
MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia

 , ,
, ,  , , , , , , , , , , and add
Show full author list
, , , , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
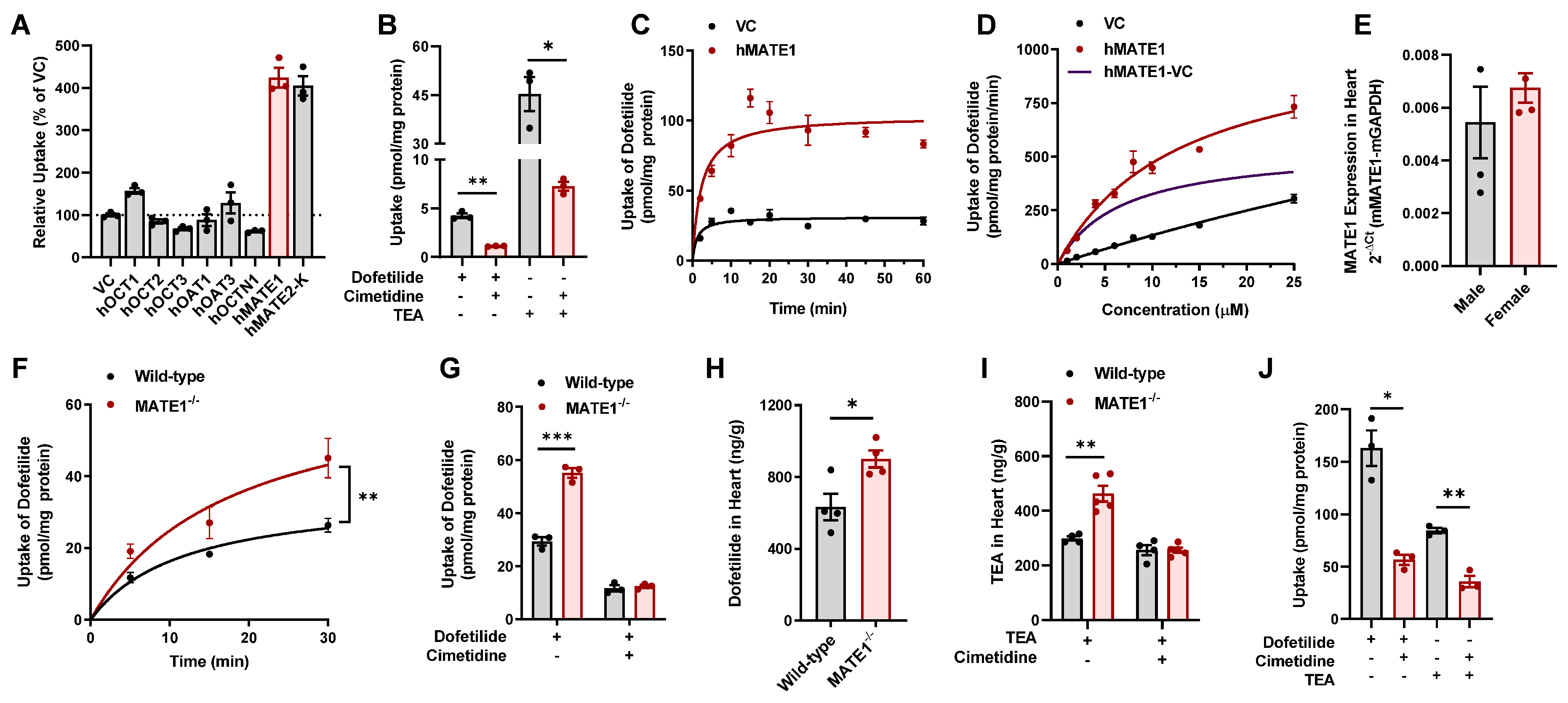
2.1. Identification of MATE1 as a High-Affinity Carrier of Dofetilide
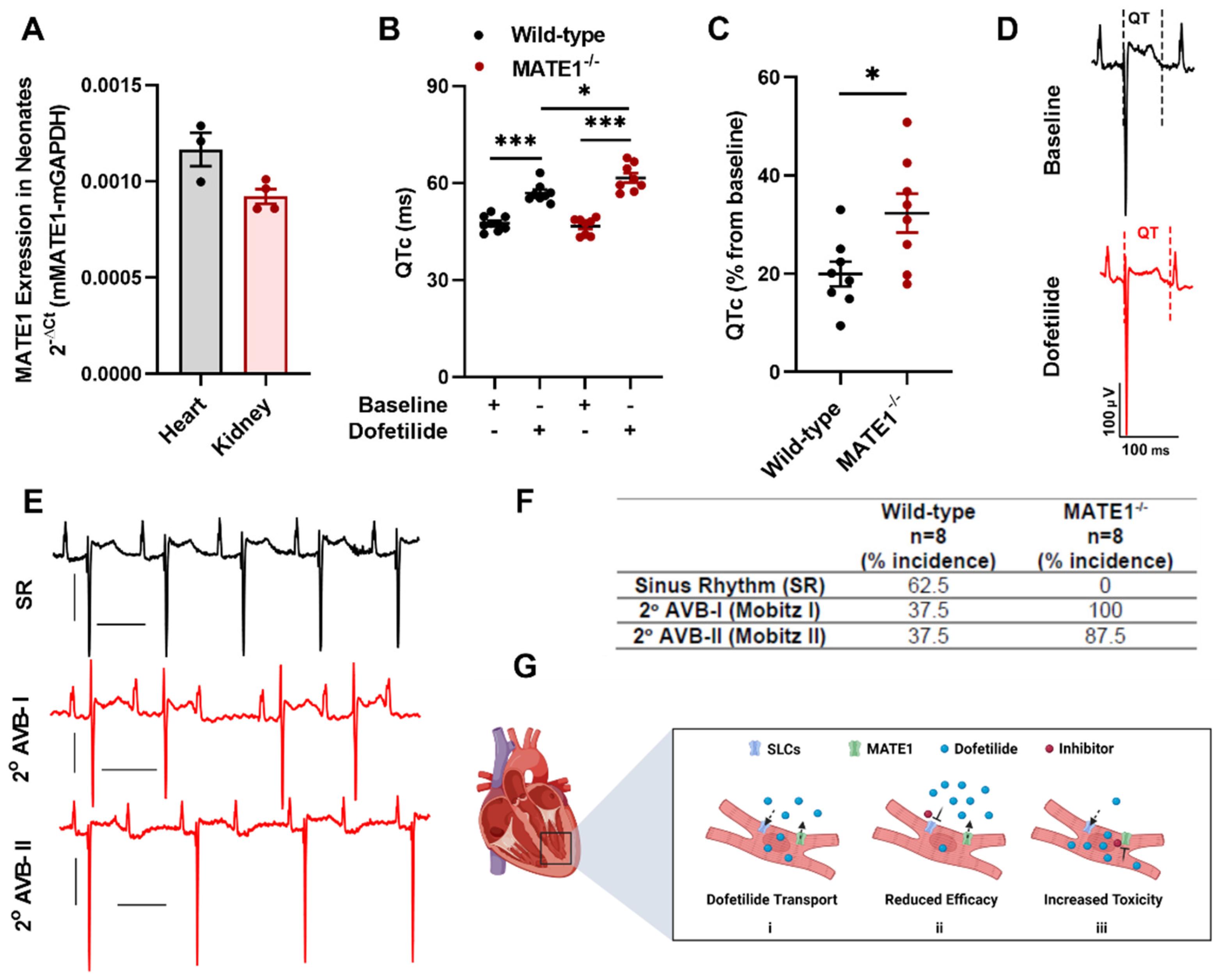
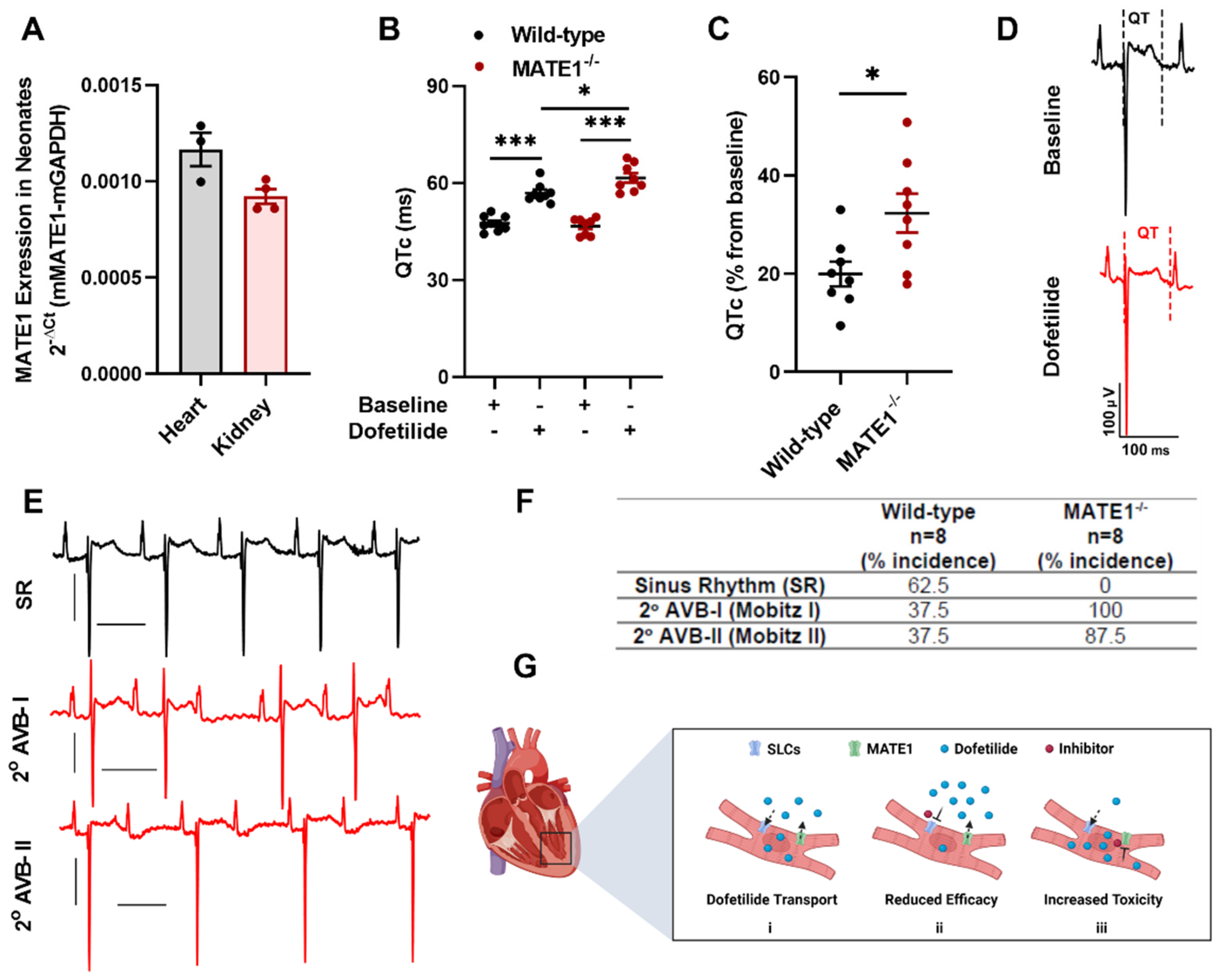
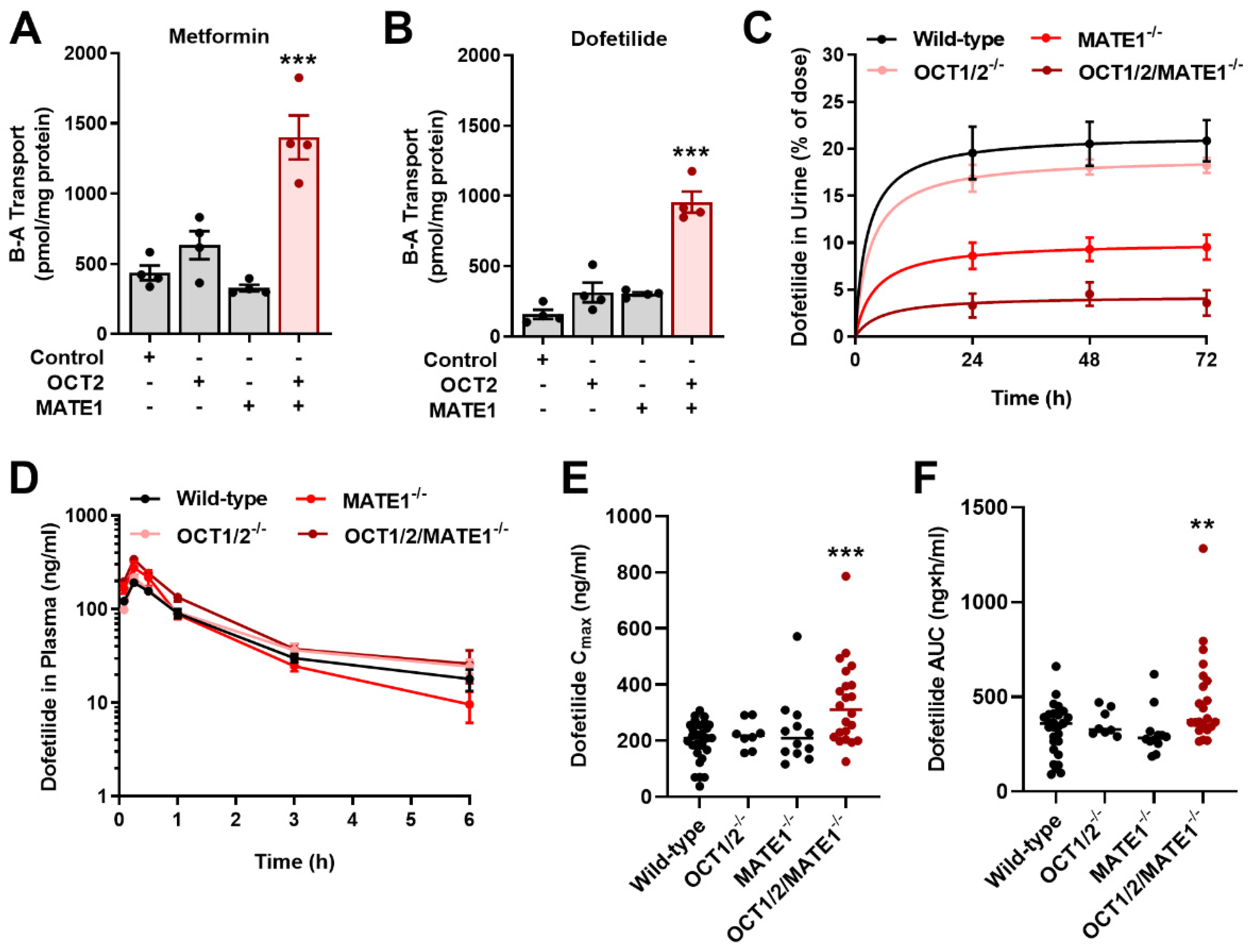
2.2. MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia
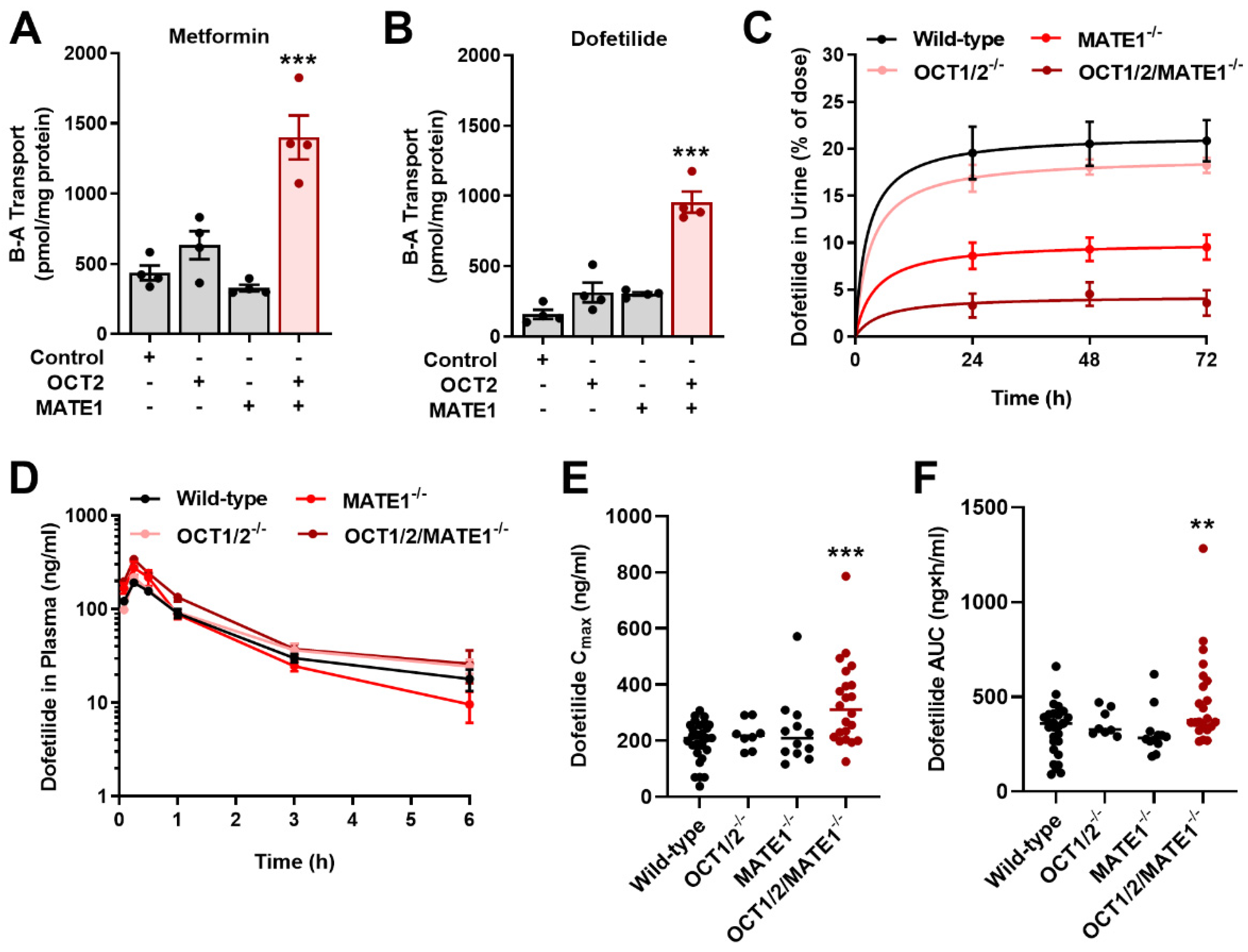
2.3. Inhibition of MATE1 Attenuates Renal Elimination of Dofetilide
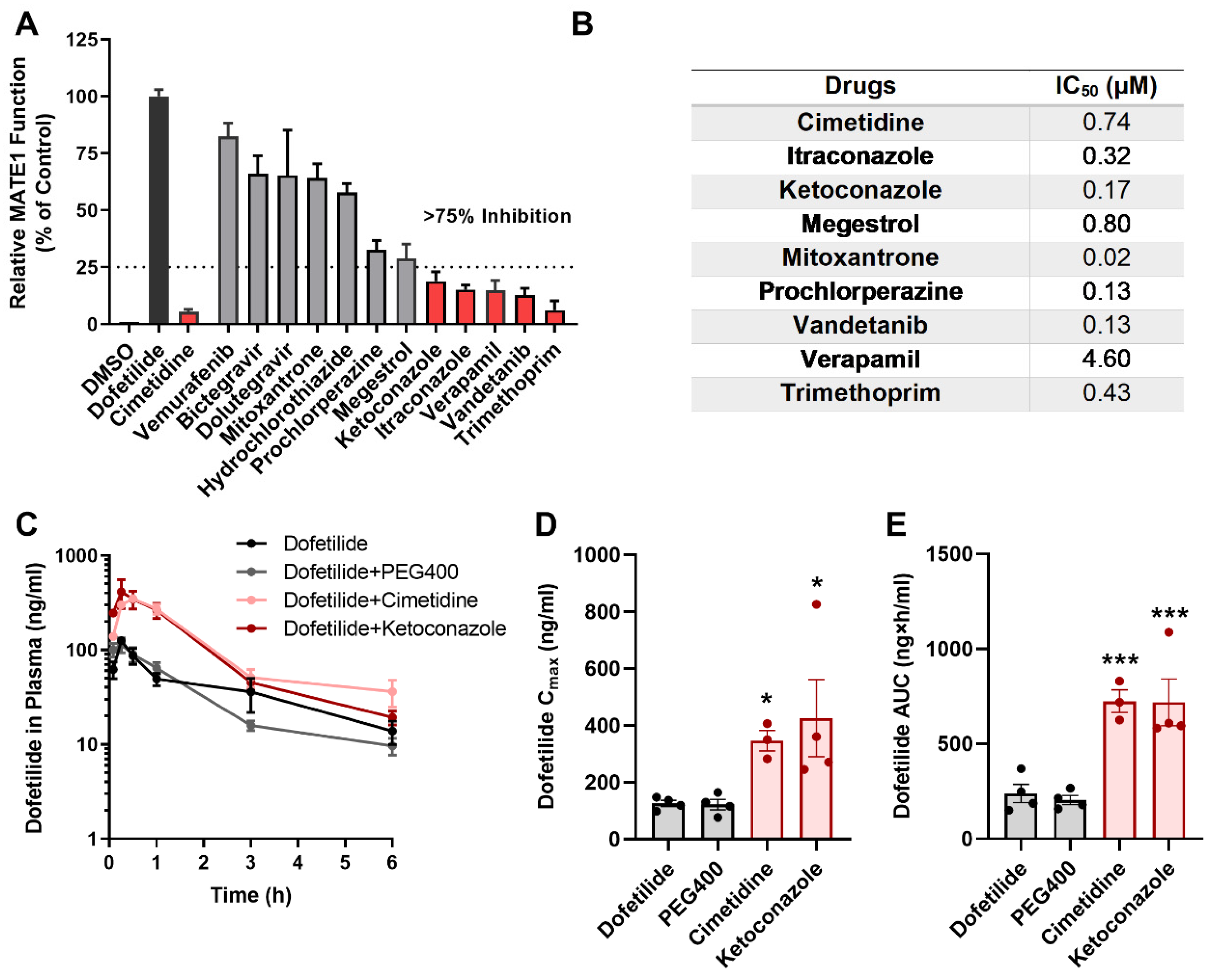
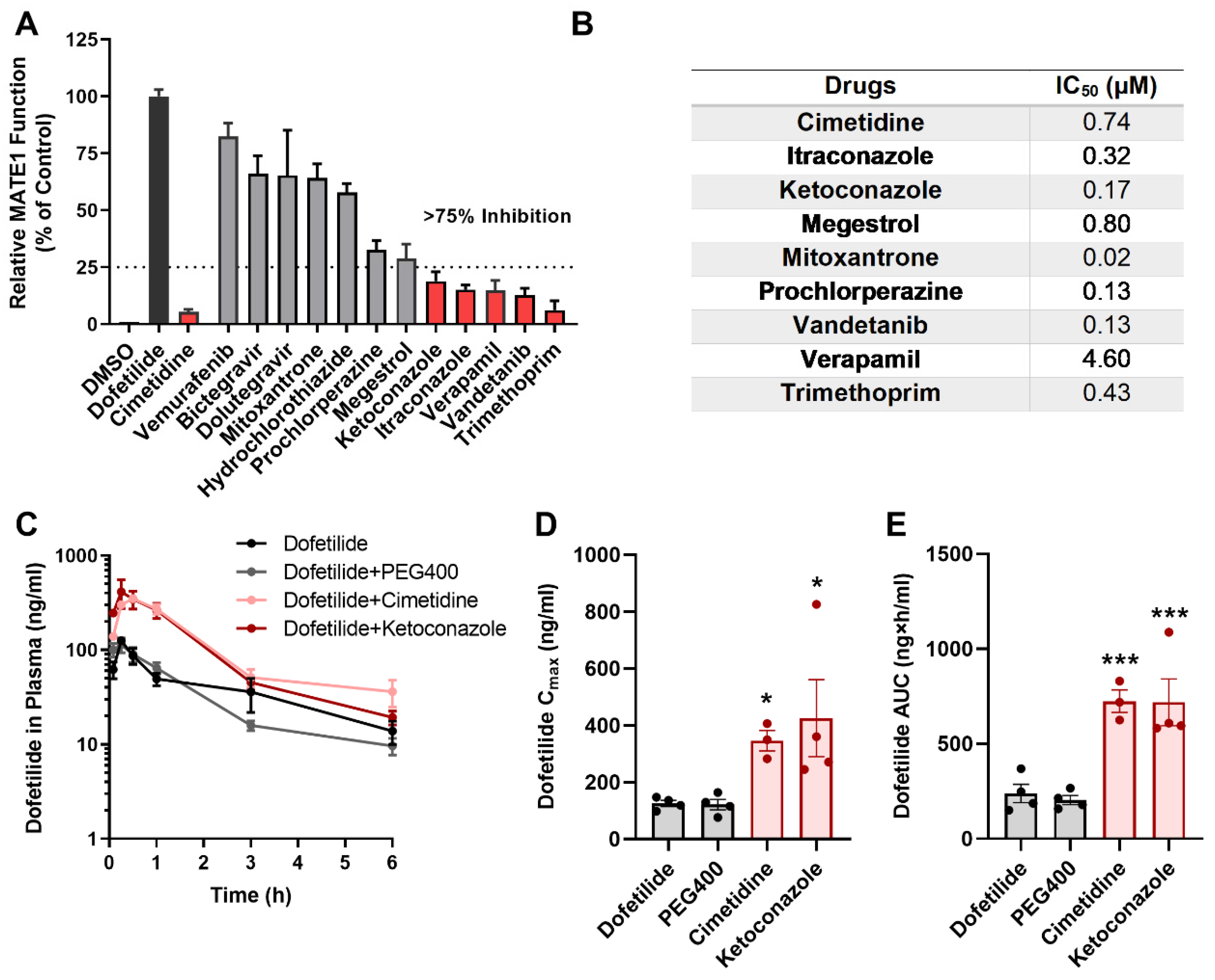
2.4. Drugs Contraindicated for Use with Dofetilide Inhibit MATE1 Function
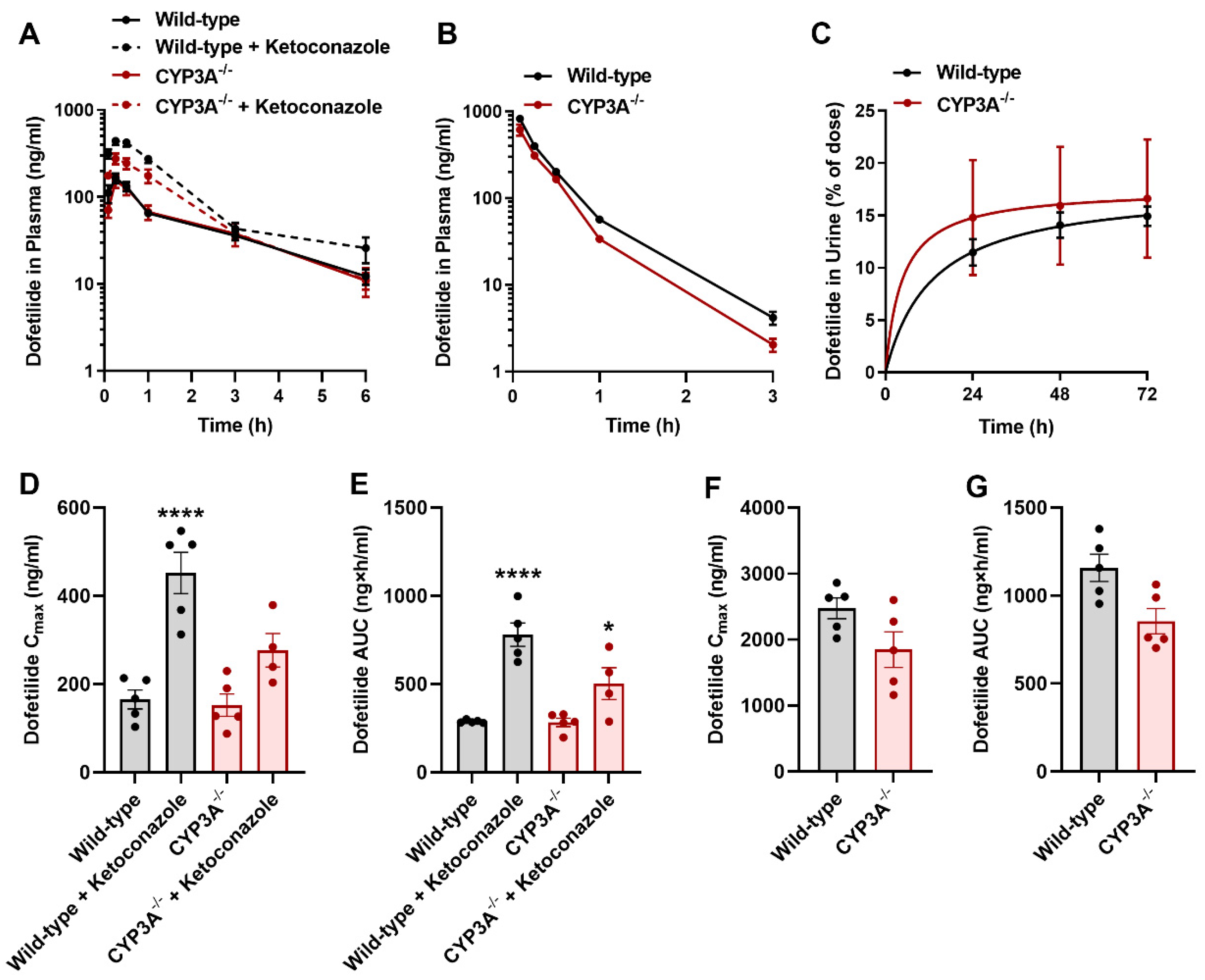
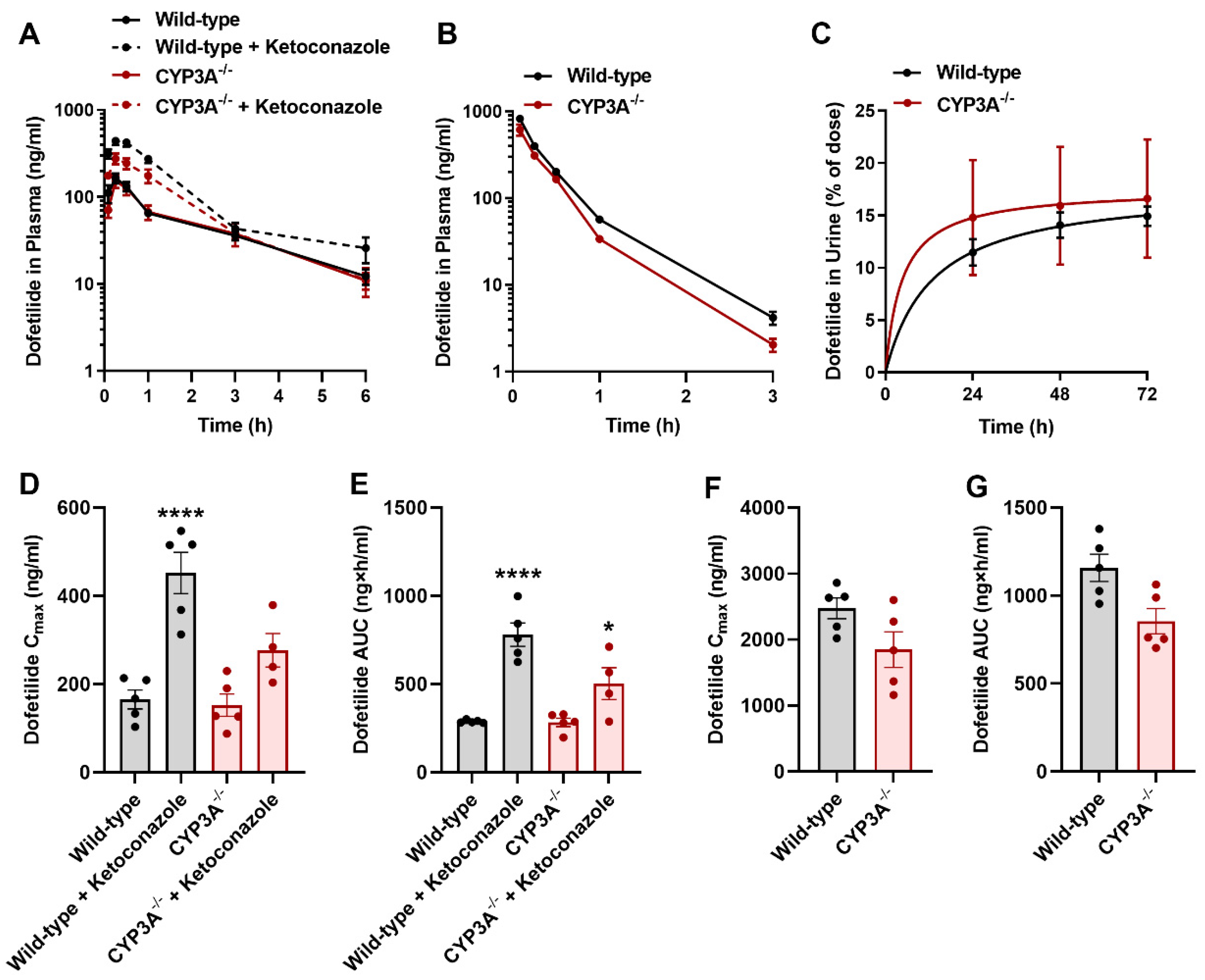
2.5. Influence of CYP3A on the Disposition of Dofetilide
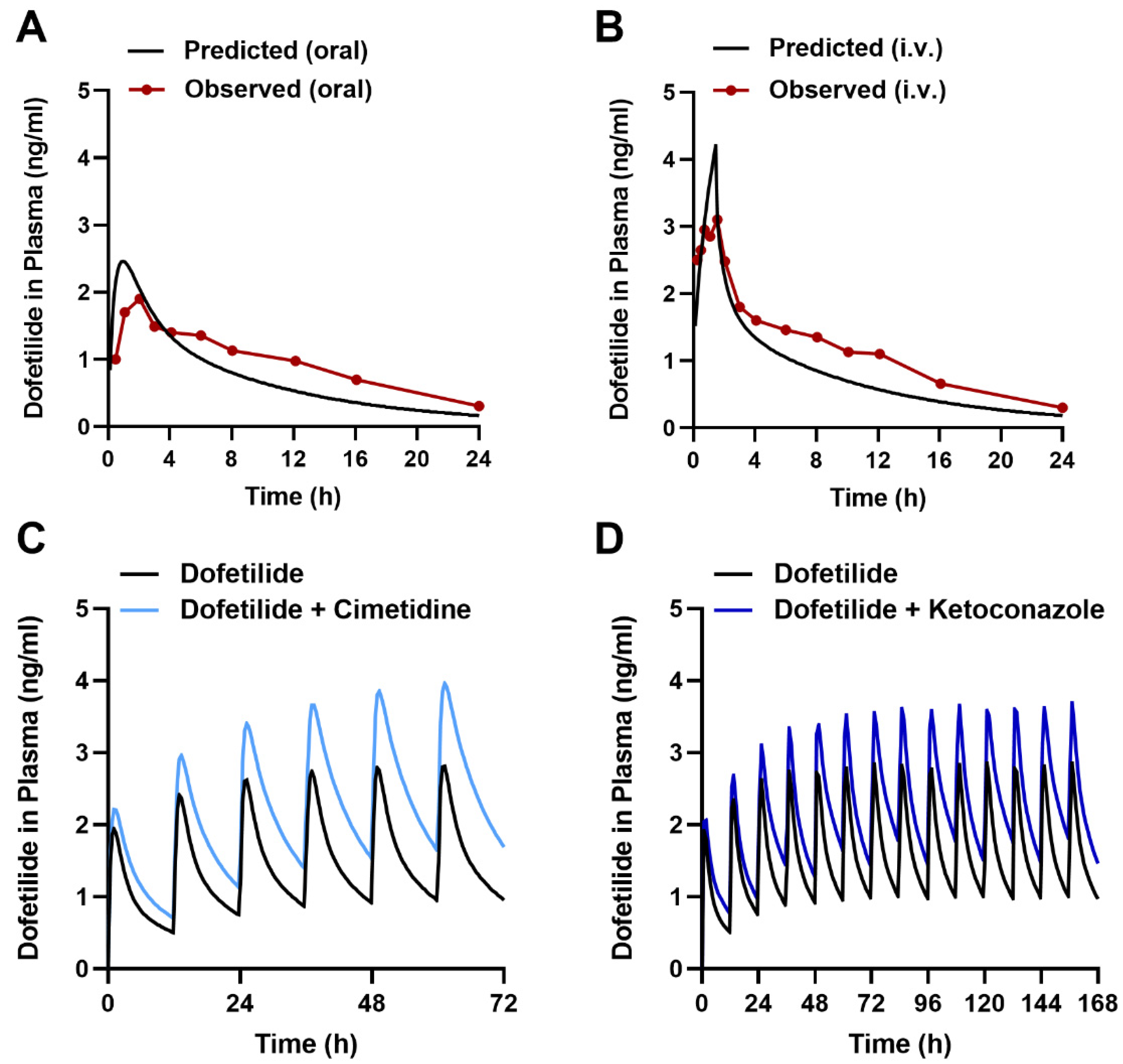
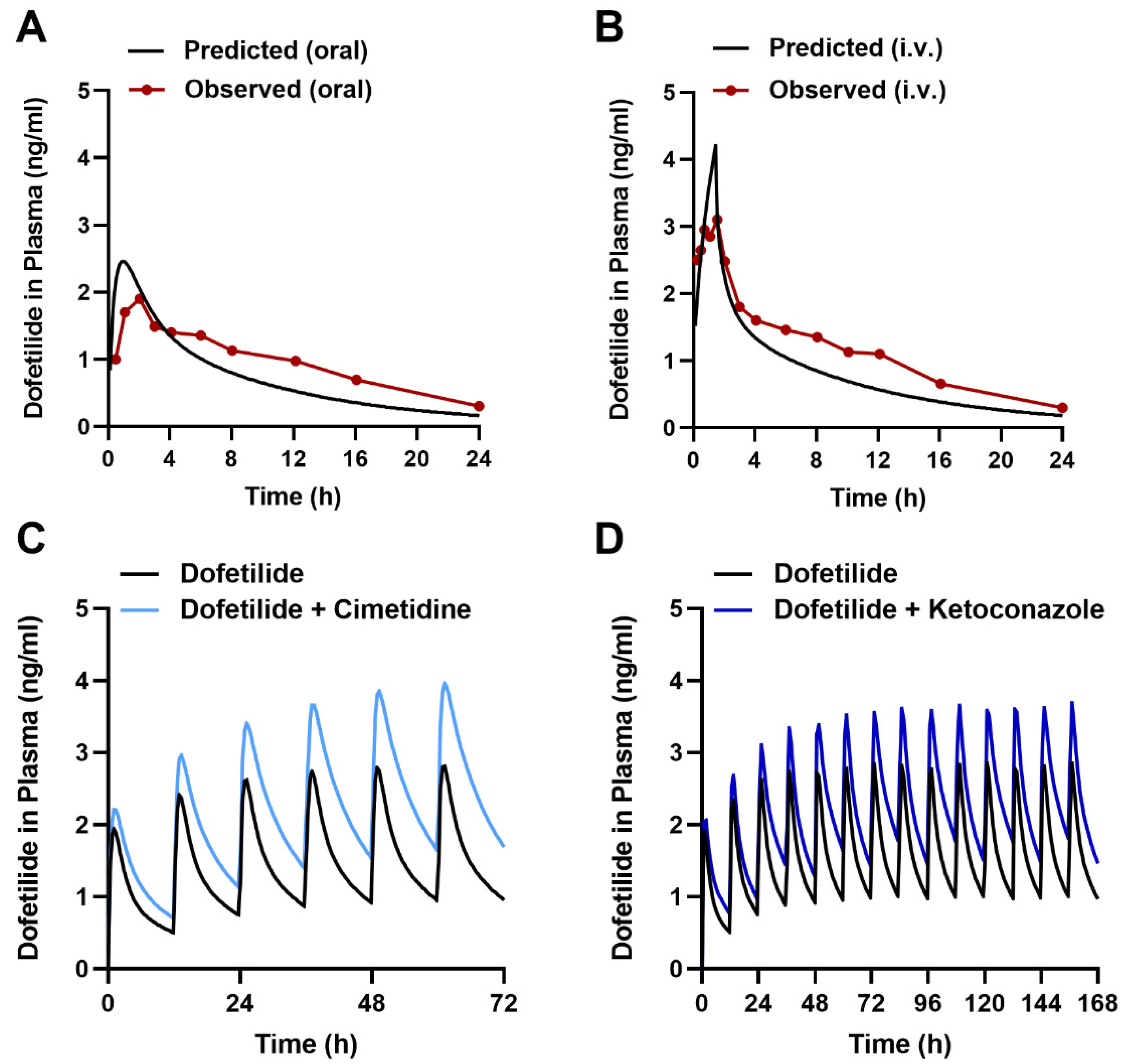
2.6. Predicting the Interaction Liability for Dofetilide in Humans
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Cellular Accumulation
3.3. Ex Vivo Cardiomyocytes Uptake
3.4. Gene Expression Analysis
3.5. Protein Analysis
3.6. Immunohistochemistry
3.7. In Vivo Electrocardiographic Recordings (ECG)
3.8. Animal Models
3.9. Pharmacokinetic Studies
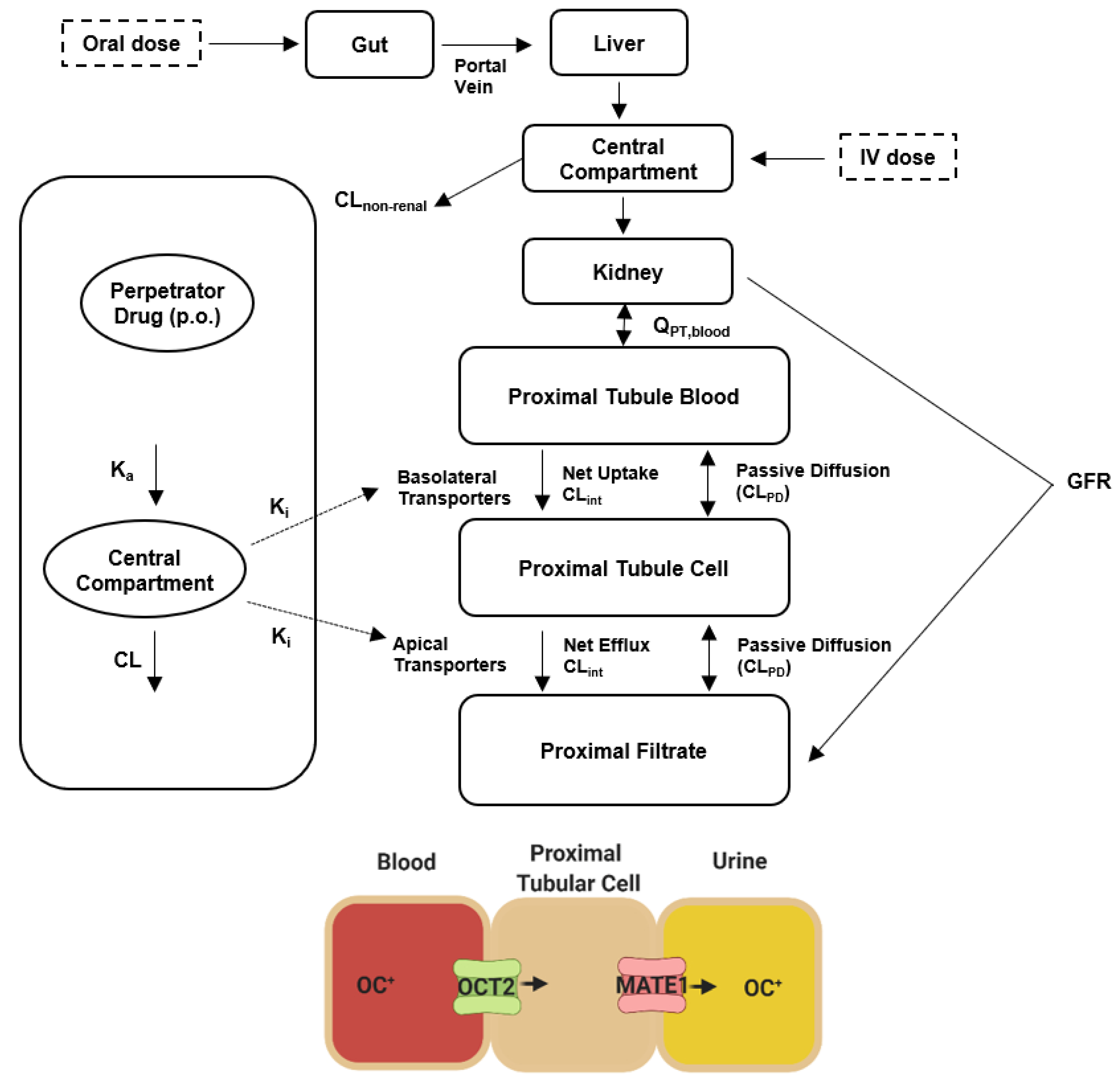
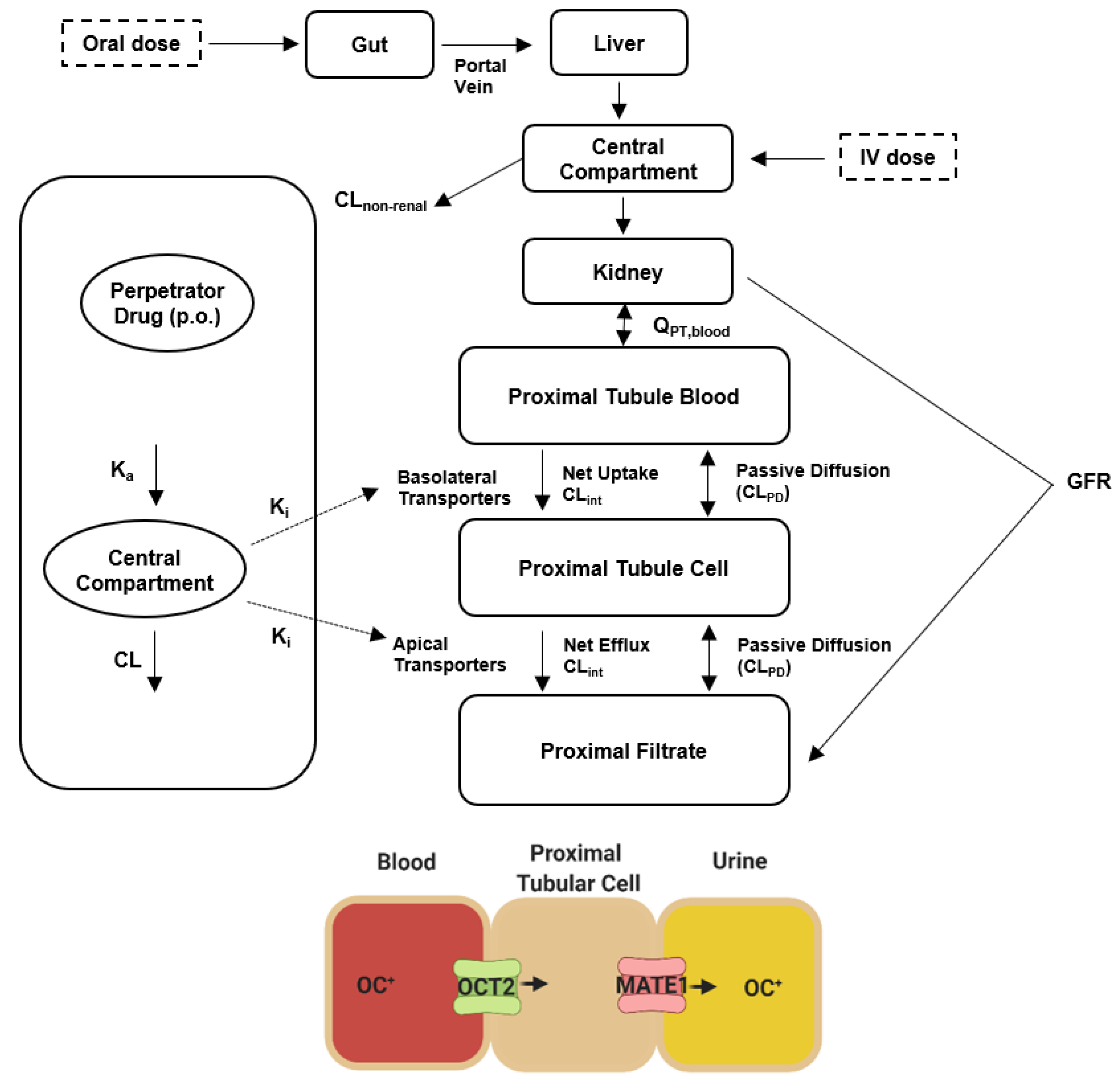
3.10. Physiologically Based Pharmacokinetic (PBPK) Modeling
3.10.1. Input Parameters
3.10.2. PBPK Modeling Strategy
3.10.3. Dofetilide Clinical Data
3.10.4. PBPK Simulations
3.11. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB1 | ATP binding cassette subfamily B member 1 |
| AFib | Atrial fibrillation |
| CYP3A | Cytochrome P450 isoform 3A |
| MATE1 | Multidrug and toxin extrusion protein 1 |
| MATE-2K | Multidrug and toxin extrusion protein 2K |
| MDCK | Madin-darby canine kidney |
| OAT1 | Organic anion transporter 1 |
| OAT3 | Organic anion transporter 3 |
| OCT1 | Organic cation transporter 1 |
| OCT2 | Organic cation transporter 2 |
| OCT3 | Organic cation transporter 3 |
| OCTN | Organic cation transporter novel 2 |
| OCTN1 | Organic cation transporter novel 1 |
| PBPK | Physiologically-based pharmacokinetic model |
| SLC | Solute carrier protein |
| TdP | Torsade de pointes |
| TEA | Tetraethylammonium |
References
- Naccarelli, G.V.; Varker, H.; Lin, J.; Schulman, K.L. Increasing Prevalence of Atrial Fibrillation and Flutter in the United States. Am. J. Cardiol. 2009, 104, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Banerjee, A.; Perel, P.; Wood, D.; Jouven, X. Atrial Fibrillation: The Current Epidemic. J. Geriatr. Cardiol. 2017, 14, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H.; Zheng, Z.J.; et al. Worldwide Epidemiology of Atrial Fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Krijthe, B.P.; Kunst, A.; Benjamin, E.J.; Lip, G.Y.; Franco, O.H.; Hofman, A.; Witteman, J.C.; Stricker, B.H.; Heeringa, J. Projections on the Number of Individuals with Atrial Fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013, 34, 2746–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Krijthe, B.P.; Aspelund, T.; Stepas, K.A.; Pencina, M.J.; Moser, C.B.; Sinner, M.F.; Sotoodehnia, N.; Fontes, J.D.; Janssens, A.C.; et al. Simple Risk Model Predicts Incidence of Atrial Fibrillation in a Racially and Geographically Diverse Population: The CHARGE-AF Consortium. J. Am. Heart Assoc. 2013, 2, e000102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, F.; Kwan, G.F.; Benjamin, E.J. Global Epidemiology of Atrial Fibrillation. Nat. Rev. Cardiol. 2014, 11, 639–654. [Google Scholar] [CrossRef]
- Savelieva, I.; Camm, J. Anti-Arrhythmic Drug Therapy for Atrial Fibrillation: Current Anti-Arrhythmic Drugs, Investigational Agents, and Innovative Approaches. Europace 2008, 10, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, L.; Castro, A.; Dinelli, M.; Alboni, P.; Pappalardo, A.; Richiardi, E.; Santini, M. Comparison of Intravenously Administered Dofetilide versus Amiodarone in the Acute Termination of Atrial Fibrillation and Flutter. A Multicentre, Randomized, Double-Blind, Placebo-Controlled Study. Eur. Heart J. 2000, 21, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Wolbrette, D.L.; Hussain, S.; Maraj, I.; Naccarelli, G.V. A Quarter of a Century Later: What Is Dofetilide’s Clinical Role Today? J. Cardiovasc. Pharmacol. Ther. 2019, 24, 3–10. [Google Scholar] [CrossRef]
- Jaiswal, A.; Goldbarg, S. Dofetilide Induced Torsade de Pointes: Mechanism, Risk Factors and Management Strategies. Indian Heart J. 2014, 66, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Pokorney, S.D.; Yen, D.C.; Campbell, K.B.; Allen LaPointe, N.M.; Sheng, S.; Thomas, L.; Bahnson, T.D.; Daubert, J.P.; Picini, J.P.; Jackson, K.P.; et al. Dofetilide Dose Reductions and Discontinuations in Women Compared with Men. Heart Rhythm. 2018, 15, 478–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenasa, F.; Shenasa, M. Dofetilide: Electrophysiologic Effect, Efficacy, and Safety in Patients with Cardiac Arrhythmias. Card Electrophysiol. Clin. 2016, 8, 423–436. [Google Scholar] [CrossRef]
- Hasannejad, H.; Takeda, M.; Narikawa, S.; Huang, X.-L.; Enomoto, A.; Taki, K.; Niwa, T.; Jung, S.H.; Onozato, M.L.; Tojo, A.; et al. Human Organic Cation Transporter 3 Mediates the Transport of Antiarrhythmic Drugs. Eur. J. Pharmacol. 2004, 499, 45–51. [Google Scholar] [CrossRef]
- Grube, M.; Meyer zu Schwabedissen, H.E.U.; Präger, D.; Haney, J.; Möritz, K.-U.; Meissner, K.; Rosskopf, D.; Eckel, L.; Böhm, M.; Jedlitschky, G.; et al. Uptake of Cardiovascular Drugs into the Human Heart: Expression, Regulation, and Function of the Carnitine Transporter OCTN2 (SLC22A5). Circulation 2006, 113, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- McBride, B.F.; Yang, T.; Liu, K.; Urban, T.J.; Giacomini, K.M.; Kim, R.B.; Roden, D.M. The Organic Cation Transporter, OCTN1, Expressed in the Human Heart, Potentiates Antagonism of the HERG Potassium Channel. J. Cardiovasc. Pharmacol. 2009, 54, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A Human Transporter Protein That Mediates the Final Excretion Step for Toxic Organic Cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiasa, M.; Matsumoto, T.; Komatsu, T.; Moriyama, Y. Wide Variety of Locations for Rodent MATE1, a Transporter Protein That Mediates the Final Excretion Step for Toxic Organic Cations. Am. J. Physiol. Cell Physiol. 2006, 291, C678–C686. [Google Scholar] [CrossRef]
- Lee, N.; Duan, H.; Hebert, M.F.; Liang, C.J.; Rice, K.M.; Wang, J. Taste of a Pill: Organic Cation Transporter-3 (OCT3) Mediates Metformin Accumulation and Secretion in Salivary Glands. J. Biol. Chem. 2014, 289, 27055–27064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, S.; Sorani, M.; Giacomini, K.M. Transport of Paraquat by Human Organic Cation Transporters and Multidrug and Toxic Compound Extrusion Family. J. Pharmacol. Exp. Ther. 2007, 322, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Terada, T.; Mizuno, T.; Katsura, T.; Shimakura, J.; Inui, K. Targeted Disruption of the Multidrug and Toxin Extrusion 1 (Mate1) Gene in Mice Reduces Renal Secretion of Metformin. Mol. Pharmacol. 2009, 75, 1280–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, M.; Terada, T.; Ueba, M.; Sato, T.; Masuda, S.; Katsura, T.; Inui, K. Involvement of Human Multidrug and Toxin Extrusion 1 in the Drug Interaction between Cimetidine and Metformin in Renal Epithelial Cells. J. Pharmacol. Exp. Ther. 2009, 329, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, A.; Inui, K. Importance of the Multidrug and Toxin Extrusion MATE/SLC47A Family to Pharmacokinetics, Pharmacodynamics/Toxicodynamics and Pharmacogenomics. Br. J. Pharmacol. 2011, 164, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Chung, H.; Park, S.-I.; Yi, S.J.; Jang, K.; Kim, A.H.; Yoon, J.; Cho, J.-Y.; Yoon, S.H.; Jang, I.-J.; et al. Inhibition of the Multidrug and Toxin Extrusion (MATE) Transporter by Pyrimethamine Increases the Plasma Concentration of Metformin but Does Not Increase Antihyperglycaemic Activity in Humans. Diabetes Obes. Metab. 2016, 18, 104–108. [Google Scholar] [CrossRef]
- Walker, D.K.; Alabaster, C.T.; Congrave, G.S.; Hargreaves, M.B.; Hyland, R.; Jones, B.C.; Reed, L.J.; Smith, D.A. Significance of Metabolism in the Disposition and Action of the Antidysrhythmic Drug, Dofetilide. In Vitro Studies and Correlation with in Vivo Data. Drug Metab. Dispos. Biol. Fate Chem. 1996, 24, 447–455. [Google Scholar] [PubMed]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde Heart Perfusion: The Langendorff Technique of Isolated Heart Perfusion. J. Mol. Cell Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef]
- Györke, S.; Lukyanenko, V.; Györke, I. Dual Effects of Tetracaine on Spontaneous Calcium Release in Rat Ventricular Myocytes. J. Physiol. 1997, 500, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, M.; Matsumoto, T.; Komatsu, T.; Omote, H.; Moriyama, Y. Functional Characterization of Testis-Specific Rodent Multidrug and Toxic Compound Extrusion 2, a Class III MATE-Type Polyspecific H+/Organic Cation Exporter. Am. J. Physiol. Cell Physiol. 2007, 293, C1437–C1444. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Leblanc, A.F.; Uddin, M.E.; Kim, J.Y.; Chen, M.; Eisenmann, E.D.; Gibson, A.A.; Li, Y.; Hong, K.W.; DiGiacomo, D.; et al. Neuronal Uptake Transporters Contribute to Oxaliplatin Neurotoxicity in Mice. J. Clin. Investig. 2020, 130, 4601–4606. [Google Scholar] [CrossRef]
- Paul Mounsey, J.; John, P. DiMarco Dofetilide. Circulation 2000, 102, 2665–2670. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.J.; Nichols, D.J.; Oliver, S.D. The Pharmacokinetics and Pharmacodynamics of Oral Dofetilide after Twice Daily and Three Times Daily Dosing. Br. J. Clin. Pharmacol. 2000, 50, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Swirp, S.; Duff, H. Age-Dependent Response of the Electrocardiogram to K(+) Channel Blockers in Mice. Am. J. Physiol. Cell Physiol. 2000, 278, C73–C80. [Google Scholar] [CrossRef] [Green Version]
- Saliba, W.I. Dofetilide (Tikosyn): A New Drug to Control Atrial Fibrillation. Cleve. Clin. J. Med. 2001, 68, 353–363. [Google Scholar] [CrossRef] [PubMed]
- McClellan, K.J.; Markham, A. Dofetilide: A Review of Its Use in Atrial Fibrillation and Atrial Flutter. Drugs 1999, 58, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Jin, Y.; Faria, T.N.; Tilford, C.A.; He, A.; Wall, D.A.; Smith, R.L.; Vig, B.S. Expression Profile of Drug and Nutrient Absorption Related Genes in Madin-Darby Canine Kidney (MDCK) Cells Grown under Differentiation Conditions. Pharmaceutics 2012, 4, 314–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.J.; Thompson, D.P.; Cramer, C.T.; Vidmar, T.J.; Scieszka, J.F. The Madin Darby Canine Kidney (MDCK) Epithelial Cell Monolayer as a Model Cellular Transport Barrier. Pharm. Res. 1989, 6, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Hammerle, S.; Suda, K.; Rothen-Rutishauser, B.; Gunthert, M.; Kramer, S.D.; Wunderli-Allenspach, H. Cell Cultures as Tools in Biopharmacy. Eur. J. Pharm. Sci. 2000, 11 (Suppl. S2), S51–S60. [Google Scholar] [CrossRef]
- Müller, F.; Weitz, D.; Mertsch, K.; König, J.; Fromm, M.F. Importance of OCT2 and MATE1 for the Cimetidine-Metformin Interaction: Insights from Investigations of Polarized Transport in Single- And Double-Transfected MDCK Cells with a Focus on Perpetrator Disposition. Mol. Pharm. 2018, 15, 3425–3433. [Google Scholar] [CrossRef]
- Gessner, A.; König, J.; Fromm, M.F. Contribution of Multidrug and Toxin Extrusion Protein 1 (MATE1) to Renal Secretion of Trimethylamine-N-Oxide (TMAO). Sci. Rep. 2018, 8, 6659. [Google Scholar] [CrossRef]
- Johnson, B.F.; Cheng, S.L.; Venitz, J. Transient Kinetic and Dynamic Interactions between Verapamil and Dofetilide, a Class III Antiarrhythmic. J. Clin. Pharmacol. 2001, 41, 1248–1256. [Google Scholar] [CrossRef]
- Diaz, A.L.; Clifton, G.D. Dofetilide: A New Class III Antiarrhythmic for the Management of Atrial Fibrillation. Prog. Cardiovasc. Nurs. 2001, 16, 126–129. [Google Scholar] [CrossRef]
- Abel, S.; Nichols, D.J.; Brearley, C.J.; Eve, M.D. Effect of Cimetidine and Ranitidine on Pharmacokinetics and Pharmacodynamics of a Single Dose of Dofetilide. Br. J. Clin. Pharmacol. 2000, 49, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Wang, J. Renal Drug Transporters and Their Significance in Drug-Drug Interactions. Acta Pharm. Sin. B 2016, 6, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay-Vacher, V.; Izzedine, H.; Karie, S.; Hulot, J.S.; Baumelou, A.; Deray, G. Renal Tubular Drug Transporters. Nephron. Physiol. 2006, 103, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lepist, E.-I.; Ray, A.S. Renal Transporter-Mediated Drug-Drug Interactions: Are They Clinically Relevant? J. Clin. Pharmacol. 2016, 56, S73–S81. [Google Scholar] [CrossRef] [Green Version]
- Teuma, C.; Perier-Muzet, M.; Pelletier, S.; Nouvier, M.; Amini-Adl, M.; Dijoud, F.; Duru, G.; Thomas, L.; Fouque, D.; Laville, M.; et al. New Insights into Renal Toxicity of the B-RAF Inhibitor, Vemurafenib, in Patients with Metastatic Melanoma. Cancer Chemother. Pharmacol. 2016, 78, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.D.; Sakhiya, V.; Fishbane, S. Nephrotoxicity of the BRAF Inhibitors Vemurafenib and Dabrafenib. JAMA Oncol. 2015, 1, 1133–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Ai, L.; Zhang, D.; Wan, L.; Zheng, T.; Yin, J.; Lu, H.; Lu, J.; Lu, F.; Liu, F.; et al. Different Effect of Testosterone and Oestrogen on Urinary Excretion of Metformin via Regulating OCTs and MATEs Expression in the Kidney of Mice. J. Cell. Mol. Med. 2016, 20, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Shchulkin, A.V.; Chernykh, I.V.; Popova, N.M.; Slepnev, A.A.; Yakusheva, E.N. Evaluation of female sex hormones influence on the protein-transporter p-glycoprotein functioning in vitro. Biomeditsinskaia Khimiia 2020, 66, 444–449. [Google Scholar] [CrossRef]
- Yamreudeewong, W.; DeBisschop, M.; Martin, L.G.; Lower, D.L. Potentially Significant Drug Interactions of Class III Antiarrhythmic Drugs. Drug Saf. 2003, 26, 421–438. [Google Scholar] [CrossRef] [PubMed]
- TIKOSYN® Clinical Pharmacology (Dofetilide)|Pfizer Medical Information—US. Available online: https://www.pfizermedicalinformation.com/en-us/tikosyn/clinical-pharmacology (accessed on 20 April 2022).
- Eisenmann, E.D.; Fu, Q.; Muhowski, E.M.; Jin, Y.; Uddin, M.E.; Garrison, D.A.; Weber, R.H.; Woyach, J.; Byrd, J.C.; Sparreboom, A.; et al. Intentional Modulation of Ibrutinib Pharmacokinetics through CYP3A Inhibition. Cancer Res. Commun. 2021, 1, 79–89. [Google Scholar] [CrossRef]
- Yamazaki, S.; Loi, C.-M.; Kimoto, E.; Costales, C.; Varma, M.V. Application of Physiologically Based Pharmacokinetic Modeling in Understanding Bosutinib Drug-Drug Interactions: Importance of Intestinal P-Glycoprotein. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-M.; Rowland, M. The Role of Physiologically Based Pharmacokinetic Modeling in Regulatory Review. Clin. Pharmacol. Ther. 2012, 91, 542–549. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science. Annu Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Chen, Y.; Gibson, C.; Heimbach, T.; Parrott, N.; Peters, S.A.; Snoeys, J.; Upreti, V.V.; Zheng, M.; Hall, S.D. Physiologically Based Pharmacokinetic Modeling in Drug Discovery and Development: A Pharmaceutical Industry Perspective. Clin. Pharmacol. Ther. 2015, 97, 247–262. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Guidance for Industry. Available online: https://www.fda.gov/media/134581/download (accessed on 2 August 2022).
- US Food and Drug Administration. In Vitro Metabolism and Transporter Mediated Drug-Drug Interaction Studies. Guidance for Industry (Draft). Available online: https://www.fda.gov/files/drugs/published/In-Vitro-Metabolism--and-Transporter--Mediated-Drug-Drug-Interaction-Studies-Guidance-for-Industry.pdf (accessed on 2 August 2022).
- Zhao, P.; Zhang, L.; Grillo, J.A.; Liu, Q.; Bullock, J.M.; Moon, Y.J.; Song, P.; Brar, S.S.; Madabushi, R.; Wu, T.C.; et al. Applications of Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation during Regulatory Review. Clin. Pharmacol. Ther. 2011, 89, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Rasmussen, H.S.; Stopher, D.A.; Walker, D.K. Pharmacokinetics and Metabolism of Dofetilide in Mouse, Rat, Dog and Man. Xenobiotica Fate Foreign Compd. Biol. Syst. 1992, 22, 709–719. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Drug Approval Package: Tikosyn (Dofetilide) NDA# 20-931; 1999. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-931_Tikosyn.cfm (accessed on 2 August 2022).
- Pfizer Pharmaceutical Production Limited Tikosyn (Dofetilide, UK-68,798) Capsules: Clinical Pharmacology and Biopharmaceutics Review(s)_Part 1.Pdf. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-931_Tikosyn_biopharmr_P1.pdf (accessed on 2 August 2022).
- Uddin, M.E.; Talebi, Z.; Chen, S.; Jin, Y.; Gibson, A.A.; Noonan, A.M.; Cheng, X.; Hu, S.; Sparreboom, A. In Vitro and In Vivo Inhibition of MATE1 by Tyrosine Kinase Inhibitors. Pharmaceutics 2021, 13, 2004. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Gibson, A.A.; Buege, M.; Ong, S.S.; Li, L.; Hu, S.; Du, G.; Sprowl, J.A.; Vasilyeva, A.; Janke, L.J.; et al. Mitigation of Acute Kidney Injury by Cell-Cycle Inhibitors That Suppress Both CDK4/6 and OCT2 Functions. Proc. Natl. Acad. Sci. USA 2015, 112, 5231–5236. [Google Scholar] [CrossRef] [Green Version]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-Induced Neurotoxicity Is Dependent on the Organic Cation Transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Zavorka Thomas, M.; Magdy, T.; Eisenmann, E.D.; Uddin, M.E.; DiGiacomo, D.F.; Pan, A.; Keiser, M.; Otter, M.; Xia, S.H.; et al. Targeting OCT3 Attenuates Doxorubicin-Induced Cardiac Injury. Proc. Natl. Acad. Sci. USA 2021, 118, e2020168118. [Google Scholar] [CrossRef]
- König, J.; Zolk, O.; Singer, K.; Hoffmann, C.; Fromm, M.F. Double-Transfected MDCK Cells Expressing Human OCT1/MATE1 or OCT2/MATE1: Determinants of Uptake and Transcellular Translocation of Organic Cations. Br. J. Pharmacol. 2011, 163, 546–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Ho, H.-T.; Velez-Cortes, F.; Lou, Q.; Valdivia, C.R.; Knollmann, B.C.; Valdivia, H.H.; Gyorke, S. Genetic Ablation of Ryanodine Receptor 2 Phosphorylation at Ser-2808 Aggravates Ca2+-Dependent Cardiomyopathy by Exacerbating Diastolic Ca2+ Release. J. Physiol. 2014, 592, 1957–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezache, L.; Struckman, H.L.; Greer-Short, A.; Baine, S.; Györke, S.; Radwański, P.B.; Hund, T.J.; Veeraraghavan, R. Vascular Endothelial Growth Factor Promotes Atrial Arrhythmias by Inducing Acute Intercalated Disk Remodeling. Sci. Rep. 2020, 10, 20463. [Google Scholar] [CrossRef] [PubMed]
- Koleske, M.; Bonilla, I.; Thomas, J.; Zaman, N.; Baine, S.; Knollmann, B.C.; Veeraraghavan, R.; Györke, S.; Radwański, P.B. Tetrodotoxin-Sensitive Navs Contribute to Early and Delayed Afterdepolarizations in Long QT Arrhythmia Models. J. Gen. Physiol. 2018, 150, 991–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, A.A.; Dech, S.J.; Nakayama, T.; Hamlin, R.L.; Bauer, J.A.; Carnes, C.A. Age and Anesthetic Effects on Murine Electrocardiography. Life Sci. 2003, 72, 2401–2412. [Google Scholar] [CrossRef]
- Li, Q.; Peng, X.; Yang, H.; Wang, H.; Shu, Y. Deficiency of Multidrug and Toxin Extrusion 1 Enhances Renal Accumulation of Paraquat and Deteriorates Kidney Injury in Mice. Mol. Pharm. 2011, 8, 2476–2483. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Ameling, S.; Noutsias, M.; Köck, K.; Triebel, I.; Bonitz, K.; Meissner, K.; Jedlitschky, G.; Herda, L.R.; Reinthaler, M.; et al. Selective Regulation of Cardiac Organic Cation Transporter Novel Type 2 (OCTN2) in Dilated Cardiomyopathy. Am. J. Pathol. 2011, 178, 2547–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.E.; Garrison, D.A.; Kim, K.; Jin, Y.; Eisenmann, E.D.; Huang, K.M.; Gibson, A.A.; Hu, Z.; Sparreboom, A.; Hu, S. Influence of YES1 Kinase and Tyrosine Phosphorylation on the Activity of OCT1. Front. Pharmacol. 2021, 12, 644342. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, A.F.; Huang, K.M.; Uddin, M.E.; Anderson, J.T.; Chen, M.; Hu, S. Murine Pharmacokinetic Studies. Bio-Protocol 2018, 8, e3056. [Google Scholar] [CrossRef]
- Uddin, M.E.; Sun, X.; Huang, K.M.; Hu, S.; Carnes, C.A.; Sparreboom, A.; Fu, Q. Development and Validation of a UPLC-MS/MS Analytical Method for Dofetilide in Mouse Plasma and Urine, and Its Application to Pharmacokinetic Study. J. Pharm. Biomed. Anal. 2019, 172, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Neuhoff, S.; Gaohua, L.; Burt, H.; Jamei, M.; Li, L.; Tucker, G.T.; Rostami-Hodjegan, A. Accounting for Transporters in Renal Clearance: Towards a Mechanistic Kidney Model (Mech KiM). In Transporters in Drug Development: Discovery, Optimization, Clinical Study and Regulation; Sugiyama, Y., Steffansen, B., Eds.; Springer: New York, NY, USA, 2013; pp. 155–177. ISBN 978-1-4614-8229-1. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uddin, M.E.; Eisenmann, E.D.; Li, Y.; Huang, K.M.; Garrison, D.A.; Talebi, Z.; Gibson, A.A.; Jin, Y.; Nepal, M.; Bonilla, I.M.; et al. MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia. Int. J. Mol. Sci. 2022, 23, 8607. https://doi.org/10.3390/ijms23158607
Uddin ME, Eisenmann ED, Li Y, Huang KM, Garrison DA, Talebi Z, Gibson AA, Jin Y, Nepal M, Bonilla IM, et al. MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia. International Journal of Molecular Sciences. 2022; 23(15):8607. https://doi.org/10.3390/ijms23158607
Chicago/Turabian StyleUddin, Muhammad Erfan, Eric D. Eisenmann, Yang Li, Kevin M. Huang, Dominique A. Garrison, Zahra Talebi, Alice A. Gibson, Yan Jin, Mahesh Nepal, Ingrid M. Bonilla, and et al. 2022. "MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia" International Journal of Molecular Sciences 23, no. 15: 8607. https://doi.org/10.3390/ijms23158607
APA StyleUddin, M. E., Eisenmann, E. D., Li, Y., Huang, K. M., Garrison, D. A., Talebi, Z., Gibson, A. A., Jin, Y., Nepal, M., Bonilla, I. M., Fu, Q., Sun, X., Millar, A., Tarasov, M., Jay, C. E., Cui, X., Einolf, H. J., Pelis, R. M., Smith, S. A., ... Sparreboom, A. (2022). MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia. International Journal of Molecular Sciences, 23(15), 8607. https://doi.org/10.3390/ijms23158607