
Insights into the Genomic Regions and Candidate Genes of Senescence-Related Traits in Upland Cotton via GWAS


Abstract
:1. Introduction
2. Results
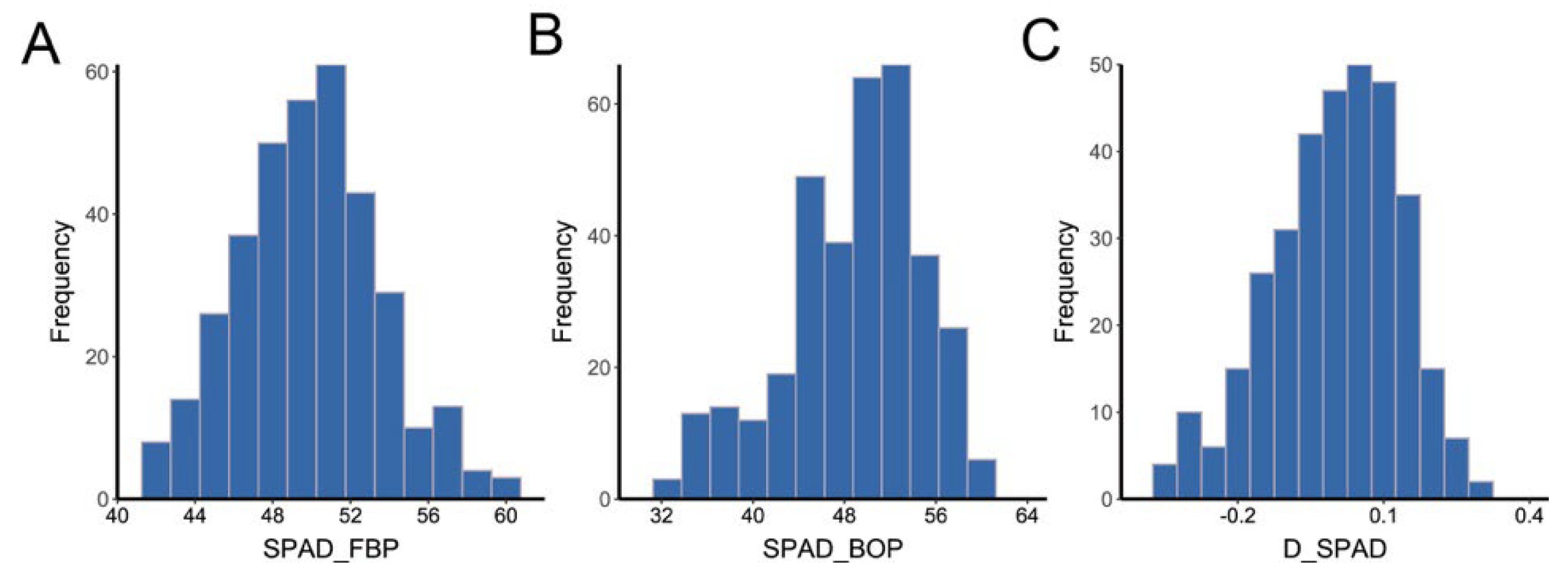
2.1. Analysis of Phenotypic Variations
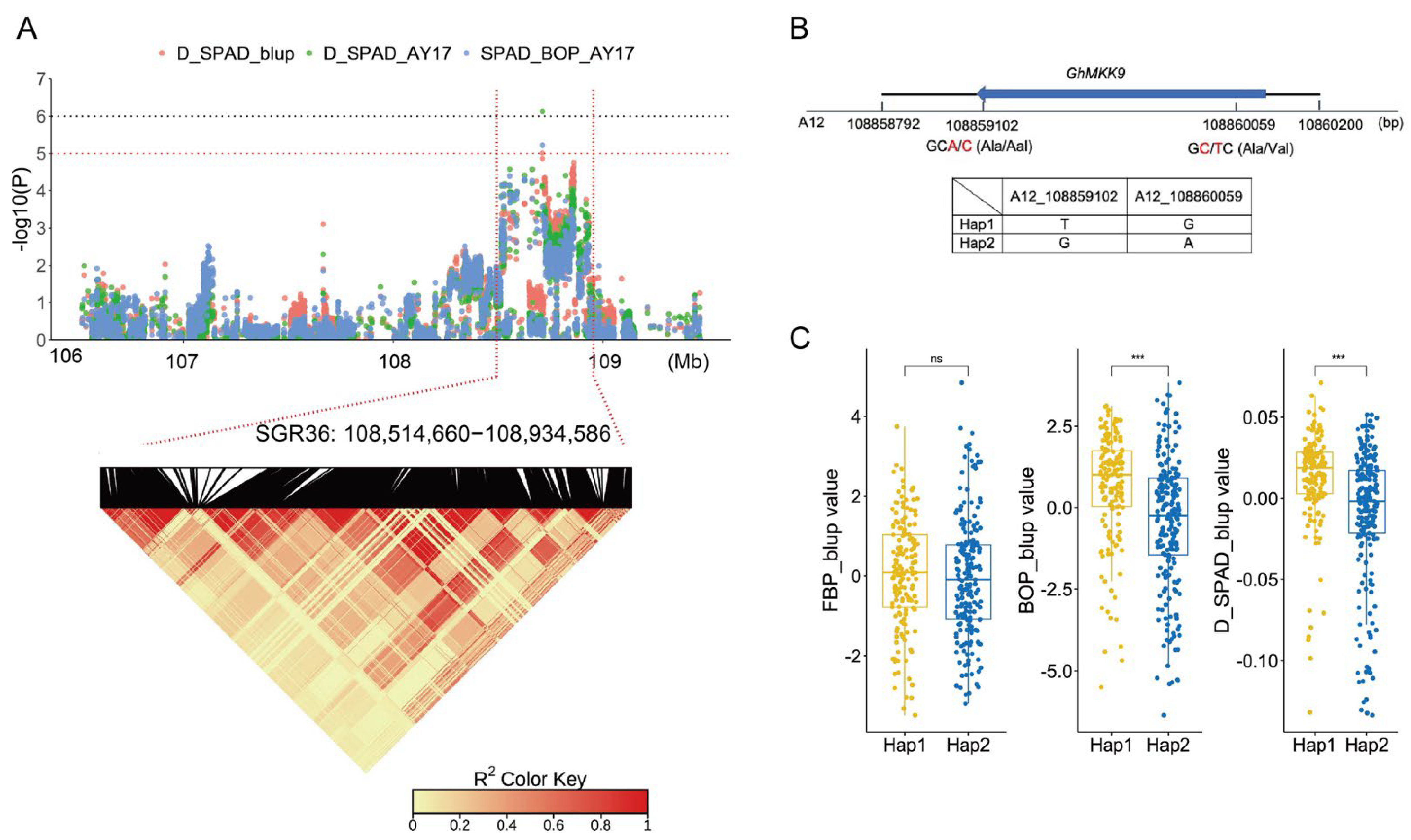
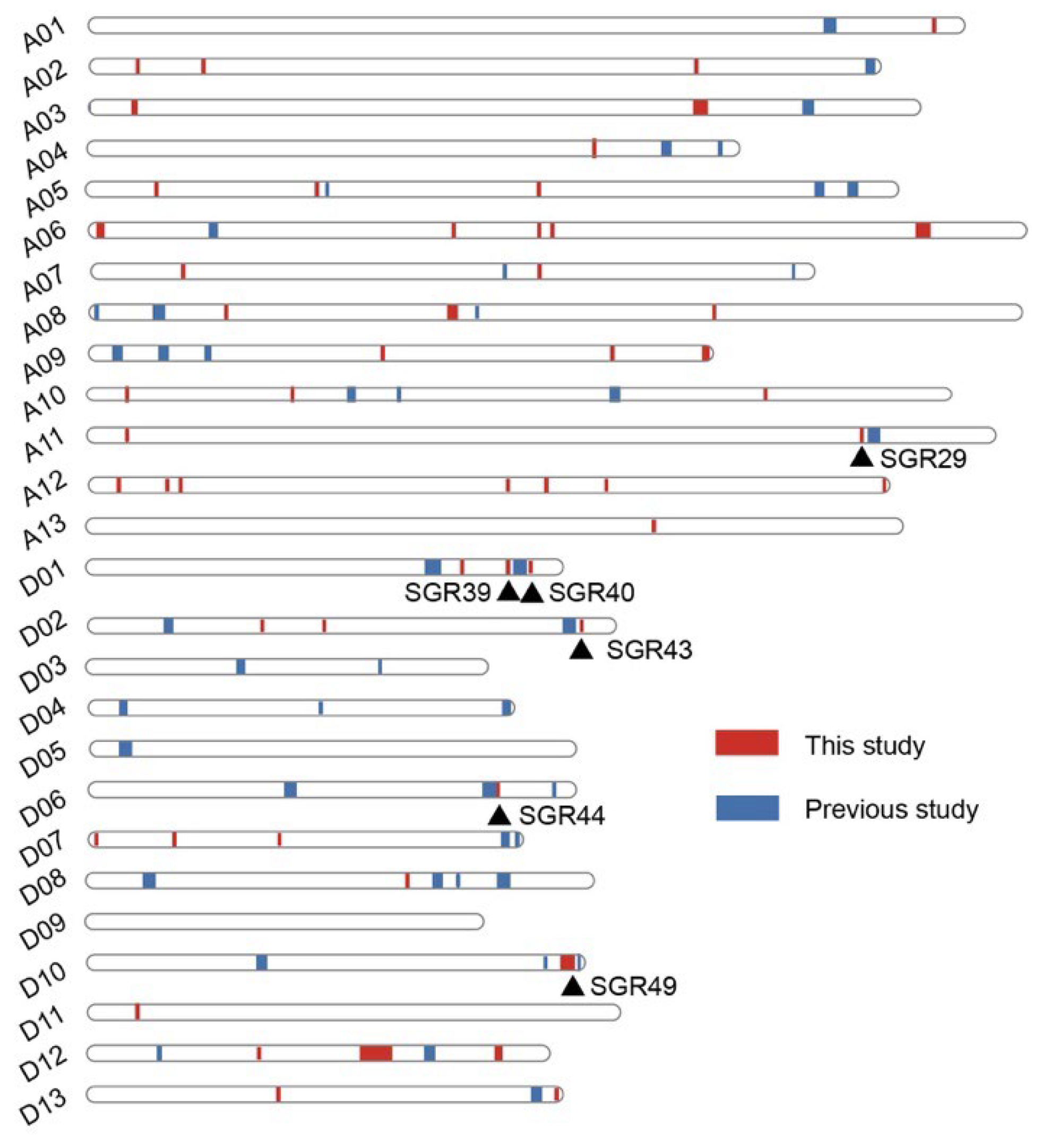
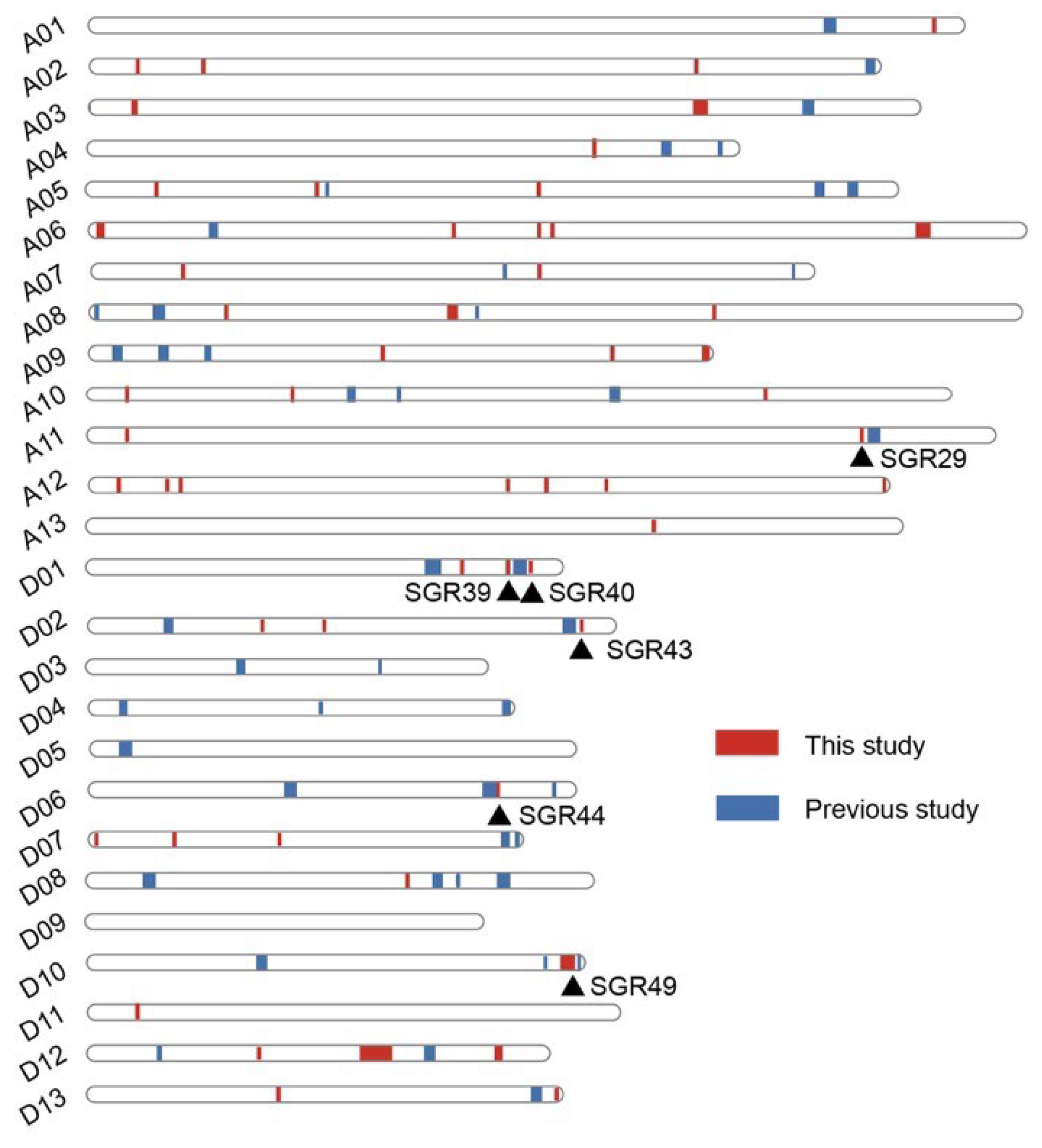
2.2. GWAS for Cotton Senescence and Identified Genomic Regions
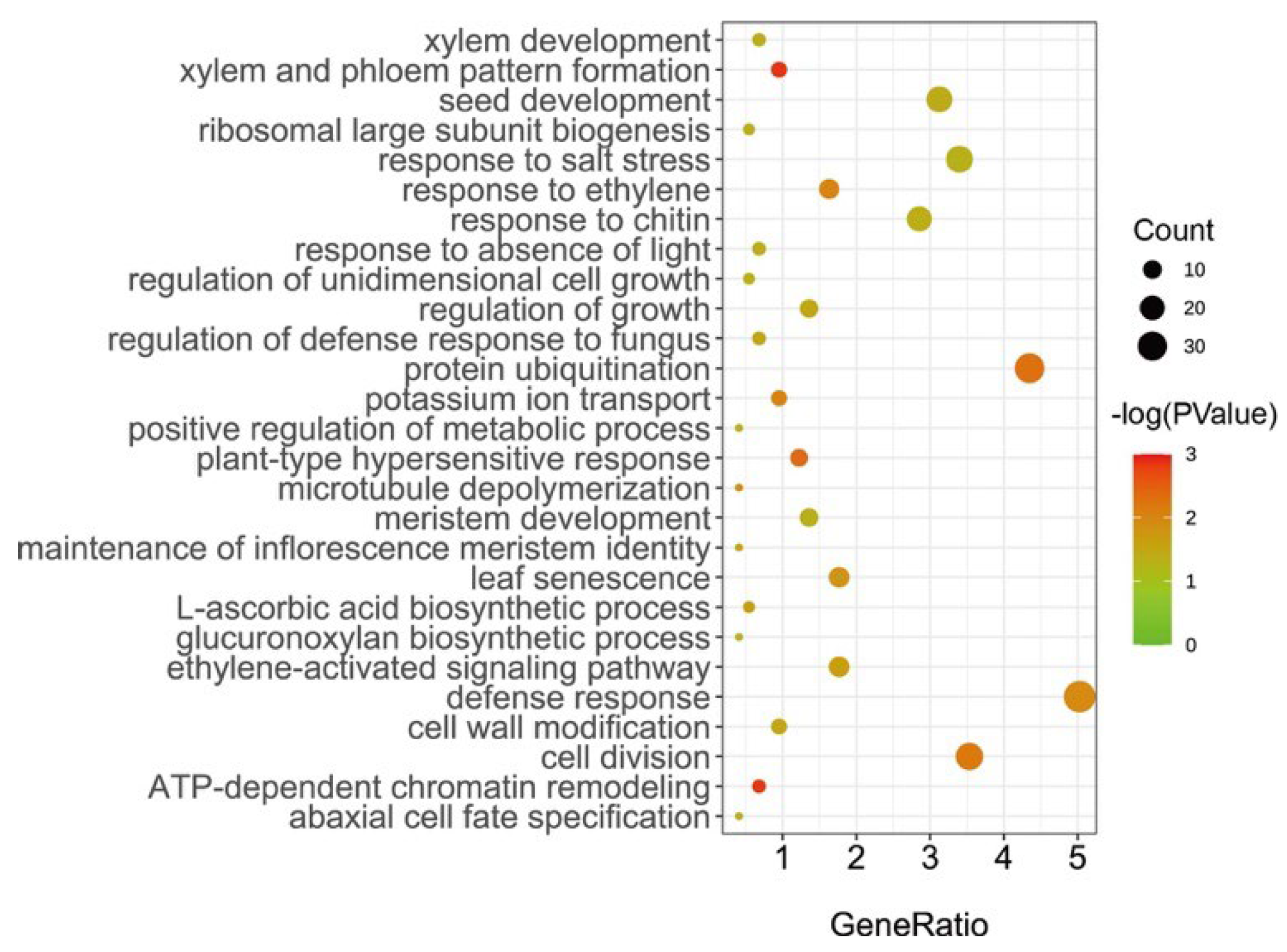
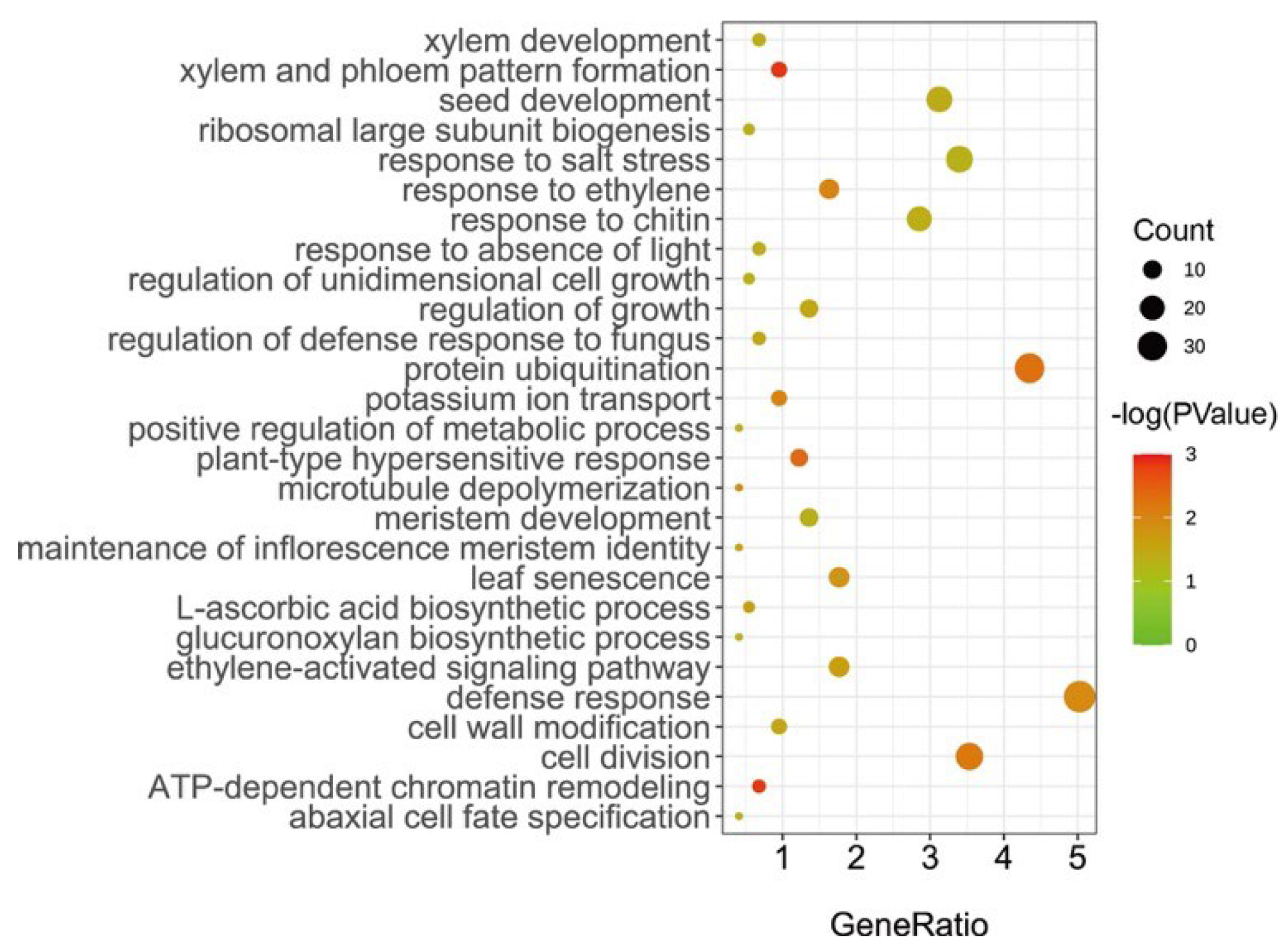
2.3. Prediction of Candidate Genes
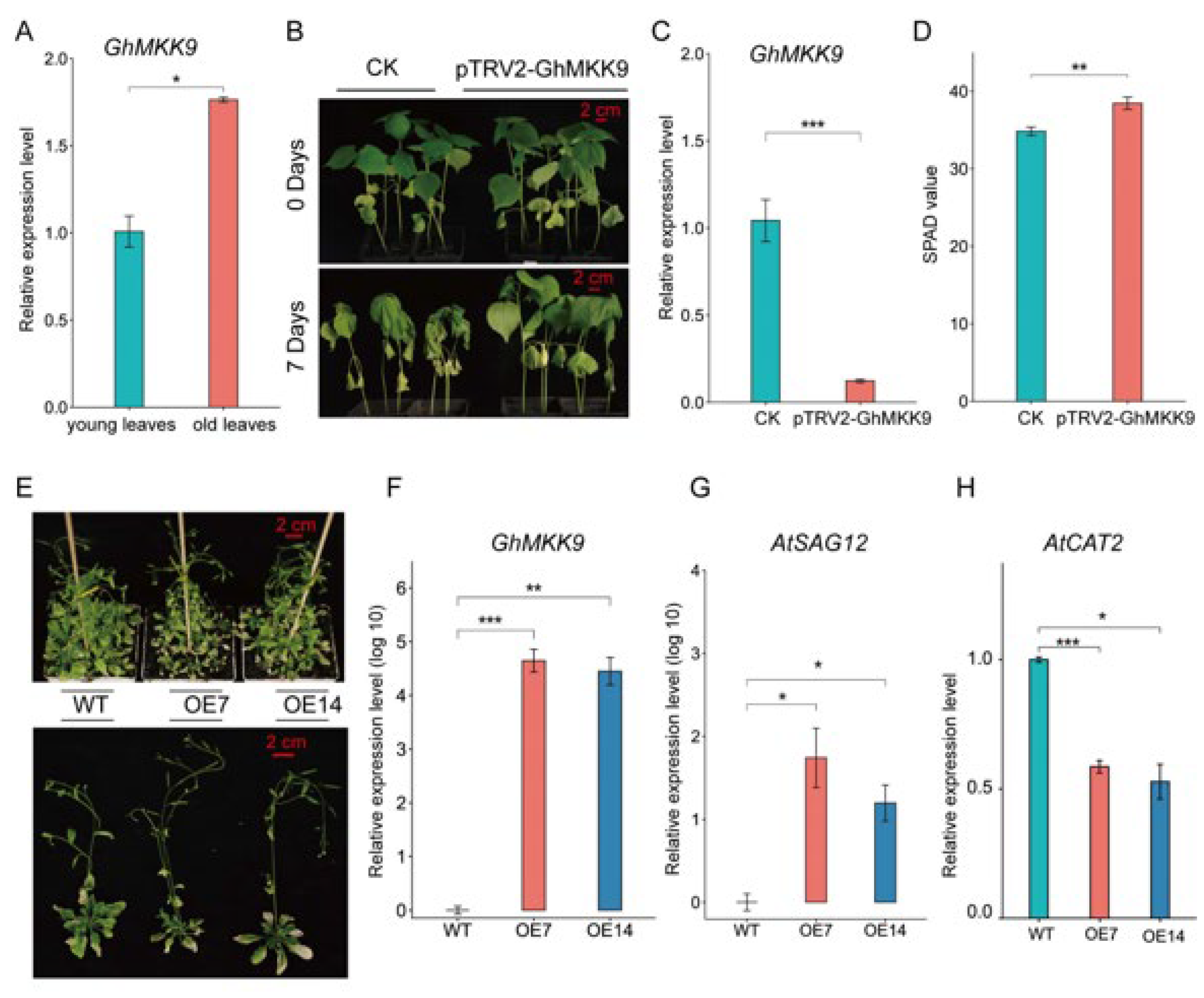
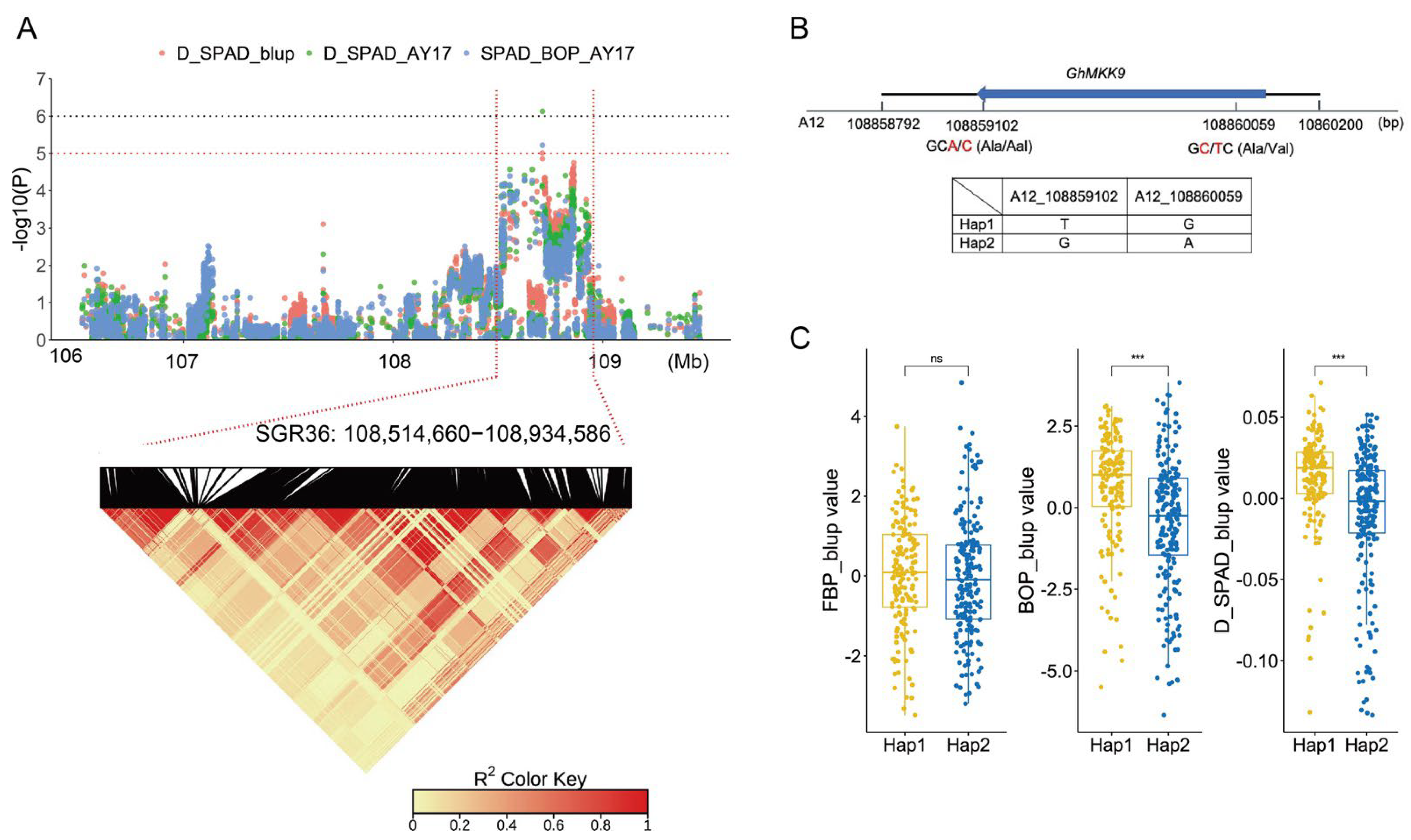
2.4. GhMKK9, A Positive Regulator of Cotton Senescence
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotyping and Data Analysis
4.3. SNP Genotyping
4.4. GWAS and Identification of Genomic Regions
4.5. Prediction of Candidate Genes
4.6. RNA Extraction and qRT-PCR
4.7. VIGS
4.8. Genetic Transformation of Arabidopsis Thaliana
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shahrajabian, M.H.; Sun, W.; Cheng, Q. Considering White Gold, Cotton, for its Fiber, Seed Oil, Traditional and Modern Health Benefits. J. Biol. Environ. Sci. 2020, 14, 25–39. [Google Scholar]
- Gallagher, J.P.; Grover, C.E.; Rex, K.; Moran, M.; Wendel, J.F. A New Species of Cotton from Wake Atoll, Gossypium Stephensii (Malvaceae). Syst. Bot. 2017, 42, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Grover, C.; Zhu, X.; Grupp, K.; Jareczek, J.; Gallagher, J.; Szadkowski, E.; Seijo, J.G.; Wendel, J. Molecular Confirmation of Species Status for the Allopolyploid Cotton Species, Gossypium Ekmanianum Wittmack. Genet. Resour. Crop Evol. 2015, 62, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.D.; Jenkins, J.N.; Deng, D.D.; McCarty, J.C.; Li, P.; Wu, J. Quantitative Trait Loci Analysis of Fiber Quality Traits Using a Random-Mated Recombinant Inbred Population in Upland Cotton (Gossypium hirsutum, L.). BMC Genom. 2014, 15, 397. [Google Scholar] [CrossRef] [Green Version]
- Hulse-Kemp, A.M.; Lemm, J.; Plieske, J.; Ashrafi, H.; Buyyarapu, R.; Fang, D.D.; Frelichowski, J.; Giband, M.; Hague, S.; Hinze, L.L.; et al. Development of a 63K SNP Array for Cotton and High-Density Mapping of Intraspecific and Interspecific Populations of Gossypium spp. G3 Genes Genomes Genet. 2015, 5, 1187–1209. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dong, H. Mechanisms and Regulation of Senescence and Maturity Performance in Cotton. Field Crops Res. 2016, 189, 1–9. [Google Scholar] [CrossRef]
- Lim, P.O.; Kim, H.J.; Nam, H.G. Leaf Senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Ren, G.; Zhang, K.; Li, Z.; Miao, Y.; Guo, H. Leaf Senescence: Progression, Regulation, and Application. Mol. Hortic. 2021, 1, 1–25. [Google Scholar] [CrossRef]
- Diaz, C.; Saliba-Colombani, V.; Loudet, O.; Belluomo, P.; Moreau, L.; Daniel-Vedele, F.; Morot-Gaudry, J.F.; Masclaux-Daubresse, C. Leaf Yellowing and Anthocyanin Accumulation are Two Genetically Independent Strategies in Response to Nitrogen Limitation in Arabidopsis Thaliana. Plant Cell Physiol. 2006, 47, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.R.; Kim, H.J.; Lim, P.O.; Nam, H.G. Leaf Senescence: Systems and Dynamics Aspects. Annu. Rev. Plant Biol. 2019, 70, 347–376. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.R.; Kim, H.J.; Nam, H.G.; Lim, P.O. Plant Leaf Senescence and Death—Regulation by Multiple Layers of Control and Implications for Aging in General. J. Cell Sci. 2013, 126, 4823–4833. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.-C.; Schippers, J.H.; Hille, J.; Dijkwel, P.P. Ethylene-Induced Leaf Senescence Depends on Age-Related Changes and OLD Genes in Arabidopsis. J. Exp. Bot. 2005, 56, 2915–2923. [Google Scholar] [CrossRef]
- Zhang, K.; Xia, X.; Zhang, Y.; Gan, S.-S. An ABA-Regulated and Golgi-Localized Protein Phosphatase Controls Water Loss during Leaf Senescence in Arabidopsis. Plant J. 2012, 69, 667–678. [Google Scholar] [CrossRef]
- Piao, W.; Kim, S.-H.; Lee, B.-D.; An, G.; Sakuraba, Y.; Paek, N.-C. Rice Transcription Factor OsMYB102 Delays Leaf Senescence by Down-Regulating Abscisic Acid Accumulation and Signaling. J. Exp. Bot. 2019, 70, 2699–2715. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Wei, H.; Li, N.; Tian, W.; Chong, K.; Wang, L. Circadian Evening Complex Represses Jasmonate-Induced Leaf Senescence in Arabidopsis. Mol. Plant 2018, 11, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ji, T.-T.; Li, T.-T.; Tian, Y.-Y.; Wang, L.-F.; Liu, W.-C. Jasmonic Acid Promotes Leaf Senescence through MYC2-Mediated Repression of CATALASE2 Expression in Arabidopsis. Plant Sci. 2020, 299, 110604. [Google Scholar] [CrossRef]
- Zhang, M.; Su, J.; Zhang, Y.; Xu, J.; Zhang, S. Conveying Endogenous and Exogenous Signals: MAPK Cascades in Plant Growth and Defense. Cell Signal. Gene Regul. 2018, 45, 1–10. [Google Scholar] [CrossRef]
- Abdelkhalik, A.F.; Shishido, R.; Nomura, K.; Ikehashi, H. QTL-Based Analysis of Leaf Senescence in an Indica/Japonica Hybrid in Rice (Oryza sativa, L.). Theor. Appl. Genet. 2005, 110, 1226–1235. [Google Scholar] [CrossRef]
- Singh, U.M.; Sinha, P.; Dixit, S.; Abbai, R.; Venkateshwarlu, C.; Chitikineni, A.; Singh, V.K.; Varshney, R.K.; Kumar, A. Unraveling Candidate Genomic Regions Responsible for Delayed Leaf Senescence in Rice. PLoS ONE 2020, 15, e0240591. [Google Scholar] [CrossRef]
- Vijayalakshmi, K.; Fritz, A.K.; Paulsen, G.M.; Bai, G.; Pandravada, S.; Gill, B.S. Modeling and Mapping QTL for Senescence-Related Traits in Winter Wheat under High Temperature. Mol. Breed. 2010, 26, 163–175. [Google Scholar] [CrossRef]
- Bogard, M.; Jourdan, M.; Allard, V.; Martre, P.; Perretant, M.R.; Ravel, C.; Heumez, E.; Orford, S.; Snape, J.; Griffiths, S.; et al. Anthesis Date Mainly Explained Correlations between Post-Anthesis Leaf Senescence, Grain Yield, and Grain Protein Concentration in a Winter Wheat Population Segregating for Flowering Time QTLs. J. Exp. Bot. 2011, 62, 3621–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, R.S.; Lopes, M.S.; Collins, N.C.; Reynolds, M.P. Modelling and Genetic Dissection of Staygreen under Heat Stress. Theor. Appl. Genet. 2016, 129, 2055–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, E.A.; Orford, S.; Lage, J.; Griffiths, S. Capturing and Selecting Senescence Variation in Wheat. Front. Plant Sci. 2021, 12, 638738. [Google Scholar] [CrossRef] [PubMed]
- Wehner, G.G.; Balko, C.C.; Enders, M.M.; Humbeck, K.K.; Ordon, F.F. Identification of Genomic Regions Involved in Tolerance to Drought Stress and Drought Stress Induced Leaf Senescence in Juvenile Barley. BMC Plant Biol. 2015, 15, 125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fengler, K.A.; Van Hemert, J.L.; Gupta, R.; Mongar, N.; Sun, J.; Allen, W.B.; Wang, Y.; Weers, B.; Mo, H.; et al. Identification and Characterization of a Novel Stay-Green QTL That Increases Yield in Maize. Plant Biotechnol. J. 2019, 17, 2272–2285. [Google Scholar] [CrossRef]
- Xu, W.; Subudhi, P.K.; Crasta, O.R.; Rosenow, D.T.; Mullet, J.E.; Nguyen, H.T. Molecular Mapping of QTLs Conferring Stay-Green in Grain Sorghum (Sorghum Bicolor L. Moench). Genome 2000, 43, 461–469. [Google Scholar] [CrossRef]
- Sanchez, A.; Subudhi, P.; Rosenow, D.; Nguyen, H. Mapping QTLs Associated with Drought Resistance in Sorghum (Sorghum bicolor L. Moench). Plant Mol. Biol. 2002, 48, 713–726. [Google Scholar] [CrossRef]
- Harris, K.; Subudhi, P.; Borrell, A.; Jordan, D.; Rosenow, D.; Nguyen, H.; Klein, P.; Klein, R.; Mullet, J. Sorghum Stay-Green QTL Individually Reduce Post-Flowering Drought-Induced Leaf Senescence. J. Exp. Bot. 2007, 58, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Kiranmayee, K.U.; Hash, C.T.; Sivasubramani, S.; Ramu, P.; Amindala, B.P.; Rathore, A.; Kishor, P.K.; Gupta, R.; Deshpande, S.P. Fine-Mapping of Sorghum Stay-Green QTL on Chromosome10 Revealed Genes Associated with Delayed Senescence. Genes 2020, 11, 1026. [Google Scholar] [CrossRef]
- Hurtado, P.X.; Schnabel, S.K.; Zaban, A.; Veteläinen, M.; Virtanen, E.; Eilers, P.H.; Van Eeuwijk, F.A.; Visser, R.G.; Maliepaard, C. Dynamics of Senescence-Related QTLs in Potato. Euphytica Neth. J. Plant Breed. 2012, 183, 289–302. [Google Scholar]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Kermanshahi, F.; Ghazizadeh, H.; Hussein, N.A.; Amerizadeh, F.; Samadi, S.; Tayefi, M.; Khodabandeh, A.K.; Moohebati, M.; Ebrahimi, M.; Esmaily, H.; et al. Association of a Genetic Variant in the AKT Gene Locus and Cardiovascular Risk Factors. Cell. Mol. Biol. 2020, 66, 57–64. [Google Scholar] [CrossRef]
- Shamari, A.-R.; Mehrabi, A.-A.; Maleki, A.; Rostami, A. Association Analysis of Tolerance to Dieback Phenomena and Trunk Form Using ISSR Markers in Quercus Brantii. Cell. Mol. Biol. 2018, 64, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Akan, G.; Kisenge, P.; Sanga, T.S.; Mbugi, E.; Adolf, I.; Turkcan, M.K.; Janabi, M.; Atalar, F. Common SNP-Based Haplotype Analysis of the 9p21. 3 Gene Locus as Predictor Coronary Artery Disease in Tanzanian Population. Cell. Mol. Biol. 2019, 65, 33–43. [Google Scholar] [CrossRef]
- Zhao, Y.; Qiang, C.; Wang, X.; Chen, Y.; Deng, J.; Jiang, C.; Sun, X.; Chen, H.; Li, J.; Piao, W.; et al. New Alleles for Chlorophyll Content and Stay-Green Traits Revealed by a Genome Wide Association Study in Rice (Oryza sativa). Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sekhon, R.S.; Saski, C.; Kumar, R.; Flinn, B.S.; Luo, F.; Beissinger, T.M.; Ackerman, A.J.; Breitzman, M.W.; Bridges, W.C.; de Leon, N.; et al. Integrated Genome-Scale Analysis Identifies Novel Genes and Networks Underlying Senescence in Maize. Plant Cell 2019, 31, 1968–1989. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, L.; Feng, Z.; Yu, S. Uncovering Novel Genomic Regions and Candidate Genes for Senescence-Related Traits by Genome-Wide Association Studies in Upland Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2021, 12, 809522. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance Component Model to Account for Sample Structure in Genome-Wide Association Studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Overmyer, K.; Tuominen, H.; Kettunen, R.; Betz, C.; Langebartels, C.; Sandermann, H., Jr.; Kangasjärvi, J. Ozone-Sensitive Arabidopsis Rcd1 Mutant Reveals Opposite Roles for Ethylene and Jasmonate Signaling Pathways in Regulating Superoxide-Dependent Cell Death. Plant Cell 2000, 12, 1849–1862. [Google Scholar] [CrossRef] [Green Version]
- Overmyer, K.; Brosché, M.; Pellinen, R.; Kuittinen, T.; Tuominen, H.; Ahlfors, R.; Keinänen, M.; Saarma, M.; Scheel, D.; Kangasjärvi, J. Ozone-Induced Programmed Cell Death in the Arabidopsis Radical-Induced Cell Death1 Mutant. Plant Physiol. 2005, 137, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Chini, A.; Fonseca, S.; Chico, J.M.; Fernández-Calvo, P.; Solano, R. The ZIM Domain Mediates Homo-and Heteromeric Interactions between Arabidopsis JAZ Proteins. Plant J. 2009, 59, 77–87. [Google Scholar] [CrossRef]
- Zhou, C.; Cai, Z.; Guo, Y.; Gan, S. An Arabidopsis Mitogen-Activated Protein Kinase Cascade, MKK9-MPK6, Plays a Role in Leaf Senescence. Plant Physiol. 2009, 150, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Noh, Y.-S.; Amasino, R.M. Identification of a Promoter Region Responsible for the Senescence-Specific Expression of SAG12. Plant Mol. Biol. 1999, 41, 181–194. [Google Scholar] [CrossRef]
- Xiao, S.; Dai, L.; Liu, F.; Wang, Z.; Peng, W.; Xie, D. COS1: An Arabidopsis coronatine insensitive1 Suppressor Essential for Regulation of Jasmonate-Mediated Plant Defense and Senescence. Plant Cell 2004, 16, 1132–1142. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, P.; Heinlein, C.; Orendi, G.; Zentgraf, U. Senescence-Specific Regulation of Catalases in Arabidopsis thaliana (L.) Heynh. Plant Cell Environ. 2006, 29, 1049–1060. [Google Scholar] [CrossRef]
- Wang, C.; Li, T.; Liu, Q.; Li, L.; Feng, Z.; Yu, S. Characterization and Functional Analysis of GhNAC82, A NAM Domain Gene, Coordinates the Leaf Senescence in Upland Cotton (Gossypium hirsutum L.). Plants 2022, 11, 1491. [Google Scholar] [CrossRef]
- Schippers, J.H.; Schmidt, R.; Wagstaff, C.; Jing, H.-C. Living to Die and Dying to Live: The Survival Strategy behind Leaf Senescence. Plant Physiol. 2015, 169, 914–930. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional Mapping and Annotation of Genetic Associations with FUMA. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Gan, S. Leaf Senescence: Signals, Execution, and Regulation. Curr. Top. Dev. Biol. 2005, 71, 83–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of Allotetraploid Cotton (Gossypium hirsutum L. Acc. TM-1) Provides a Resource for Fiber Improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, K.; Shinozaki, K.; Tena, G.; Sheen, J.; Henry, Y.; Champion, A.; Kreis, M.; Zhang, S.; Hirt, H.; Wilson, C.; et al. Mitogen-Activated Protein Kinase Cascades in Plants: A New Nomenclature. Trends Plant Sci. 2002, 7, 301–308. [Google Scholar] [CrossRef]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, S. Mitogen-Activated Protein Kinase Cascades in Plant Signaling. J. Integr. Plant Biol. 2022, 64, 301–341. [Google Scholar] [CrossRef]
- Yin, Z.; Zhu, W.; Zhang, X.; Chen, X.; Wang, W.; Lin, H.; Wang, J.; Ye, W. Molecular Characterization, Expression and Interaction of MAPK, MAPKK and MAPKKK Genes in Upland Cotton. Genomics 2021, 113, 1071–1086. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Wang, Y.; Liu, H.; Lei, L.; Yang, H.; Liu, G.; Ren, D. Activation of MAPK Kinase 9 Induces Ethylene and Camalexin Biosynthesis and Enhances Sensitivity to Salt Stress in Arabidopsis. J. Biol. Chem. 2008, 283, 26996–27006. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Q.; Wang, Q.; Feng, M.; Li, Y.; Meng, Y.; Zhang, Y.; Liu, G.; Ma, Z.; Wu, H.; et al. RhMKK9, a Rose MAP KINASE KINASE Gene, Is Involved in Rehydration-Triggered Ethylene Production in Rose Gynoecia. BMC Plant Biol. 2017, 17, 51. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Ma, N.; Zhang, Q.; You, Q.; Li, N.; Ali Khan, M.; Liu, X.; Wu, L.; Su, Z.; Gao, J. Precise Spatio-Temporal Modulation of ACC Synthase by MPK 6 Cascade Mediates the Response of Rose Flowers to Rehydration. Plant J. 2014, 79, 941–950. [Google Scholar] [CrossRef]
- Yoo, S.-D.; Cho, Y.-H.; Tena, G.; Xiong, Y.; Sheen, J. Dual Control of Nuclear EIN3 by Bifurcate MAPK Cascades in C2H4 Signalling. Nature 2008, 451, 789–795. [Google Scholar] [CrossRef]
- Shen, L.; Zhuang, B.; Wu, Q.; Zhang, H.; Nie, J.; Jing, W.; Yang, L.; Zhang, W. Phosphatidic Acid Promotes the Activation and Plasma Membrane Localization of MKK7 and MKK9 in Response to Salt Stress. Plant Sci. 2019, 287, 110190. [Google Scholar] [CrossRef]
- Alzwiy, I.A.; Morris, P.C. A Mutation in the Arabidopsis MAP Kinase Kinase 9 Gene Results in Enhanced Seedling Stress Tolerance. Plant Sci. 2007, 173, 302–308. [Google Scholar] [CrossRef]
- Li, L.; Zhang, C.; Huang, J.; Liu, Q.; Wei, H.; Wang, H.; Liu, G.; Gu, L.; Yu, S. Genomic Analyses Reveal the Genetic Basis of Early Maturity and Identification of Loci and Candidate Genes in Upland Cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2021, 19, 109–123. [Google Scholar] [CrossRef]
- Arnon, D.I. Copper Enzymes in Isolated Chloroplasts. Polyphenoloxidase in Beta Vulgaris. Plant Physiol. 1949, 24, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Tieman, D.M.; Jiao, C.; Xu, Y.; Chen, K.; Fei, Z.; Giovannoni, J.J.; Klee, H.J. Chilling-Induced Tomato Flavor Loss Is Associated with Altered Volatile Synthesis and Transient Changes in DNA Methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 12580–12585. [Google Scholar] [CrossRef] [Green Version]
- Harrell, F.E.; Dupont, C. Hmisc: Harrell Miscellaneous, Version 4.5-0. 2021. Available online: https://cran.r-project.org/package=Hmisc (accessed on 1 December 2021).
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix, Version 0.84. 2021. Available online: https://githubcom/taiyun/corrplot (accessed on 3 January 2022).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 March 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago, L.M.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic Diversifications of Five Gossypium Allopolyploid Species and Their Impact on Cotton Improvement. Nat. Genet. 2020, 52, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:13033997. [Google Scholar]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. RMVP: A Memory-Efficient, Visualization-Enhanced, and Parallel-Accelerated Tool for Genome-Wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 618–628. [Google Scholar] [CrossRef]
- Shin, J.-H.; Blay, S.; McNeney, B.; Graham, J. LDheatmap: An r Function for Graphical Display of Pairwise Linkage Disequilibria between Single Nucleotide Polymorphisms. J. Stat. Softw. 2006, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yan, P.; Zeng, Y.; Shen, W.; Tuo, D.; Li, X.; Zhou, P. Nimble Cloning: A Simple, Versatile, and Efficient System for Standardized Molecular Cloning. Front. Bioeng. Biotechnol. 2020, 7, 460. [Google Scholar] [CrossRef]
- Gao, X.; Britt Jr, R.C.; Shan, L.; He, P. Agrobacterium-Mediated Virus-Induced Gene Silencing Assay in Cotton. J. Vis. Exp. JoVE 2011, 54, 2938. [Google Scholar] [CrossRef] [Green Version]
- Bent, A. Arabidopsis Thaliana Floral Dip Transformation Method. Agrobacterium Protoc. 2006, 343, 87–104. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SGR | Chr | Start (bp) | End (bp) | Trait |
|---|---|---|---|---|
| SGR1 | A01 | 115,568,753 | 115,568,865 | Ratio_ab_FBP |
| SGR2 | A02 | 6,556,304 | 6,653,102 | SPAD_BOP_AY17, D_SPAD_blup, D_SPAD_AY17 |
| SGR3 | A02 | 155,73,340 | 15,573,343 | D_total_ab, D_chla |
| SGR4 | A02 | 82,886,748 | 82,925,332 | SPAD_BOP_AY16, D_SPAD_AY16 |
| SGR5 | A03 | 5,877,926 | 6,672,551 | D_SPAD_blup, SPAD_BOP_AY17 |
| SGR6 | A03 | 82,626,806 | 84,562,267 | SPAD_BOP_AY16, D_SPAD_AY16 |
| SGR7 | A03 | 113,682,897 | 113,683,008 | SPAD_BOP_AY16, D_SPAD_AY16 |
| SGR8 | A04 | 69,311,017 | 69,315,686 | Ratio_ab_BOP |
| SGR9 | A05 | 9,695,614 | 9,695,614 | Ratio_ab_FBP |
| SGR10 | A05 | 31,628,900 | 31,628,910 | SPAD_BOP_AY17, D_SPAD_AY17 |
| SGR11 | A05 | 61,940,854 | 61,940,864 | D_chlb, D_ratio_ab |
| SGR12 | A06 | 1,128,396 | 2,179,285 | Chlb_BOP, D_chlb, Total_ab_BOP, Ratio_ab_BOP, D_ratio_ab |
| SGR13 | A06 | 49,902,382 | 49,941,259 | D_ratio_ab, D_chla, D_total_ab, Chla_BOP, Total_ab_BOP |
| SGR14 | A06 | 61,589,693 | 61,589,748 | Ratio_ab_FBP |
| SGR15 | A06 | 63,390,768 | 63,390,795 | D_chla, Chla_BOP, Total_ab_BOP |
| SGR16 | A06 | 113,048,017 | 115,046,117 | D_SPAD_HG17, Ratio_ab_BOP |
| SGR17 | A07 | 12,602,257 | 12,602,280 | SPAD_BOP_AY17, D_SPAD_AY17 |
| SGR18 | A07 | 61,313,631 | 61,313,636 | Chla_BOP, Total_ab_BOP |
| SGR19 | A08 | 18,726,393 | 18,727,072 | D_SPAD_AY16, SPAD_BOP_AY16 |
| SGR20 | A08 | 48,942,300 | 50,338,088 | SPAD_BOP_AY17, D_SPAD_AY17 |
| SGR21 | A08 | 85,414,259 | 85,415,664 | Ratio_ab_BOP, D_SPAD_HG17 |
| SGR22 | A09 | 40,190,612 | 40,190,642 | D_SPAD_AY17, D_SPAD_blup |
| SGR23 | A09 | 71,543,578 | 71,543,578 | Ratio_ab_FBP |
| SGR24 | A09 | 83,855,283 | 84,822,913 | D_SPAD_AY16, SPAD_BOP_AY16 |
| SGR25 | A10 | 5,521,694 | 5,526,321 | D_SPAD_AY16, SPAD_BOP_AY16 |
| SGR26 | A10 | 27,875,320 | 28,364,565 | BOP_blup, D_SPAD_blup |
| SGR27 | A10 | 92,752,436 | 92,766,905 | SPAD_FBP_HG16, Ratio_ab_FBP |
| SGR28 | A11 | 5,387,130 | 5,588,967 | D_SPAD_blup, D_SPAD_AY17, SPAD_BOP_AY17 |
| SGR29 | A11 | 106,469,948 | 106,477,914 | D_SPAD_AY16, SPAD_BOP_AY16 |
| SGR30 | A12 | 4,116,205 | 4,126,358 | Chlb_FBP, Total_ab_FBP |
| SGR31 | A12 | 10,742,829 | 10,742,829 | FBP_blup |
| SGR32 | A12 | 12,591,095 | 12,594,279 | D_SPAD_blup, D_SPAD_AY17, SPAD_BOP_AY17 |
| SGR33 | A12 | 57,300,054 | 57,307,338 | D_SPAD_AY16, SPAD_BOP_AY16 |
| SGR34 | A12 | 62,551,488 | 62,556,405 | D_ratio_ab, D_ratio_ab |
| SGR35 | A12 | 70,740,474 | 70,740,474 | D_SPAD_blup, D_ratio_ab, D_SPAD_AY17 |
| SGR36 | A12 | 108,514,660 | 108,934,586 | SPAD_BOP_AY17, D_SPAD_AY17, D_SPAD_blup |
| SGR37 | A13 | 77,580,822 | 77,580,838 | BOP_blup, SPAD_BOP_AY16 |
| SGR38 | D01 | 51,414,839 | 51,415,298 | BOP_blup, D_SPAD_blup |
| SGR39 | D01 | 57,675,353 | 57,675,364 | Ratio_ab_FBP, Chla_FBP |
| SGR40 | D01 | 60,671,107 | 60,671,202 | D_SPAD_blup, D_SPAD_AY17 |
| SGR41 | D02 | 23,837,463 | 23,837,463 | Ratio_ab_FBP |
| SGR42 | D02 | 32,314,480 | 32,314,480 | D_SPAD_AY17, SPAD_BOP_AY17 |
| SGR43 | D02 | 67,476,213 | 67,476,218 | D_ratio_ab |
| SGR44 | D06 | 55,810,689 | 56,177,650 | Ratio_ab_BOP, D_ratio_ab, D_SPAD_HG17 |
| SGR45 | D07 | 510,384 | 510,546 | Ratio_ab_BOP |
| SGR46 | D07 | 10,915,964 | 11,432,705 | SPAD_FBP_HG17, SPAD_FBP_HG17 |
| SGR47 | D07 | 26,125,297 | 26,125,366 | Ratio_ab_FBP, D_ratio_ab, Chlb_FBP |
| SGR48 | D08 | 43,939,104 | 43,939,104 | Chlb_FBP |
| SGR49 | D10 | 65,317,192 | 67,265,400 | SPAD_BOP_AY17, BOP_blup, D_SPAD_AY17, FBP_blup, SPAD_FBP_HG16 |
| SGR50 | D11 | 9,657,009 | 10,060,413 | FBP_blup, SPAD_FBP_HG16 |
| SGR51 | D12 | 23,540,471 | 23,540,583 | D_SPAD_AY16 |
| SGR52 | D12 | 37,359,379 | 41,685,124 | D_total_ab, Chla_BOP, BOP_blup, SPAD_BOP_HG16, SPAD_FBP_HG16 |
| SGR53 | D12 | 55,716,433 | 56,755,391 | Ratio_ab_BOP, D_ratio_ab |
| SGR54 | D13 | 26,198,781 | 26,198,804 | D_SPAD_AY17, SPAD_BOP_AY17 |
| SGR55 | D13 | 64,827,390 | 64,827,424 | Ratio_ab_FBP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Feng, Z.; Huang, C.; Wen, J.; Li, L.; Yu, S. Insights into the Genomic Regions and Candidate Genes of Senescence-Related Traits in Upland Cotton via GWAS. Int. J. Mol. Sci. 2022, 23, 8584. https://doi.org/10.3390/ijms23158584
Liu Q, Feng Z, Huang C, Wen J, Li L, Yu S. Insights into the Genomic Regions and Candidate Genes of Senescence-Related Traits in Upland Cotton via GWAS. International Journal of Molecular Sciences. 2022; 23(15):8584. https://doi.org/10.3390/ijms23158584
Chicago/Turabian StyleLiu, Qibao, Zhen Feng, Chenjue Huang, Jia Wen, Libei Li, and Shuxun Yu. 2022. "Insights into the Genomic Regions and Candidate Genes of Senescence-Related Traits in Upland Cotton via GWAS" International Journal of Molecular Sciences 23, no. 15: 8584. https://doi.org/10.3390/ijms23158584
APA StyleLiu, Q., Feng, Z., Huang, C., Wen, J., Li, L., & Yu, S. (2022). Insights into the Genomic Regions and Candidate Genes of Senescence-Related Traits in Upland Cotton via GWAS. International Journal of Molecular Sciences, 23(15), 8584. https://doi.org/10.3390/ijms23158584