
Beyond NMDA Receptors: Homeostasis at the Glutamate Tripartite Synapse and Its Contributions to Cognitive Dysfunction in Schizophrenia



Abstract
1. Introduction
2. Measuring Cognition in Humans and Rodents: Tasks and Circuitry
2.1. Working Memory
2.2. Recognition Memory
2.3. Cognitive Flexibility
2.4. Response Inhibition

{kind=link}
| Cognitive Capacity | Human Measurement # | Tasks Commonly Used in Rodents | Brain Circuitry |
|---|---|---|---|
| Memory | |||
| WM | Digit and spatial span subtests, WMS-III:33 [8]; spatial span test, CANTAB [9]; DNMS, CANTAB [12]. | Alternation in a T- or a Y-maze [10]; DNMS tasks [11]. | DLPFC/mPFC, PFC connectivity with the hippocampus and MDT [14,15,16]. |
| Episodic memory (visual/visuospatial/social recognition) | RISE task [18]; VRM, ET tasks, CANTAB [9]. | NOR, OLT [19] social interaction [20]. | Prelimbic mPFC [21], Perirhinal cortex [22], hippocampal circuity [23]. |
| Cognitive Flexibility | |||
| Reversal/Attentional Set Shifting | WCST; CANTAB IED [9]. | Appetitive: Birrel–Brown task [25]. Aversive: Water T-maze with reversal and attentional set-shifting components [26]. | Reversal: OFC-ventral, dorsal striatum [27]. Set-shifting: mPFC—MDT [28], ventral striatum [29]. |
| Response inhibition | |||
| Response Inhibition | AX-CPT: 35 [30]. | SST [31]. | Fronto-basal ganglia circuity [32]. |
3. Cognitive Dysfunction and Glutamate in SZ
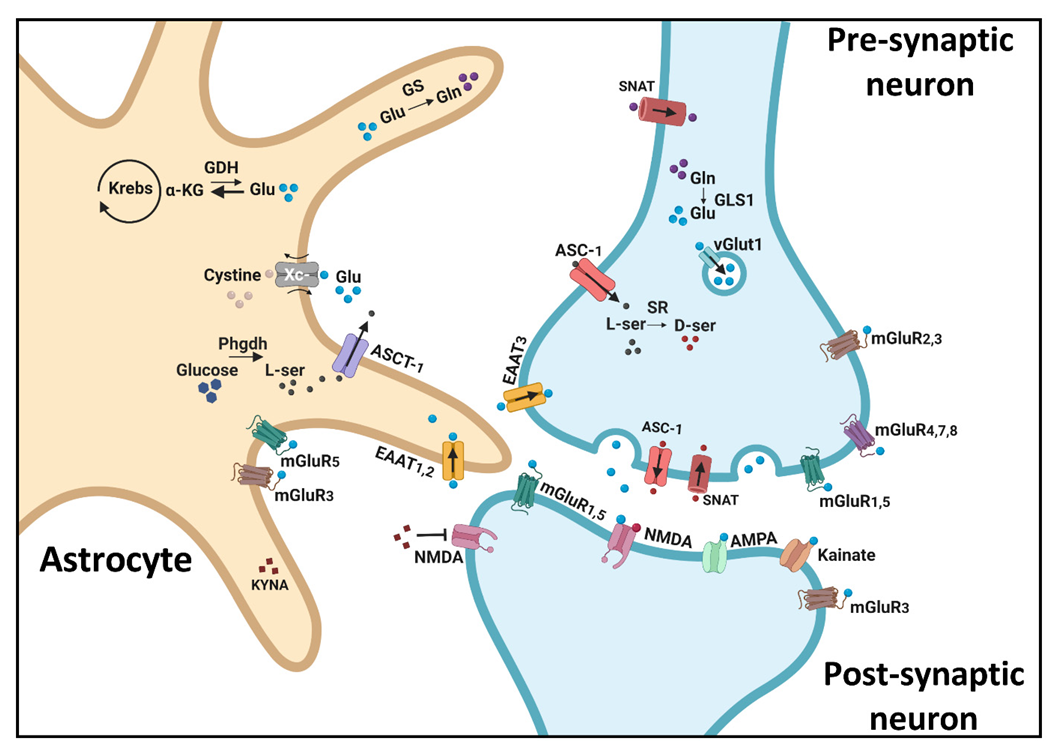
4. The Glutamate Tripartite Synapse: Involvement in Cognitive Capacities

4.1. The NMDA Receptor
4.2. AMPA and Kainate Receptors
4.3. Metabotropic Glutamate Receptors (mGluRs)
4.4. Glutamate Synthesis and Release Mechanisms
4.5. Glutamate Reuptake
4.6. Indirect Astrocytic Effects on Glutamate Dynamics
| Tripartite Synapse Component | Memory | Cognitive Flexibility | Response Inhibition | Effects on Other Components of the Tripartite Synapse | ||
|---|---|---|---|---|---|---|
| WM | Recognition Memory | Reversal Learning | Attentional Set-Shifting | |||
| NMDAR antagonists (PCP/ketamine/MK-801) | Radial arm maze, DNMS, delayed alternation task deficits [65]. | NOR deficits [66]. | Operant task deficits [66]. | EDSS deficits in ASST [66]. | 5CSRTT deficits [67,68]. | Ketamine, MK-801: increased mPFC glutamate release [80,81]. Ketamine: reduced hippocampal GLT-1 expression [87,88]. Adolescence MK-801: increased PrL Vglut1 mRNA expression [83]; increased hippocampal Vglut1 protein expression [82]. |
| Genetically induced NMDAR hypofunction | NR1-KD mice: Y-maze deficits [69]. | NR1-KD mice: radial arm maze perseverative errors [73]. | mPFC/dCA3 NR1 deletion: 5CSRTT deficits [74]. | |||
| Glycine site manipulations | SrrY269 mice: OLT deficits; D-serine reversal [71]. GlyT1-KO mice: improved NOR and OLT [77]. DAO−/− mice: improved MWM [75]. SR−/− mice: impaired memory of order of events in object recognition and odor sequence tests [70]. | Dao1G181R mice: improved reversal learning in the MWM [76]. | SrrY269 mice: reduced D-serine levels [71]. | |||
| AMPAR | Gria−/− mice: spatial WM deficits in the T and Y maze [98]. CNQX infusion to PPC: TUNL impairment [100]. Ampakine CX516: procognitive influence in a DNMS task [108]. | Ampakines (CX546/CX516): reversal of PCP-induced NOR deficits [107]. | GluR-A−/− mice: appetitive elevated plus-maze task, impairment [102]. | NBQX mPFC injection: EDSS deficits in the Birrell and Brown ASST [104]. | NASPM PFC infusion: SST impairment [105]. | GluA1flox/floxCamKCreER mice: GluA2 redistribution [99]. |
| Kainate receptors | CNQX infusion to PPC: TUNL impairment [100]. | UBP-302 perirhinal cortex infusion: NOR impairment [101]. | GluK2 KO: MWM deficits [103]. | |||
| mGluRs | mGluR7 KO: 4/8-arm maze task impairment [119]. LY354740: reversal of PCP-induced deficits in the discrete-trial delayed alternation task [120]. SAR218645 NR1neo−/− mice: reversal of Y-maze deficits [125]. VU6004909 pretreatment to MK-801, mice: reversal of MK-801-induced Y-maze deficits [130]. | LY379268: reversal of MK-801-induced NOR deficits [121]. LY379268 co-administration with clozapine: reversal of PCP-induced NOR deficits [123]. SAR218645: reversal of MK-801-induced NOR deficits in rats [125]. CDPPB: reversal of PCP-induced NOR deficits [127]. LSP4-2022: reversal of MK-801-induced NOR deficits [128]. | mGluR4 KO mice: MWM impairment [117]. | LY487379: procognitive effect in the Birrell and Brown ASST [124]. CDPPB: reversal of MK-801-induced EDSS deficits in 4 arm maze ASST [126]. | LY379268: exacerbation of PCP-induced 5CSRTT deficits [122]. | Astrocytic activation: increased mGluR2,3 and 5 expression [132]. VU6004909 pretreatment to MK-801, rats: reversal of MK-801-induced cortical hyperactivity [130]. |
| Glutamate synthesis and release mechanisms | vGlut1+/− mice: T-maze impairment [141]. dnSNARE mice: MWM deficits [152]. | MSO, mice: OLT impairment [135]. vGlut1+/− mice: NOR impairment [140]; social recognition impairment [141]. dorsal hippocampus vGlut1 depletion, mice: NOR impairment [145]. Nestin-Cre+;Glud1−/− mice: NOR, OLT and social recognition deficits [149]. TeNT astrocytic expression, mice: NOR impairment [150]. dnSNARE mice: NOR deficits [152]. | vGlut1+/− mice: MWM deficits [142]; visual discrimination task deficits [143]. Nestin-Cre+;Glud1−/− mice: IDSS deficits in the water T-maze [148]. | Nestin-Cre+;Glud1−/− mice: EDSS deficits in the water T-maze [148]. | vGlut1+/− mice: spatial extinction learning deficits [144]. | MSO, mice: decreases CA3 sEPSC, reduced functional synapses and decreased glutamatergic neurotransmission [135]. GLS1 het’ mice: resiliency to ketamine-induced PFC activation; reduced PFC and hippocampal glutamate levels; increased glutamine levels [136]. vGlut1+/− mice: reduced hippocampal glutamate levels [168].Nestin-Cre+;Glud1−/− mice: enhanced pyramidal neuron activity, hippocampal glutamate levels, astrocytic glutamate transporters and NMDA and AMPA receptor subunits expression [148]. hGFAP-dnSNARE mice: reduced glutamate release from astrocytes [151]. |
| Glutamate reuptake | Ceftriaxone, APP/PS1 mice: reversal of MWM deficits [164].Transgenic/ pharmacological EAAT2 restoration, APPSw,Ind mice: reversal of Y-maze impairments [163]. | EAAT1−/− mice: social recognition impairment [158]. DHK, mice: NOR deficits [159]. EAAT+/− mice: resilience to PCP-induced NOR deficits [161]. DHK infusion, rats: MWM impairment [162]. Transgenic/ pharmacological EAAT2 restoration, APPSw,Ind mice: reversal of NOR deficits [163]. | In vitro ceftriaxone, cultured neurons and astrocytes: increased glutamate reuptake [160]. Ceftriaxone, APP/PS1 mice: upregulated GS activity [164]. | |||
| Indirect astrocytic effects on glutamate dynamics | Gfa2-A2AR KO mice: Y-maze and radial arm maze deficits; reversal of deficits by DHK or GluA1,2 endocytosis blockade [165]. | Gfa2-A2AR KO mice: altered GLT-1 activity, increased glutamate release, NMDAR expression abnormalities and increased AMPAR internalization [165]. | ||||
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Squire, L.R.; Dede, A.J.O. Conscious and Unconscious Memory Systems. Cold Spring Harb. Perspect. Med. 2015, 7, a021667. [Google Scholar] [CrossRef] [PubMed]
- Shohamy, D.; Turk-Browne, N.B. Mechanisms for Widespread Hippocampal Involvement in Cognition. J. Exp. Psychol. Gen. 2013, 142, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Diamond, A. Executive Functions. Annu. Rev. Psychol. 2013, 64, 135–168. [Google Scholar] [CrossRef] [PubMed]
- Mcteague, L.M.; Goodkind, M.S.; Etkin, A. Transdiagnostic Impairment of Cognitive Control in Mental Illness. J. Psychiatr. Res. 2016, 83, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.S.; Keefe, R.S.E. Schizophrenia Is a Cognitive Illness: Time for a Change in Focus. JAMA Psychiatry 2013, 70, 1107–1112. [Google Scholar] [CrossRef]
- Salimi, K.; Jarskog, L.F.; Lieberman, J.A. Antipsychotic Drugs for First-Episode Schizophrenia: A Comparative Review. CNS Drugs 2009, 23, 837–855. [Google Scholar] [CrossRef] [PubMed]
- Merritt, K.; McGuire, P.; Egerton, A. Relationship between Glutamate Dysfunction and Symptoms and Cognitive Function in Psychosis. Front. Psychiatry 2013, 4, 151. [Google Scholar] [CrossRef] [PubMed]
- Giraldo-Chica, M.; Rogers, B.P.; Damon, S.M.; Landman, B.A.; Woodward, N.D. Prefrontal-Thalamic Anatomical Connectivity and Executive Cognitive Function in Schizophrenia. Biol. Psychiatry 2018, 83, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.H.; Robbins, T.W.; Leeson, V.C.; Sahakian, B.J.; Joyce, E.M.; Blackwell, A.D. Assessing Cognitive Function in Clinical Trials of Schizophrenia. Neurosci. Biobehav. Rev. 2010, 34, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J.; Rawlins, J.N.P. T-Maze Alternation in the Rodent. Nat. Protoc. 2006, 1, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dudchenko, P.A. An Overview of the Tasks Used to Test Working Memory in Rodents. Neurosci. Biobehav. Rev. 2004, 28, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Sahakian, B.J.; Owen, A.M. Computerized Assessment in Neuropsychiatry Using CANTAB: Discussion Paper. J. R. Soc. Med. 1992, 85, 399–402. [Google Scholar]
- Uylings, H.B.M.; Groenewegen, H.J.; Kolb, B. Do Rats Have a Prefrontal Cortex? Behav. Brain Res. 2003, 146, 3–17. [Google Scholar] [CrossRef]
- Bolkan, S.S.; Stujenske, J.M.; Parnaudeau, S.; Spellman, T.J.; Rauffenbart, C.; Abbas, A.I.; Harris, A.Z.; Gordon, J.A.; Kellendonk, C. Thalamic Projections Sustain Prefrontal Activity during Working Memory Maintenance. Nat. Neurosci. 2017, 20, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.I.; Sundiang, M.J.M.; Henoch, B.; Morton, M.P.; Bolkan, S.S.; Park, A.J.; Harris, A.Z.; Kellendonk, C.; Gordon, J.A. Somatostatin Interneurons Facilitate Hippocampal-Prefrontal Synchrony and Prefrontal Spatial Encoding. Neuron 2018, 100, 926–939.e3. [Google Scholar] [CrossRef] [PubMed]
- Barbey, A.K.; Koenigs, M.; Grafman, J. Dorsolateral Prefrontal Contributions to Human Working Memory. Cortex 2013, 49, 1195–1205. [Google Scholar] [CrossRef]
- Ennaceur, A. One-Trial Object Recognition in Rats and Mice: Methodological and Theoretical Issues. Behav. Brain Res. 2010, 215, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ragland, J.D.; Ranganath, C.; Barch, D.M.; Gold, J.M.; Haley, B.; MacDonald, A.W.; Silverstein, S.M.; Strauss, M.E.; Yonelinas, A.P.; Carter, C.S. Relational and Item-Specific Encoding (RISE): Task Development and Psychometric Characteristics. Schizophr. Bull. 2012, 38, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.; Biala, G. The Novel Object Recognition Memory: Neurobiology, Test Procedure, and Its Modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef] [PubMed]
- van der Kooij, M.A.; Sandi, C. Social Memories in Rodents: Methods, Mechanisms and Modulation by Stress. Neurosci. Biobehav. Rev. 2012, 36, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Farahbakhsh, Z.; Siciliano, C. De-Stressing the T Cells in Need: Protection of Transfer RNAs from Fragmentation Avoids Overstressing T Cells. Science 2021, 372, 684–685. [Google Scholar] [CrossRef]
- Winters, B.D.; Saksida, L.M.; Bussey, T.J. Object Recognition Memory: Neurobiological Mechanisms of Encoding, Consolidation and Retrieval. Neurosci. Biobehav. Rev. 2008, 32, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Barker, G.R.I.; Warburton, E.C. When Is the Hippocampus Involved in Recognition Memory? J. Neurosci. 2011, 31, 10721–10731. [Google Scholar] [CrossRef]
- Dajani, D.R.; Uddin, L.Q. Demystifying Cognitive Flexibility: Implications for Clinical and Developmental Neuroscience. Trends Neurosci. 2015, 38, 571–578. [Google Scholar] [CrossRef]
- Birrell, J.M.; Brown, V.J. Medial Frontal Cortex Mediates Perceptual Attentional Set Shifting in the Rat. J. Neurosci. 2000, 20, 4320–4324. [Google Scholar] [CrossRef] [PubMed]
- Lander, S.S.; Linder-Shacham, D.; Gaisler-Salomon, I. Differential Effects of Social Isolation in Adolescent and Adult Mice on Behavior and Cortical Gene Expression. Behav. Brain Res. 2017, 316, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.; Brigman, J.L.; Radke, A.K.; Rudebeck, P.H.; Holmes, A. The Neural Basis of Reversal Learning: An Updated Perspective. Neuroscience 2017, 345, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Parnaudeau, S.; Bolkan, S.S.; Kellendonk, C. The Mediodorsal Thalamus: An Essential Partner of the Prefrontal Cortex for Cognition. Biol. Psychiatry 2018, 83, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Van Schouwenburg, M.R.; Den Ouden, H.E.M.; Cools, R. The Human Basal Ganglia Modulate Frontal-Posterior Connectivity during Attention Shifting. J. Neurosci. 2010, 30, 9910–9918. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Minzenberg, M.J.; Ursu, S.; Walters, R.; Carter Wendelken, B.; Daniel Ragland, J.; Carter, C.S. Association of Dorsolateral Prefrontal Cortex Dysfunction with Disrupted Coordinated Brain Activity in Schizophrenia: Relationship with Impaired Cognition, Behavioral Disorganization, and Global Function. Am. J. Psychiatry 2008, 165, 1006–1014. [Google Scholar] [CrossRef]
- Eagle, D.M.; Robbins, T.W. Inhibitory Control in Rats Performing a Stop-Signal Reaction-Time Task: Effects of Lesions of the Medial Striatum and d-Amphetamine. Behav. Neurosci. 2003, 117, 1302–1317. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Leventhal, D.K.; Mallet, N.; Chen, F.; Berke, J.D. Canceling Actions Involves a Race between Basal Ganglia Pathways. Nat. Neurosci. 2013, 16, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.C.; Murray, A. Assessing Cognitive Impairment in Patients with Schizophrenia. J. Clin. Psychiatry 2016, 77, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M.; Day, F.; Tsagaraki, H.; Valli, I.; McLean, M.A.; Lythgoe, D.J.; O’Gorman, R.L.; Barker, G.J.; McGuire, P.K. Glutamate Dysfunction in People with Prodromal Symptoms of Psychosis: Relationship to Gray Matter Volume. Biol. Psychiatry 2009, 66, 533–539. [Google Scholar] [CrossRef] [PubMed]
- MacCabe, J.H. Population-Based Cohort Studies on Premorbid Cognitive Function in Schizophrenia. Epidemiol. Rev. 2008, 30, 77–83. [Google Scholar] [CrossRef]
- Ohi, K.; Sumiyoshi, C.; Fujino, H.; Yasuda, Y.; Yamamori, H.; Fujimoto, M.; Shiino, T.; Sumiyoshi, T.; Hashimoto, R. Genetic Overlap between General Cognitive Function and Schizophrenia: A Review of Cognitive GWASs. Int. J. Mol. Sci. 2018, 19, 3822. [Google Scholar] [CrossRef]
- Ohi, K.; Hashimoto, R.; Ikeda, M.; Yamamori, H.; Yasuda, Y.; Fujimoto, M.; Umeda-Yano, S.; Fukunaga, M.; Fujino, H.; Watanabe, Y.; et al. Glutamate Networks Implicate Cognitive Impairments in Schizophrenia: Genome-Wide Association Studies of 52 Cognitive Phenotypes. Schizophr. Bull. 2015, 41, 909–918. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite Synapses: Glia, the Unacknowledged Partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate–Glutamine Cycle. In Advances in Neurobiology; Springer: Cham, Switzerland, 2016; Volume 13, pp. 1–7. [Google Scholar]
- Fremeau, R.T.; Voglmaier, S.; Seal, R.P.; Edwards, R.H. VGLUTs Define Subsets of Excitatory Neurons and Suggest Novel Roles for Glutamate. Trends Neurosci. 2004, 27, 98–103. [Google Scholar] [CrossRef] [PubMed]
- D’Antoni, S.; Berretta, A.; Bonaccorso, C.M.; Bruno, V.; Aronica, E.; Nicoletti, F.; Catania, M.V. Metabotropic Glutamate Receptors in Glial Cells. Neurochem. Res. 2008, 33, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The Glutamate/GABA-Glutamine Cycle: Aspects of Transport, Neurotransmitter Homeostasis and Ammonia Transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- Baker, D.A.; Xi, Z.X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The Origin and Neuronal Function of in Vivo Nonsynaptic Glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Notarangelo, F.M.; Wu, H.Q.; Bruno, J.P.; Schwarcz, R. Astrocytes as Pharmacological Targets in the Treatment of Schizophrenia: Focus on Kynurenic Acid. In Handbook of Behavioral Neuroscience; Elsevier B.V.: Amsterdam, The Netherlands, 2016; Volume 23, pp. 423–443. [Google Scholar]
- Wonodi, I.; Schwarcz, R. Cortical Kynurenine Pathway Metabolism: A Novel Target for Cognitive Enhancement in Schizophrenia. Schizophr. Bull. 2010, 36, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Neyton, J. NMDA Receptor Subunits: Function and Pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Neame, S.; Safory, H.; Radzishevsky, I.; Touitou, A.; Marchesani, F.; Marchetti, M.; Kellner, S.; Berlin, S.; Foltyn, V.N.; Engelender, S.; et al. The NMDA Receptor Activation by D-Serine and Glycine Is Controlled by an Astrocytic Phgdh-Dependent Serine Shuttle. Proc. Natl. Acad. Sci. USA 2019, 116, 20736–20742. [Google Scholar] [CrossRef]
- Wolosker, H.; Balu, D.T. D-Serine as the Gatekeeper of NMDA Receptor Activity: Implications for the Pharmacologic Management of Anxiety Disorders. Transl. Psychiatry 2020, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, E.; Zubedat, S.; Radzishevsky, I.; Valenta, A.C.; Rechnitz, O.; Sason, H.; Sajrawi, C.; Bodner, O.; Konno, K.; Esaki, K.; et al. ASCT1 (Slc1a4) Transporter Is a Physiologic Regulator of Brain D-Serine and Neurodevelopment. Proc. Natl. Acad. Sci. USA 2018, 115, 9628–9633. [Google Scholar] [CrossRef]
- Wolosker, H.; Balu, D.T.; Coyle, J.T. The Rise and Fall of the D-Serine-Mediated Gliotransmission Hypothesis. Trends Neurosci. 2016, 39, 712–721. [Google Scholar] [CrossRef]
- Wolosker, H.; Blackshaw, S.; Snyder, S.H. Serine Racemase: A Glial Enzyme Synthesizing D-Serine to Regulate Glutamate-N-Methyl-D-Aspartate Neurotransmission. Proc. Natl. Acad. Sci. USA 1999, 96, 13409–13414. [Google Scholar] [CrossRef] [PubMed]
- Kartvelishvily, E.; Shleper, M.; Balan, L.; Dumin, E.; Wolosker, H. Neuron-Derived D-Serine Release Provides a Novel Means to Activate N-Methyl-D-Aspartate Receptors. J. Biol. Chem. 2006, 281, 14151–14162. [Google Scholar] [CrossRef]
- Rosenberg, D.; Artoul, S.; Segal, A.C.; Kolodney, G.; Radzishevsky, I.; Dikopoltsev, E.; Foltyn, V.N.; Inoue, R.; Mori, H.; Billard, J.M.; et al. Neuronal D-Serine and Glycine Release via the Asc-1 Transporter Regulates NMDA Receptor-Dependent Synaptic Activity. J. Neurosci. 2013, 33, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; Folorunso, O.O.; Barragan, E.V.; Berciu, C.; Harvey, T.L.; Coyle, J.T.; Balu, D.T.; Gray, J.A. Postsynaptic Serine Racemase Regulates NMDA Receptor Function. J. Neurosci. 2020, 40, 9564–9575. [Google Scholar] [CrossRef] [PubMed]
- Bodner, O.; Radzishevsky, I.; Foltyn, V.N.; Touitou, A.; Valenta, A.C.; Rangel, I.F.; Panizzutti, R.; Kennedy, R.T.; Billard, J.M.; Wolosker, H. D-Serine Signaling and NMDAR-Mediated Synaptic Plasticity Are Regulated by System A-Type of Glutamine/ D-Serine Dual Transporters. J. Neurosci. 2020, 40, 6489–6502. [Google Scholar] [CrossRef]
- Verrall, L.; Walker, M.; Rawlings, N.; Benzel, I.; Kew, J.N.C.; Harrison, P.J.; Burnet, P.W.J. D-Amino Acid Oxidase and Serine Racemase in Human Brain: Normal Distribution and Altered Expression in Schizophrenia. Eur. J. Neurosci. 2007, 26, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Bliss, T.V.P. Memories of NMDA Receptors and LTP. Trends Neurosci. 1995, 18, 54–56. [Google Scholar] [CrossRef]
- Morris, R.G.M. NMDA Receptors and Memory Encoding. Neuropharmacology 2013, 74, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Javitt, D.C. N-Methyl-D-Aspartate (NMDA) Receptor Dysfunction or Dysregulation: The Final Common Pathway on the Road to Schizophrenia? Brain Res. Bull. 2010, 83, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-Induced Exacerbation of Psychotic Symptoms and Cognitive Impairment in Neuroleptic-Free Schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S. Subanesthetic Effects of the Noncompetitive NMDA Antagonist, Ketamine, in Humans: Psychotomimetic, Perceptual, Cognitive, and Neuroendocrine Responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Neale, B.M.; Daly, M.J. Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Ripke, S.; Walters, J.T.; O’Donovan, M.C. Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Weickert, C.S.; Fung, S.J.; Catts, V.S.; Schofield, P.R.; Allen, K.M.; Moore, L.T.; Newell, K.A.; Pellen, D.; Huang, X.F.; Catts, S.V.; et al. Molecular Evidence of N-Methyl-D-Aspartate Receptor Hypofunction in Schizophrenia. Mol. Psychiatry 2013, 18, 1185–1192. [Google Scholar] [CrossRef]
- Barak, S.; Weiner, I. Putative Cognitive Enhancers in Preclinical Models Related to Schizophrenia: The Search for an Elusive Target. Pharmacol. Biochem. Behav. 2011, 99, 164–189. [Google Scholar] [CrossRef] [PubMed]
- Neill, J.C.; Barnes, S.; Cook, S.; Grayson, B.; Idris, N.F.; McLean, S.L.; Snigdha, S.; Rajagopal, L.; Harte, M.K. Animal Models of Cognitive Dysfunction and Negative Symptoms of Schizophrenia: Focus on NMDA Receptor Antagonism. Pharmacol. Ther. 2010, 128, 419–432. [Google Scholar] [CrossRef]
- Paine, T.A.; Tomasiewicz, H.C.; Zhang, K.; Carlezon, W.A. Sensitivity of the Five-Choice Serial Reaction Time Task to the Effects of Various Psychotropic Drugs in Sprague-Dawley Rats. Biol. Psychiatry 2007, 62, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Enderlin, M.; Haman, M.; Fletcher, P.J. The 5-HT2A Receptor Antagonist M100,907 Attenuates Motor and “impulsive-Type” Behaviours Produced by NMDA Receptor Antagonism. Psychopharmacology 2003, 170, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, A.J. NR1 Knockdown Mice as a Representative Model of the Glutamate Hypothesis of Schizophrenia. Prog. Brain Res. 2009, 179, 51–58. [Google Scholar] [CrossRef]
- Devito, L.M.; Balu, D.T.; Kanter, B.R.; Lykken, C.; Basu, A.C.; Coyle, J.T.; Eichenbaum, H. Serine Racemase Deletion Disrupts Memory for Order and Alters Cortical Dendritic Morphology. Genes Brain Behav. 2011, 10, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Labrie, V.; Fukumura, R.; Rastogi, A.; Fick, L.J.; Wang, W.; Boutros, P.C.; Kennedy, J.L.; Semeralul, M.O.; Lee, F.H.; Baker, G.B.; et al. Serine Racemase Is Associated with Schizophrenia Susceptibility in Humans and in a Mouse Model. Hum. Mol. Genet. 2009, 18, 3227–3243. [Google Scholar] [CrossRef]
- Coyle, J.T.; Balu, D.; Wolosker, H. D-Serine, the Shape-Shifting NMDA Receptor Co-Agonist. Neurochem. Res. 2020, 45, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Dzirasa, K.; Ramsey, A.J.; Takahashi, D.Y.; Stapleton, J.; Potes, J.M.; Williams, J.K.; Gainetdinov, R.R.; Sameshima, K.; Caron, M.G.; Nicolelis, M.A.L. Hyperdopaminergia and NMDA Receptor Hypofunction Disrupt Neural Phase Signaling. J. Neurosci. 2009, 29, 8215–8224. [Google Scholar] [CrossRef]
- Finlay, J.M.; Dunham, G.A.; Isherwood, A.M.; Newton, C.J.; Nguyen, T.V.; Reppar, P.C.; Snitkovski, I.; Paschall, S.A.; Greene, R.W. Effects of Prefrontal Cortex and Hippocampal NMDA NR1-Subunit Deletion on Complex Cognitive and Social Behaviors. Brain Res. 2015, 1600, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Watanabe, M.; Yamaguchi, S.; Konno, R.; Hori, Y. Spatial Learning and Long-Term Potentiation of Mutant Mice Lacking D-Amino-Acid Oxidase. Neurosci. Res. 2005, 53, 34–38. [Google Scholar] [CrossRef]
- Labrie, V.; Duffy, S.; Wang, W.; Barger, S.W.; Baker, G.B.; Roder, J.C. Genetic Inactivation of D-Amino Acid Oxidase Enhances Extinction and Reversal Learning in Mice. Learn. Mem. 2009, 16, 28–37. [Google Scholar] [CrossRef]
- Singer, P.; Boison, D.; Möhler, H.; Feldon, J.; Yee, B.K. Enhanced Recognition Memory Following Glycine Transporter 1 Deletion in Forebrain Neurons. Behav. Neurosci. 2007, 121, 815–825. [Google Scholar] [CrossRef]
- Goff, D.C. Drug Development in Schizophrenia: Are Glutamatergic Targets Still Worth Aiming At? Curr. Opin. Psychiatry 2015, 28, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T. The NMDA Receptor and Schizophrenia. From Pathophysiology to Treatment. In Advances in Pharmacology; Academic Press Inc.: Cambridge, MA, USA, 2016; Volume 76, pp. 351–382. ISBN 9780128097458. [Google Scholar]
- Roenker, N.L.; Gudelsky, G.A.; Ahlbrand, R.; Horn, P.S.; Richtand, N.M. Evidence for Involvement of Nitric Oxide and GABAB Receptors in MK-801-Stimulated Release of Glutamate in Rat Prefrontal Cortex. Neuropharmacology 2012, 63, 575–581. [Google Scholar] [CrossRef][Green Version]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of Glutamatergic Neurotransmission by Ketamine: A Novel Step in the Pathway from NMDA Receptor Blockade to Dopaminergic and Cognitive Disruptions Associated with the Prefrontal Cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Ma, Y.N.; Sun, Y.X.; Wang, T.; Wang, H.; Zhang, Y.; Su, Y.A.; Li, J.T.; Si, T.M. Subchronic MK-801 Treatment during Adolescence Induces Long-Term, Not Permanent, Excitatory-Inhibitory Imbalance in the Rat Hippocampus. Eur. J. Pharmacol. 2020, 867, 172807. [Google Scholar] [CrossRef] [PubMed]
- Bauminger, H.; Zaidan, H.; Akirav, I.; Gaisler-Salomon, I. Anandamide Hydrolysis Inhibition Reverses the Long-Term Behavioral and Gene Expression Alterations Induced by MK-801 in Male Rats: Differential CB1 and CB2 Receptor-Mediated Effects. Schizophr. Bull. 2022, 48, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Onaolapo, A.Y.; Ayeni, O.J.; Ogundeji, M.O.; Ajao, A.; Owolabi, A.R.; Onaolapo, O.J. Subchronic Ketamine Alters Behaviour, Metabolic Indices and Brain Morphology in Adolescent Rats: Involvement of Oxidative Stress, Glutamate Toxicity and Caspase-3-Mediated Apoptosis. J. Chem. Neuroanat. 2019, 96, 22–33. [Google Scholar] [CrossRef]
- Homayoun, H.; Moghaddam, B. NMDA Receptor Hypofunction Produces Opposite Effects on Prefrontal Cortex Interneurons and Pyramidal Neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef] [PubMed]
- Bygrave, A.M.; Masiulis, S.; Nicholson, E.; Berkemann, M.; Barkus, C.; Sprengel, R.; Harrison, P.J.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Knockout of NMDA-Receptors from Parvalbumin Interneurons Sensitizes to Schizophrenia-Related Deficits Induced by MK-801. Transl. Psychiatry 2016, 6, e778. [Google Scholar] [CrossRef]
- Featherstone, R.E.; Liang, Y.; Saunders, J.A.; Tatard-Leitman, V.M.; Ehrlichman, R.S.; Siegel, S.J. Subchronic Ketamine Treatment Leads to Permanent Changes in EEG, Cognition and the Astrocytic Glutamate Transporter EAAT2 in Mice. Neurobiol. Dis. 2012, 47, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Dodman, K.; Featherstone, R.E.; Bang, J.; Liang, Y.; Siegel, S.J. Ceftriaxone Reverses Ketamine-Induced Lasting EEG and Astrocyte Alterations in Juvenile Mice. Drug Alcohol Depend. 2015, 156, 14–20. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The Role of Glutamate Transporters in the Pathophysiology of Neuropsychiatric Disorders. npj Schizophr. 2017, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Keinanen, K.; Wisden, W.; Sommer, B.; Werner, P.; Herb, A.; Verdoorn, T.A.; Sakmann, B.; Seeburgt, P.H. A Family of AMPA-Selective Glutamate Receptors. Science 1990, 249, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.; Mulle, C. Kainate Receptors. Cell Tissue Res. 2006, 326, 457–482. [Google Scholar] [CrossRef]
- Blanco-Suarez, E.; Liu, T.F.; Kopelevich, A.; Allen, N.J. Astrocyte-Secreted Chordin-like 1 Drives Synapse Maturation and Limits Plasticity by Increasing Synaptic GluA2 AMPA Receptors. Neuron 2018, 100, 1116–1132.e13. [Google Scholar] [CrossRef]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte Glypicans 4 and 6 Promote Formation of Excitatory Synapses via GluA1 AMPA Receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, B.; Du, T.; Wang, F.; Hertz, L. Does Conventional Anti-Bipolar and Antidepressant Drug Therapy Reduce NMDA-Mediated Neuronal Excitation by Downregulating Astrocytic GluK2 Function? Pharmacol. Biochem. Behav. 2012, 100, 712–725. [Google Scholar] [CrossRef]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.H.; Holmans, P.A.; Al, E. Biological Insights from 108 Schizophrenia-Associated Genetic Loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Davis, K.L.; Haroutunian, V. Abnormal Kainate Receptor Expression in Prefrontal Cortex in Schizophrenia. Neuropsychopharmacology 2001, 24, 545–552. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Jahans-Price, T.; Wolff, A.R.; Sprengel, R.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Hippocampal–Prefrontal Coherence Mediates Working Memory and Selective Attention at Distinct Frequency Bands and Provides a Causal Link between Schizophrenia and Its Risk Gene GRIA1. Transl. Psychiatry 2019, 9, 142. [Google Scholar] [CrossRef]
- Inta, D.; Vogt, M.A.; Elkin, H.; Weber, T.; Lima-Ojeda, J.M.; Schneider, M.; Luoni, A.; Riva, M.A.; Gertz, K.; Hellmann-Regen, J.; et al. Phenotype of Mice with Inducible Ablation of GluA1 AMPA Receptors during Late Adolescence: Relevance for Mental Disorders. Hippocampus 2014, 24, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.A.; Roebuck, A.J.; Greba, Q.; Howland, J.G. Performance of the Trial-Unique, Delayed Non-Matching-to-Location (TUNL) Task Depends on AMPA/Kainate, but Not NMDA, Ionotropic Glutamate Receptors in the Rat Posterior Parietal Cortex. Neurobiol. Learn. Mem. 2019, 159, 16–23. [Google Scholar] [CrossRef]
- Barker, G.R.I.; Warburton, E.C.; Koder, T.; Dolman, N.P.; More, J.C.A.; Aggleton, J.P.; Bashir, Z.I.; Auberson, Y.P.; Jane, D.E.; Brown, M.W. The Different Effects on Recognition Memory of Perirhinal Kainate and NMDA Glutamate Receptor Antagonism: Implications for Underlying Plasticity Mechanisms. J. Neurosci. 2006, 26, 3561–3566. [Google Scholar] [CrossRef]
- Bannerman, D.M.; Deacon, R.M.J.; Seeburg, P.H.; Rawlins, J.N.P. GluR-A-Deficient Mice Display Normal Acquisition of a Hippocampus-Dependent Spatial Reference Memory Task but Are Impaired during Spatial Reversal. Behav. Neurosci. 2003, 117, 866–870. [Google Scholar] [CrossRef]
- Micheau, J.; Vimeney, A.; Normand, E.; Mulle, C.; Riedel, G. Impaired Hippocampus-Dependent Spatial Flexibility and Sociability Represent Autism-like Phenotypes in GluK2 Mice. Hippocampus 2014, 24, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Jett, J.D.; Bulin, S.E.; Hatherall, L.C.; McCartney, C.M.; Morilak, D.A. Deficits in Cognitive Flexibility Induced by Chronic Unpredictable Stress Are Associated with Impaired Glutamate Neurotransmission in the Rat Medial Prefrontal Cortex. Neuroscience 2017, 346, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Zhang, Y.Q.; Zhang, X.H. Prefrontal AMPA Receptors Are Involved in the Effect of Methylphenidate on Response Inhibition in Rats. Acta Pharmacol. Sin. 2018, 39, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, M.; DeMartinis, N.; Huguenel, B.; Gaudreault, F.; Bednar, M.M.; Shaffer, C.L.; Gupta, S.; Cahill, J.; Sherif, M.A.; Mancuso, J.; et al. Attenuation of Ketamine-Induced Impairment in Verbal Learning and Memory in Healthy Volunteers by the AMPA Receptor Potentiator PF-04958242. Mol. Psychiatry 2017, 22, 1633–1640. [Google Scholar] [CrossRef]
- Damgaard, T.; Larsen, D.B.; Hansen, S.L.; Grayson, B.; Neill, J.C.; Plath, N. Positive Modulation of Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid (AMPA) Receptors Reverses Sub-Chronic PCP-Induced Deficits in the Novel Object Recognition Task in Rats. Behav. Brain Res. 2010, 207, 144–150. [Google Scholar] [CrossRef]
- Hampson, R.E.; Rogers, G.; Lynch, G.; Deadwyler, S.A. Facilitative Effects of the Ampakine CX516 on Short-Term Memory in Rats: Enhancement of Delayed-Nonmatch-to-Sample Performance. J. Neurosci. 1998, 18, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. AmpakineTM CX546 Bolsters Energetic Response of Astrocytes: A Novel Target for Cognitive-Enhancing Drugs Acting as α-Amino-3-Hydroxy-5-Methyl- 4-Isoxazolepropionic Acid (AMPA) Receptor Modulators. J. Neurochem. 2005, 92, 668–677. [Google Scholar] [CrossRef]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The Tripartite Glutamatergic Synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Bruno, V.; Catania, M.V.; Battaglia, G.; Copani, A.; Barbagallo, G.; Ceñ, V.; Sanchez-Prieto, J.; Spano, P.F.; Pizzi, M. Group-I Metabotropic Glutamate Receptors: Hypotheses to Explain Their Dual Role in Neurotoxicity and Neuroprotection. Neuropharmacology 1999, 38, 1477–1484. [Google Scholar] [CrossRef]
- Egan, M.F.; Straub, R.E.; Goldberg, T.E.; Yakub, I.; Callicott, J.H.; Hariri, A.R.; Mattay, V.S.; Bertolino, A.; Hyde, T.M.; Shannon-Weickert, C.; et al. Variation in GRM3 Affects Cognition, Prefrontal Glutamate, and Risk for Schizophrenia. Proc. Natl. Acad. Sci. USA 2004, 101, 12604–12609. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Abi-Saab, W.; Perry, E.; D’Souza, D.C.; Liu, N.; Gueorguieva, R.; McDougall, L.; Hunsberger, T.; Belger, A.; Levine, L.; et al. Preliminary Evidence of Attenuation of the Disruptive Effects of the NMDA Glutamate Receptor Antagonist, Ketamine, on Working Memory by Pretreatment with the Group II Metabotropic Glutamate Receptor Agonist, LY354740, in Healthy Human Subjects. Psychopharmacology 2005, 179, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.T.; Zhang, L.; Martenyi, F.; Lowe, S.L.; Jackson, K.A.; Andreev, B.V.; Avedisova, A.S.; Bardenstein, L.M.; Gurovich, I.Y.; Morozova, M.A.; et al. Activation of MGlu2/3 Receptors as a New Approach to Treat Schizophrenia: A Randomized Phase 2 Clinical Trial. Nat. Med. 2007, 13, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, V.L.; Millen, B.A.; Andersen, S.; Kinon, B.J.; LaGrandeur, L.; Lindenmayer, J.P.; Gomez, J.C. Pomaglumetad Methionil: No Significant Difference as an Adjunctive Treatment for Patients with Prominent Negative Symptoms of Schizophrenia Compared to Placebo. Schizophr. Res. 2013, 150, 434–441. [Google Scholar] [CrossRef]
- Gerlai, R.; Roder, J.C.; Hampson, D.R. Altered Spatial Learning and Memory in Mice Lacking the MGluR4 Subtype of Metabotropic Glutamate Receptor. Behav. Neurosci. 1998, 112, 525–532. [Google Scholar] [CrossRef]
- Klar, R.; Walker, A.G.; Ghose, D.; Grueter, B.A.; Engers, D.W.; Hopkins, C.R.; Lindsley, C.W.; Xiang, Z.; Conn, P.J.; Niswender, C.M. Activation of Metabotropic Glutamate Receptor 7 Is Required for Induction of Long-Term Potentiation at SCCA1 Synapses in the Hippocampus. J. Neurosci. 2015, 35, 7600–7615. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C.; Schmid, S.; Pilz, P.K.D.; Sansig, G.; Van Der Putten, H.; Plappert, C.F. Lack of the Metabotropic Glutamate Receptor Subtype 7 Selectively Impairs Short-Term Working Memory but Not Long-Term Memory. Behav. Brain Res. 2004, 154, 473–481. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, W.B. Reversal of Phencyclidine Effects by a Group II Metabotropic Glutamate Receptor Agonist in Rats. Science 1998, 281, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Wierońska, J.M.; Sławińska, A.; Stachowicz, K.; Łasoń-Tyburkiewicz, M.; Gruca, P.; Papp, M.; Pilc, A. The Reversal of Cognitive, but Not Negative or Positive Symptoms of Schizophrenia, by the MGlu2/3 Receptor Agonist, LY379268, Is 5-HT1A Dependent. Behav. Brain Res. 2013, 256, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Amitai, N.; Markou, A. Effects of Metabotropic Glutamate Receptor 2/3 Agonism and Antagonism on Schizophrenia-like Cognitive Deficits Induced by Phencyclidine in Rats. Eur. J. Pharmacol. 2010, 639, 67–80. [Google Scholar] [CrossRef]
- Horiguchi, M.; Huang, M.; Meltzer, H.Y. Interaction of MGlu 2/3 Agonism with Clozapine and Lurasidone to Restore Novel Object Recognition in Subchronic Phencyclidine-Treated Rats. Psychopharmacology 2011, 217, 13–24. [Google Scholar] [CrossRef]
- Nikiforuk, A.; Popik, P.; Drescher, K.U.; Van Gaalen, M.; Relo, A.L.; Mezler, M.; Marek, G.; Schoemaker, H.; Gross, G.; Bespalov, A. Effects of a Positive Allosteric Modulator of Group II Metabotropic Glutamate Receptors, LY487379, on Cognitive Flexibility and Impulsive-like Responding in Rats. J. Pharmacol. Exp. Ther. 2010, 335, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Pichat, P.; Boulay, D.; Naimoli, V.; Potestio, L.; Featherstone, R.; Sahni, S.; Defex, H.; Desvignes, C.; Slowinski, F.; et al. The MGluR2 Positive Allosteric Modulator, SAR218645, Improves Memory and Attention Deficits in Translational Models of Cognitive Symptoms Associated with Schizophrenia. Sci. Rep. 2016, 6, 35320. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.R.; Moghaddam, B. Activation of Type 5 Metabotropic Glutamate Receptors Attenuates Deficits in Cognitive Flexibility Induced by NMDA Receptor Blockade. Eur. J. Pharmacol. 2010, 639, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Horio, M.; Fujita, Y.; Hashimoto, K. Therapeutic Effects of Metabotropic Glutamate Receptor 5 Positive Allosteric Modulator CDPPB on Phencyclidine-Induced Cognitive Deficits in Mice. Fundam. Clin. Pharmacol. 2013, 27, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.; Acher, F.; Marciniak, M.; Łasoń-Tyburkiewicz, M.; Gruca, P.; Papp, M.; Pilc, A.; Wierońska, J.M. Involvement of GABAB Receptor Signaling in Antipsychotic-like Action of the Novel Orthosteric Agonist of the MGlu4 Receptor, LSP4-2022. Curr. Neuropharmacol. 2016, 14, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Cartmell, J.; Schoepp, D.D. Regulation of Neurotransmitter Release by Metabotropic Glutamate Receptors. J. Neurochem. 2000, 75, 889–907. [Google Scholar] [CrossRef] [PubMed]
- Maksymetz, J.; Byun, N.E.; Luessen, D.J.; Li, B.; Barry, R.L.; Gore, J.C.; Niswender, C.M.; Lindsley, C.W.; Joffe, M.E.; Conn, P.J. MGlu1 Potentiation Enhances Prelimbic Somatostatin Interneuron Activity to Rescue Schizophrenia-like Physiological and Cognitive Deficits. Cell Rep. 2021, 37, 109950. [Google Scholar] [CrossRef]
- Fiacco, T.A.; McCarthy, K.D. Intracellular Astrocyte Calcium Waves in Situ Increase the Frequency of Spontaneous AMPA Receptor Currents in CA1 Pyramidal Neurons. J. Neurosci. 2004, 24, 722–732. [Google Scholar] [CrossRef]
- Ferraguti, F.; Corti, C.; Valerio, E.; Mion, S.; Xuereb, J. Activated Astrocytes in Areas of Kainate-Induced Neuronal Injury Upregulate the Expression of the Metabotropic Glutamate Receptors 2/3 and 5. Exp. Brain Res. 2001, 137, 1–11. [Google Scholar] [CrossRef]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial Dysfunction in Schizophrenia: Evidence for Compromised Brain Metabolism and Oxidative Stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef]
- Hu, W.; Macdonald, M.L.; Elswick, D.E.; Sweet, R.A. The Glutamate Hypothesis of Schizophrenia: Evidence from Human Brain Tissue Studies. Ann. N. Y. Acad. Sci. 2015, 1338, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Kim, S.; Jung, D.H.; Baek, J.H.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Lee, D.K.; et al. Insufficient Glutamine Synthetase Activity during Synaptogenesis Causes Spatial Memory Impairment in Adult Mice. Sci. Rep. 2019, 9, 252. [Google Scholar] [CrossRef] [PubMed]
- Gaisler-Salomon, I.; Miller, G.M.; Chuhma, N.; Lee, S.; Zhang, H.; Ghoddoussi, F.; Lewandowski, N.; Fairhurst, S.; Wang, Y.; Conjard-Duplany, A.; et al. Glutaminase-Deficient Mice Display Hippocampal Hypoactivity, Insensitivity to pro-Psychotic Drugs and Potentiated Latent Inhibition: Relevance to Schizophrenia. Neuropsychopharmacology 2009, 34, 2305–2322. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, S.L.; Harrison, P.J. Decreased Expression of Vesicular Glutamate Transporter 1 and Complexin II MRNAs in Schizophrenia: Further Evidence for a Synaptic Pathology Affecting Glutamate Neurons. Schizophr. Res. 2005, 73, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.J.; Sivagnanasundaram, S.; Weickert, C.S. Lack of Change in Markers of Presynaptic Terminal Abundance alongside Subtle Reductions in Markers of Presynaptic Terminal Plasticity in Prefrontal Cortex of Schizophrenia Patients. Biol. Psychiatry 2011, 69, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Oni-Orisan, A.; Kristiansen, L.V.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Altered Vesicular Glutamate Transporter Expression in the Anterior Cingulate Cortex in Schizophrenia. Biol. Psychiatry 2008, 63, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Tordera, R.M.; Totterdell, S.; Wojcik, S.M.; Brose, N.; Elizalde, N.; Lasheras, B.; Del Rio, J. Enhanced Anxiety, Depressive-like Behaviour and Impaired Recognition Memory in Mice with Reduced Expression of the Vesicular Glutamate Transporter 1 (VGLUT1). Eur. J. Neurosci. 2007, 25, 281–290. [Google Scholar] [CrossRef]
- Inta, D.; Vogt, M.A.; Perreau-Lenz, S.; Schneider, M.; Pfeiffer, N.; Wojcik, S.M.; Spanagel, R.; Gass, P. Sensorimotor Gating, Working and Social Memory Deficits in Mice with Reduced Expression of the Vesicular Glutamate Transporter VGLUT1. Behav. Brain Res. 2012, 228, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Balschun, D.; Moechars, D.; Callaerts-Vegh, Z.; Vermaercke, B.; Van Acker, N.; Andries, L.; D’Hooge, R. Vesicular Glutamate Transporter VGLUT1 Has a Role in Hippocampal Long-Term Potentiation and Spatial Reversal Learning. Cereb. Cortex 2010, 20, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Granseth, B.; Andersson, F.K.; Lindström, S.H. The Initial Stage of Reversal Learning Is Impaired in Mice Hemizygous for the Vesicular Glutamate Transporter (VGluT1). Genes Brain Behav. 2015, 14, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Callaerts-Vegh, Z.; Moechars, D.; Van Acker, N.; Daneels, G.; Goris, I.; Leo, S.; Naert, A.; Meert, T.; Balschun, D.; D’Hooge, R. Haploinsufficiency of VGluT1 but Not VGluT2 Impairs Extinction of Spatial Preference and Response Suppression. Behav. Brain Res. 2013, 245, 13–21. [Google Scholar] [CrossRef] [PubMed]
- King, M.V.; Kurian, N.; Qin, S.; Papadopoulou, N.; Westerink, B.H.; Cremers, T.I.; Epping-Jordan, M.P.; Le Poul, E.; Ray, D.E.; Fone, K.C.; et al. Lentiviral Delivery of a Vesicular Glutamate Transporter 1 (VGLUT1)-Targeting Short Hairpin RNA Vector into the Mouse Hippocampus Impairs Cognition. Neuropsychopharmacology 2014, 39, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Waagepetersen, H.S.; Georgopoulos, P.; Sonnewald, U.; Plaitakis, A.; Schousboe, A. Differential Expression of Glutamate Dehydrogenase in Cultured Neurons and Astrocytes from Mouse Cerebellum and Cerebral Cortex. J. Neurosci. Res. 2001, 66, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Burbaeva, G.S.; Boksha, I.S.; Turishcheva, M.S.; Vorobyeva, E.A.; Savushkina, O.K.; Tereshkina, E.B. Glutamine Synthetase and Glutamate Dehydrogenase in the Prefrontal Cortex of Patients with Schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 675–680. [Google Scholar] [CrossRef]
- Lander, S.S.; Khan, U.; Lewandowski, N.; Chakraborty, D.; Provenzano, F.A.; Mingote, S.; Chornyy, S.; Frigerio, F.; Maechler, P.; Kaphzan, H.; et al. Glutamate Dehydrogenase-Deficient Mice Display Schizophrenia-like Behavioral Abnormalities and CA1-Specific Hippocampal Dysfunction. Schizophr. Bull. 2019, 45, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Lander, S.S.; Chornyy, S.; Safory, H.; Gross, A.; Wolosker, H.; Gaisler-Salomon, I. Glutamate Dehydrogenase Deficiency Disrupts Glutamate Homeostasis in Hippocampus and Prefrontal Cortex and Impairs Recognition Memory. Genes Brain Behav. 2020, 19, e12636. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Ghetti, A.; Pinto-Duarte, A.; Wang, X.; Dziewczapolski, G.; Galimi, F.; Huitron-Resendiz, S.; Piña-Crespo, J.C.; Roberts, A.J.; Verma, I.M.; et al. Astrocytes Contribute to Gamma Oscillations and Recognition Memory. Proc. Natl. Acad. Sci. USA 2014, 111, E3343–E3352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pangršič, T.; Kreft, M.; Kržan, M.; Li, N.; Sul, J.Y.; Halassa, M.; Van Bockstaele, E.; Zorec, R.; Haydon, P.G. Fusion-Related Release of Glutamate from Astrocytes. J. Biol. Chem. 2004, 279, 12724–12733. [Google Scholar] [CrossRef]
- Sardinha, V.M.; Guerra-Gomes, S.; Caetano, I.; Tavares, G.; Martins, M.; Reis, J.S.; Correia, J.S.; Teixeira-Castro, A.; Pinto, L.; Sousa, N.; et al. Astrocytic Signaling Supports Hippocampal–Prefrontal Theta Synchronization and Cognitive Function. Glia 2017, 65, 1944–1960. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Melone, M.; Vallejo-Illarramendi, A.; Conti, F. Increased Expression of the Astrocytic Glutamate Transporter GLT-1 in the Prefrontal Cortex of Schizophrenics. Glia 2005, 49, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Augood, S.J.; Arai, H.; Mckenna, P.J.; Emson, P.C. Expression of the Human Excitatory Amino Acid Transporter 2 and Metabotropic Glutamate Receptors 3 and 5 in the Prefrontal Cortex from Normal Individuals and Patients with Schizophrenia. Mol. Brain Res. 1998, 56, 207–217. [Google Scholar] [CrossRef]
- Shan, D.; Lucas, E.K.; Drummond, J.B.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal Expression of Glutamate Transporters in Temporal Lobe Areas in Elderly Patients with Schizophrenia. Schizophr. Res. 2013, 144, 1–8. [Google Scholar] [CrossRef]
- Spangaro, M.; Bosia, M.; Zanoletti, A.; Bechi, M.; Mariachiara, B.; Pirovano, A.; Lorenzi, C.; Bramanti, P.; Smeraldi, E.; Cavallaro, R. Exploring Effects of EAAT Polymorphisms on Cognitive Functions in Schizophrenia. Pharmacogenomics 2014, 15, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.M.; Tanaka, K.; Saksida, L.M.; Bussey, T.J.; Heilig, M.; Holmes, A. Assessment of Glutamate Transporter GLAST (EAAT1)-Deficient Mice for Phenotypes Relevant to the Negative and Executive/Cognitive Symptoms of Schizophrenia. Neuropsychopharmacology 2009, 34, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.W.; Yu, X.D.; Cen, L.; Xiao, Z.Y. Glutamate Transporter GLT1 Inhibitor Dihydrokainic Acid Impairs Novel Object Recognition Memory Performance in Mice. Physiol. Behav. 2019, 199, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Beller, J.A.; Gurkoff, G.G.; Berman, R.F.; Lyeth, B.G. Pharmacological Enhancement of Glutamate Transport Reduces Excitotoxicity in Vitro. Restor. Neurol. Neurosci. 2011, 29, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Hida, H.; Mori, K.; Yoshimi, A.; Kitagaki, S.; Yamada, K.; Hiraoka, Y.; Aida, T.; Tanaka, K.; Ozaki, N.; et al. Functional Roles of the Glial Glutamate Transporter (GLAST) in Emotional and Cognitive Abnormalities of Mice after Repeated Phencyclidine Administration. Eur. Neuropsychopharmacol. 2019, 29, 914–924. [Google Scholar] [CrossRef]
- Bechtholt-Gompf, A.J.; Walther, H.V.; Adams, M.A.; Carlezon, W.A.; Ngür, D.; Cohen, B.M. Blockade of Astrocytic Glutamate Uptake in Rats Induces Signs of Anhedonia and Impaired Spatial Memory. Neuropsychopharmacology 2010, 35, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kong, Q.; Lin, Y.; Stouffer, N.; Schulte, D.A.; Lai, L.; Liu, Q.; Chang, L.C.; Dominguez, S.; Xing, X.; et al. Restored Glial Glutamate Transporter EAAT2 Function as a Potential Therapeutic Approach for Alzheimer’s Disease. J. Exp. Med. 2015, 212, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xian, X.; Li, L.; Yao, X.; Hu, Y.; Zhang, M.; Li, W. Ceftriaxone Improves Cognitive Function and Upregulates GLT-1-Related Glutamate-Glutamine Cycle in APP/PS1 Mice. J. Alzheimer’s Dis. 2018, 66, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Santos-Rodrigues, A.D.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; Agostinho, P. Adenosine A 2A Receptors Modulate Glutamate Uptake in Cultured Astrocytes and Gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Shen, H.Y.; Augusto, E.; Wang, Y.; Wei, C.J.; Wang, Y.T.; Agostinho, P.; Boison, D.; Cunha, R.A.; Chen, J.F. Deletion of Adenosine A2A Receptors from Astrocytes Disrupts Glutamate Homeostasis Leading to Psychomotor and Cognitive Impairment: Relevance to Schizophrenia. Biol. Psychiatry 2015, 78, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Freche, D.; Dallérac, G.; Ghézali, G.; Escartin, C.; Ezan, P.; Cohen-Salmon, M.; Benchenane, K.; Abudara, V.; Dufour, A.; et al. Connexin 30 Sets Synaptic Strength by Controlling Astroglial Synapse Invasion. Nat. Neurosci. 2014, 17, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S.M.; Rhee, J.S.; Herzog, E.; Sigler, A.; Jahn, R.; Takamori, S.; Brose, N.; Rosenmund, C. An Essential Role for Vesicular Glutamate Transporter 1 (VGLUT1) in Postnatal Development and Control of Quantal Size. Proc. Natl. Acad. Sci. USA 2004, 101, 7158–7163. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauminger, H.; Gaisler-Salomon, I. Beyond NMDA Receptors: Homeostasis at the Glutamate Tripartite Synapse and Its Contributions to Cognitive Dysfunction in Schizophrenia. Int. J. Mol. Sci. 2022, 23, 8617. https://doi.org/10.3390/ijms23158617
Bauminger H, Gaisler-Salomon I. Beyond NMDA Receptors: Homeostasis at the Glutamate Tripartite Synapse and Its Contributions to Cognitive Dysfunction in Schizophrenia. International Journal of Molecular Sciences. 2022; 23(15):8617. https://doi.org/10.3390/ijms23158617
Chicago/Turabian StyleBauminger, Hagar, and Inna Gaisler-Salomon. 2022. "Beyond NMDA Receptors: Homeostasis at the Glutamate Tripartite Synapse and Its Contributions to Cognitive Dysfunction in Schizophrenia" International Journal of Molecular Sciences 23, no. 15: 8617. https://doi.org/10.3390/ijms23158617
APA StyleBauminger, H., & Gaisler-Salomon, I. (2022). Beyond NMDA Receptors: Homeostasis at the Glutamate Tripartite Synapse and Its Contributions to Cognitive Dysfunction in Schizophrenia. International Journal of Molecular Sciences, 23(15), 8617. https://doi.org/10.3390/ijms23158617