Antiestrogen Toremifene Protects against Alcoholic Liver Injury in Female Rats. J. Hepatol. 2001, 35, 46–52. [Google Scholar] [CrossRef]
- Colantoni, A.; Emanuele, M.A.; Kovacs, E.J.; Villa, E.; Van Thiel, D.H. Hepatic Estrogen Receptors and Alcohol Intake. Mol. Cell. Endocrinol. 2002, 193, 101–104. [Google Scholar] [CrossRef]
- Banerjee, A.; Rose, R.; Johnson, G.A.; Burghardt, R.C.; Ramaiah, S.K. The Influence of Estrogen on Hepatobiliary Osteopontin (SPP1) Expression in a Female Rodent Model of Alcoholic Steatohepatitis. Toxicol. Pathol. 2009, 37, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Russell, W.K.; Jayaraman, A.; Ramaiah, S.K. Identification of Proteins to Predict the Molecular Basis for the Observed Gender Susceptibility in a Rat Model of Alcoholic Steatohepatitis by 2-D Gel Proteomics. Proteomics 2008, 8, 4327–4337. [Google Scholar] [CrossRef]
- Tadic, S.D.; Elm, M.S.; Li, H.-S.; Van Londen, G.J.; Subbotin, V.M.; Whitcomb, D.C.; Eagon, P.K. Sex Differences in Hepatic Gene Expression in a Rat Model of Ethanol-Induced Liver Injury. J. Appl. Physiol. 2002, 93, 1057–1068. [Google Scholar] [CrossRef]
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J.C.; Bode, C. Increased Intestinal Permeability to Macromolecules and Endotoxemia in Patients with Chronic Alcohol Abuse in Different Stages of Alcohol-Induced Liver Disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Holmes, E.W.; Patel, M.; Iber, F.; Fields, J.Z.; Pethkar, S. Leaky Gut in Alcoholic Cirrhosis: A Possible Mechanism for Alcohol-Induced Liver Damage. Am. J. Gastroenterol. 1999, 94, 200–207. [Google Scholar] [CrossRef]
- Kirpich, I.A.; McClain, C.J.; Vatsalya, V.; Schwandt, M.; Phillips, M.; Falkner, K.C.; Zhang, L.; Harwell, C.; George, D.T.; Umhau, J.C. Liver Injury and Endotoxemia in Male and Female Alcohol-Dependent Individuals Admitted to an Alcohol Treatment Program. Alcohol. Clin. Exp. Res. 2017, 41, 747–757. [Google Scholar] [CrossRef]
- Gabbia, D.; Roverso, M.; Guido, M.; Sacchi, D.; Scaffidi, M.; Carrara, M.; Orso, G.; Russo, F.P.; Floreani, A.; Bogialli, S.; et al. Western Diet-Induced Metabolic Alterations Affect Circulating Markers of Liver Function before the Development of Steatosis. Nutrients 2019, 11, 1602. [Google Scholar] [CrossRef] [PubMed]
- Colognesi, M.; Gabbia, D.; De Martin, S. Depression and Cognitive Impairment-Extrahepatic Manifestations of NAFLD and NASH. Biomedicines 2020, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.-H. Regulation of Glucose Metabolism from a Liver-Centric Perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef]
- Bazhan, N.; Jakovleva, T.; Feofanova, N.; Denisova, E.; Dubinina, A.; Sitnikova, N.; Makarova, E. Sex Differences in Liver, Adipose Tissue, and Muscle Transcriptional Response to Fasting and Refeeding in Mice. Cells 2019, 8, E1529. [Google Scholar] [CrossRef]
- Sorrentino, D.; Zhou, S.L.; Kokkotou, E.; Berk, P.D. Sex Differences in Hepatic Fatty Acid Uptake Reflect a Greater Affinity of the Transport System in Females. Am. J. Physiol. 1992, 263, G380–G385. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, N.; Rico-Bautista, E.; Fisher, R.M.; Wu, X.; Cheung, L.; Flores-Morales, A.; Tybring, G.; Norstedt, G.; Tollet-Egnell, P. Female-Predominant Expression of Fatty Acid Translocase/CD36 in Rat and Human Liver. Endocrinology 2004, 145, 1972–1979. [Google Scholar] [CrossRef]
- Meda, C.; Barone, M.; Mitro, N.; Lolli, F.; Pedretti, S.; Caruso, D.; Maggi, A.; Della Torre, S. Hepatic ERα Accounts for Sex Differences in the Ability to Cope with an Excess of Dietary Lipids. Mol. Metab. 2020, 32, 97–108. [Google Scholar] [CrossRef]
- Goossens, G.H.; Jocken, J.W.E.; Blaak, E.E. Sexual Dimorphism in Cardiometabolic Health: The Role of Adipose Tissue, Muscle and Liver. Nat. Rev. Endocrinol. 2021, 17, 47–66. [Google Scholar] [CrossRef]
- Trapani, L.; Segatto, M.; Pallottini, V. Regulation and Deregulation of Cholesterol Homeostasis: The Liver as a Metabolic “Power Station”. World J. Hepatol. 2012, 4, 184–190. [Google Scholar] [CrossRef]
- D’Eon, T.M.; Souza, S.C.; Aronovitz, M.; Obin, M.S.; Fried, S.K.; Greenberg, A.S. Estrogen Regulation of Adiposity and Fuel Partitioning. Evidence of Genomic and Non-Genomic Regulation of Lipogenic and Oxidative Pathways. J. Biol. Chem. 2005, 280, 35983–35991. [Google Scholar] [CrossRef]
- Bruno, S.; Maisonneuve, P.; Castellana, P.; Rotmensz, N.; Rossi, S.; Maggioni, M.; Persico, M.; Colombo, A.; Monasterolo, F.; Casadei-Giunchi, D.; et al. Incidence and Risk Factors for Non-Alcoholic Steatohepatitis: Prospective Study of 5408 Women Enrolled in Italian Tamoxifen Chemoprevention Trial. BMJ 2005, 330, 932. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.L.; Rifkind, B.M.; Sempos, C.T.; Carroll, M.D.; Bachorik, P.S.; Briefel, R.R.; Gordon, D.J.; Burt, V.L.; Brown, C.D.; Lippel, K. Declining Serum Total Cholesterol Levels among US Adults. The National Health and Nutrition Examination Surveys. JAMA 1993, 269, 3002–3008. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Marzorati, S.; Anaya, J.-M.; Gershwin, M.E. Primary Biliary Cholangitis: A Comprehensive Overview. Hepatol. Int. 2017, 11, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Purohit, T.; Cappell, M.S. Primary Biliary Cirrhosis: Pathophysiology, Clinical Presentation and Therapy. World J. Hepatol. 2015, 7, 926–941. [Google Scholar] [CrossRef]
- Smyk, D.S.; Rigopoulou, E.I.; Pares, A.; Billinis, C.; Burroughs, A.K.; Muratori, L.; Invernizzi, P.; Bogdanos, D.P. Sex Differences Associated with Primary Biliary Cirrhosis. Clin. Dev. Immunol. 2012, 2012, 610504. [Google Scholar] [CrossRef]
- Marchioni Beery, R.M.; Vaziri, H.; Forouhar, F. Primary Biliary Cirrhosis and Primary Sclerosing Cholangitis: A Review Featuring a Women’s Health Perspective. J. Clin. Transl. Hepatol. 2014, 2, 266–284. [Google Scholar] [CrossRef]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary Biliary Cholangitis: Pathogenesis and Therapeutic Opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int. J. Environ. Res. Public Health 2020, 17, E2620. [Google Scholar] [CrossRef]
- Chung, B.K.; Karlsen, T.H.; Folseraas, T. Cholangiocytes in the Pathogenesis of Primary Sclerosing Cholangitis and Development of Cholangiocarcinoma. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.G.; Lindor, K.D. Primary Sclerosing Cholangitis. Can. J. Gastroenterol. 2008, 22, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Toy, E.; Balasubramanian, S.; Selmi, C.; Li, C.-S.; Bowlus, C.L. The Prevalence, Incidence and Natural History of Primary Sclerosing Cholangitis in an Ethnically Diverse Population. BMC Gastroenterol. 2011, 11, 83. [Google Scholar] [CrossRef]
- Sayaf, K.; Zanotto, I.; Russo, F.P.; Gabbia, D.; De Martin, S. The Nuclear Receptor PXR in Chronic Liver Disease. Cells 2021, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Andersen, I.M.; Tengesdal, G.; Lie, B.A.; Boberg, K.M.; Karlsen, T.H.; Hov, J.R. Effects of Coffee Consumption, Smoking, and Hormones on Risk for Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. 2014, 12, 1019–1028. [Google Scholar] [CrossRef]
- Wronka, K.M.; Bik, E.; Milkiewicz, P. Outcome of Pregnancy in Patients with Primary Sclerosing Cholangitis. Dig. Liver Dis. 2022, 54, 509–514. [Google Scholar] [CrossRef]
- Pollheimer, M.J.; Halilbasic, E.; Fickert, P.; Trauner, M. Pathogenesis of Primary Sclerosing Cholangitis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 727–739. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, H. The Pathogenesis of Autoimmune Hepatitis. Front. Lab. Med. 2018, 2, 36–39. [Google Scholar] [CrossRef]
- Oliveira, L.C.; Porta, G.; Marin, M.L.C.; Bittencourt, P.L.; Kalil, J.; Goldberg, A.C. Autoimmune Hepatitis, HLA and Extended Haplotypes. Autoimmun. Rev. 2011, 10, 189–193. [Google Scholar] [CrossRef]
- Al-Chalabi, T.; Underhill, J.A.; Portmann, B.C.; McFarlane, I.G.; Heneghan, M.A. Impact of Gender on the Long-Term Outcome and Survival of Patients with Autoimmune Hepatitis. J. Hepatol. 2008, 48, 140–147. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular Carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the Diagnosis and Management of Intrahepatic Cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx Gene of Hepatitis B Virus Induces Liver Cancer in Transgenic Mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef]
- Sugamori, K.S.; Brenneman, D.; Sanchez, O.; Doll, M.A.; Hein, D.W.; Pierce, W.M.; Grant, D.M. Reduced 4-Aminobiphenyl-Induced Liver Tumorigenicity but Not DNA Damage in Arylamine N-Acetyltransferase Null Mice. Cancer Lett. 2012, 318, 206–213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vesselinovitch, S.D.; Mihailovich, N.; Wogan, G.N.; Lombard, L.S.; Rao, K.V. Aflatoxin B 1, a Hepatocarcinogen in the Infant Mouse. Cancer Res. 1972, 32, 2289–2291. [Google Scholar]
- Nakatani, T.; Roy, G.; Fujimoto, N.; Asahara, T.; Ito, A. Sex Hormone Dependency of Diethylnitrosamine-Induced Liver Tumors in Mice and Chemoprevention by Leuprorelin. Jpn. J. Cancer Res. 2001, 92, 249–256. [Google Scholar] [CrossRef]
- Xu, X.; Hou, Y.; Yin, X.; Bao, L.; Tang, A.; Song, L.; Li, F.; Tsang, S.; Wu, K.; Wu, H.; et al. Single-Cell Exome Sequencing Reveals Single-Nucleotide Mutation Characteristics of a Kidney Tumor. Cell 2012, 148, 886–895. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef]
- Li, Z.; White, P.; Tuteja, G.; Rubins, N.; Sackett, S.; Kaestner, K.H. Foxa1 and Foxa2 Regulate Bile Duct Development in Mice. J. Clin. Invest. 2009, 119, 1537–1545. [Google Scholar] [CrossRef]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Götz, M.E.; Tohidnezhad, M.; Brandenburg, L.-O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 Induces Interleukin-6 (IL-6) Expression via an Antioxidant Response Element within the IL-6 Promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sugamori, K.S.; Tung, A.; McPherson, J.P.; Grant, D.M. N-Hydroxylation of 4-Aminobiphenyl by CYP2E1 Produces Oxidative Stress in a Mouse Model of Chemically Induced Liver Cancer. Toxicol. Sci. 2015, 144, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Tamoxifen in Treatment of Hepatocellular Carcinoma: A Randomised Controlled Trial. CLIP Group (Cancer of the Liver Italian Programme). Lancet 1998, 352, 17–20. [CrossRef]
- Chou, W.C.; Su, I.J.; Tien, H.F.; Liang, D.C.; Wang, C.H.; Chang, Y.C.; Cheng, A.L. Clinicopathologic, Cytogenetic, and Molecular Studies of 13 Chinese Patients with Ki-1 Anaplastic Large Cell Lymphoma. Special Emphasis on the Tumor Response to 13-Cis Retinoic Acid. Cancer 1996, 78, 1805–1812. [Google Scholar] [CrossRef]
- Lasset, C.; Bonadona, V.; Chauvin, F.; Mignotte, H.; Brémond, A. Risk of Endometrial Cancer in Premenopausal Women on Tamoxifen. Lancet 1998, 352, 1476. [Google Scholar] [CrossRef]
- Dorak, M.T.; Karpuzoglu, E. Gender Differences in Cancer Susceptibility: An Inadequately Addressed Issue. Front. Genet. 2012, 3, 268. [Google Scholar] [CrossRef]
- Buettner, N.; Thimme, R. Sexual Dimorphism in Hepatitis B and C and Hepatocellular Carcinoma. Semin. Immunopathol. 2019, 41, 203–211. [Google Scholar] [CrossRef]
- Scotland, R.S.; Stables, M.J.; Madalli, S.; Watson, P.; Gilroy, D.W. Sex Differences in Resident Immune Cell Phenotype Underlie More Efficient Acute Inflammatory Responses in Female Mice. Blood 2011, 118, 5918–5927. [Google Scholar] [CrossRef]
- Lee, J.H.; Wang, C.; Kim, C.H. FoxP3+ Regulatory T Cells Restrain Splenic Extramedullary Myelopoiesis via Suppression of Hemopoietic Cytokine-Producing T Cells. J. Immunol. 2009, 183, 6377–6386. [Google Scholar] [CrossRef]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and Number of Cancer Cases and Deaths Attributable to Potentially Modifiable Risk Factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- Wang, C.; Dehghani, B.; Li, Y.; Kaler, L.J.; Proctor, T.; Vandenbark, A.A.; Offner, H. Membrane Estrogen Receptor Regulates Experimental Autoimmune Encephalomyelitis through Up-Regulation of Programmed Death 1. J. Immunol. 2009, 182, 3294–3303. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv369. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Artomov, M.; Goggins, W.; Daly, M.; Tsao, H. Gender Disparity and Mutation Burden in Metastatic Melanoma. J. Natl. Cancer Inst. 2015, 107, djv221. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-T.; Dai, Y.-S.; Chou, Y.-B.; Juan, Y.-H.; Wang, C.-C.; Chiang, B.-L. Regulatory T Cells Negatively Regulate Neovasculature of Airway Remodeling via DLL4-Notch Signaling. J. Immunol. 2009, 183, 4745–4754. [Google Scholar] [CrossRef]
- Wang, S.; Cowley, L.A.; Liu, X.-S. Sex Differences in Cancer Immunotherapy Efficacy, Biomarkers, and Therapeutic Strategy. Molecules 2019, 24, 3214. [Google Scholar] [CrossRef]
- Jepsen, P.; Ott, P.; Andersen, P.K.; Sørensen, H.T.; Vilstrup, H. Risk for Hepatocellular Carcinoma in Patients with Alcoholic Cirrhosis: A Danish Nationwide Cohort Study. Ann. Intern. Med. 2012, 156, 841–847, W295. [Google Scholar] [CrossRef]
- Balabaud, C.; Al-Rabih, W.R.; Chen, P.-J.; Evason, K.; Ferrell, L.; Hernandez-Prera, J.C.; Huang, S.-F.; Longerich, T.; Park, Y.N.; Quaglia, A.; et al. Focal Nodular Hyperplasia and Hepatocellular Adenoma around the World Viewed through the Scope of the Immunopathological Classification. Int. J. Hepatol. 2013, 2013, 268625. [Google Scholar] [CrossRef]
- Scalori, A.; Tavani, A.; Gallus, S.; La Vecchia, C.; Colombo, M. Oral Contraceptives and the Risk of Focal Nodular Hyperplasia of the Liver: A Case-Control Study. Am. J. Obstet. Gynecol. 2002, 186, 195–197. [Google Scholar] [CrossRef]


{kind=link}
| Liver Disease | Relative Incidence Male:Female Ratio | Mechanisms of Sex Differences | Refs. |
|---|---|---|---|
| Acute liver injury | |||
| DILI (according to RUCAM) | 1:2 | Sex-related different bioavailability and excretion of drugs e.g., due to sex hormone activity that affect CYP and P-gp expression Difference in genetic backgrounds | [47,49,51,52] |
| Chronic liver disease | |||
| Viral hepatitis | Conflicting results, Female generally have higher rate of symptoms but increased viral clearance | Females display more efficient innate, humoral and cell-mediated immune response (higher cytotoxic T cells, higher CD4+/CD8+ ratio and higher CD4+ T cells), as well as more TLRs | [70,71,72,84] |
| ALD | 1:2 | Estrogen-induced activation of KCs after alcohol administration in female rats increases hepatocyte inflammation and necrosis Alcohol exposure in female decreases upregulation of hepatoprotective genes, and genes involved in compensatory pathways, inflammation and oxidative stress | [98,99,100,103,159] |
| NAFLD | n.a. | Higher FA clearance and synthesis in females (increased FA transport protein expression) Higher LDL-cholesterol in men and postmenopausal women E2 seems to be protective for NAFLD | [107,110,112,114,115] |
| PBC | 1:10 | Estrogen-dependent alteration of HLA expression, cytokine release and cholangiocyte proliferation | [34,120,123] |
| PSC | 2.6:1 | Few evidences suggest a correlation between a good female reproductive health and childbearing and delay of PSC development | [128,129] |
| AIH | 1:3.5 | Female hormone-related modulation of immune system improving AIH-induced inflammation | [42,72] |
| Benign hepatic cancerous lesions | 1:5–15 (depending on types) | Estrogens improve the outcome of benign lesions | [160,161] |
| HCC | 3–4:1 | Estrogen-modulated IL6 decrease in females improves HCC progression, through regulating NRF2-antioxidant response Increased expression of TLRs involved in the innate immune response, Higher rate of CD4+ cells in females with respect to males Males better respond than female to checkpoint blockade therapy since sex hormones control PD1-PDL1 expression. | [144,145,150,151,154,155,156,157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayaf, K.; Gabbia, D.; Russo, F.P.; De Martin, S. The Role of Sex in Acute and Chronic Liver Damage. Int. J. Mol. Sci. 2022, 23, 10654. https://doi.org/10.3390/ijms231810654
Sayaf K, Gabbia D, Russo FP, De Martin S. The Role of Sex in Acute and Chronic Liver Damage. International Journal of Molecular Sciences. 2022; 23(18):10654. https://doi.org/10.3390/ijms231810654
Chicago/Turabian StyleSayaf, Katia, Daniela Gabbia, Francesco Paolo Russo, and Sara De Martin. 2022. "The Role of Sex in Acute and Chronic Liver Damage" International Journal of Molecular Sciences 23, no. 18: 10654. https://doi.org/10.3390/ijms231810654
APA StyleSayaf, K., Gabbia, D., Russo, F. P., & De Martin, S. (2022). The Role of Sex in Acute and Chronic Liver Damage. International Journal of Molecular Sciences, 23(18), 10654. https://doi.org/10.3390/ijms231810654