
Computer-Aided (In Silico) Modeling of Cytochrome P450-Mediated Food–Drug Interactions (FDI)

Abstract
1. Introduction
2. Structure-Based Methods
2.1. Modeling the Mechanisms of CYP Action and Enzyme Inhibition
2.2. Prediction of Candidate Inhibitors
2.3. Protein–Ligand Interactions
3. Ligand-Based Methods
4. Databases
4.1. In Vitro Inhibition Data
4.2. Dietary Compounds
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lewis, D.F.; Ito, Y. Human CYPs Involved in Drug Metabolism: Structures, Substrates and Binding Affinities. Expert Opin. Drug Metab. Toxicol. 2010, 6, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical Importance of the Cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Rodeiro, I.; Donato, M.T.; Lahoz, A.; Garrido, G.; Delgado, R.; Gómez Lechón, M.J. Interactions of Polyphenols with the P450 System: Possible Implications on Human Therapeutics. Mini. Rev. Med. Chem. 2008, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Galetin, A.; Gertz, M.; Houston, J.B. Contribution of Intestinal Cytochrome P450-Mediated Metabolism to Drug-Drug Inhibition and Induction Interactions. Drug Metab. Pharmacokinet. 2010, 25, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tucker, G.T.; Rostami-Hodjegan, A. Cytochrome P450 3A Expression and Activity in the Human Small Intestine. Clin. Pharmacol. Ther. 2004, 76, 391. [Google Scholar] [CrossRef] [PubMed]
- Gavhane, Y.N.; Yadav, A.V. Loss of Orally Administered Drugs in GI Tract. Saudi Pharm. J. 2012, 20, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.C.D.; Ring, N.; Amaral, K.; Doshi, U.; Li, A.P. Human Enterocytes as an in Vitro Model for the Evaluation of Intestinal Drug Metabolism: Characterization of Drug-Metabolizing Enzyme Activities of Cryopreserved Human Enterocytes from Twenty-Four Donors. Drug Metab. Dispos. 2017, 45, 686. [Google Scholar] [CrossRef] [PubMed]
- Siissalo, S.; Heikkinen, A. In Vitro Methods to Study the Interplay of Drug Metabolism and Efflux in the Intestine. Curr. Drug Metab. 2013, 14, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Harrison, A.; Houston, J.B.; Galetin, A. Prediction of Human Intestinal First-Pass Metabolism of 25 CYP3A Substrates from in Vitro Clearance and Permeability Data. Drug Metab. Dispos. 2010, 38, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Basheer, L.; Kerem, Z. Interactions between CYP3A4 and Dietary Polyphenols. Oxid. Med. Cell. Longev. 2015, 2015, 854015. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Di, L. In Vitro and in Vivo Methods to Assess Pharmacokinetic Drug– Drug Interactions in Drug Discovery and Development. Biopharm. Drug Dispos. 2020, 41, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Wienkers, L.C.; Heath, T.G. Predicting in Vivo Drug Interactions from in Vitro Drug Discovery Data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef]
- Bailey, D.G.; Dresser, G.; Arnold, J.M.O. Grapefruit-Medication Interactions: Forbidden Fruit or Avoidable Consequences? CMAJ 2013, 185, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Altman, H. Food–Drug Interaction: Grapefruit Juice Augments Drug Bioavailability—Mechanism, Extent and Relevance. Eur. J. Clin. Nutr. 2004, 58, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Detampel, P.; Beck, M.; Krähenbühl, S.; Huwyler, J. Drug Interaction Potential of Resveratrol. Drug Metab. Rev. 2012, 44, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Choi, D.H.; Choi, J.S. Effects of Resveratrol on the Pharmacokinetics of Diltiazem and Its Major Metabolite, Desacetyldiltiazem, in Rats. Cardiovasc. Ther. 2008, 26, 269–275. [Google Scholar] [CrossRef]
- Veerman, G.D.M.; Hussaarts, K.G.A.M.; Jansman, F.G.A.; Koolen, S.W.L.; van Leeuwen, R.W.F.; Mathijssen, R.H.J. Clinical Implications of Food–Drug Interactions with Small-Molecule Kinase Inhibitors. Lancet Oncol. 2020, 21, e265–e279. [Google Scholar] [CrossRef]
- Cao, X.; Gibbs, S.T.; Fang, L.; Miller, H.A.; Landowski, C.P.; Shin, H.C.; Lennernas, H.; Zhong, Y.; Amidon, G.L.; Yu, L.X.; et al. Why Is It Challenging to Predict Intestinal Drug Absorption and Oral Bioavailability in Human Using Rat Model? Pharm. Res. 2006, 23, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.; Groothuis, G.; de Kanter, R. Comparison of Mouse and Rat Cytochrome P450-Mediated Metabolism in Liver and Intestine. Drug Metab. Dispos. 2006, 34, 1047. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Loi, C.-M.; Brodfuehrer, J.; El-Kattan, A. Impact of Physiological, Physicochemical and Biopharmaceutical Factors in Absorption and Metabolism Mechanisms on the Drug Oral Bioavailability of Rats and Humans. Expert Opin. Drug Metab. Toxicol. 2007, 3, 469–489. [Google Scholar] [CrossRef]
- Awortwe, C.; Fasinu, P.S.; Rosenkranz, B. Application of Caco-2 Cell Line in Herb-Drug Interaction Studies: Current Approaches and Challenges. J. Pharm. Pharm. Sci. 2014, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gillam, E.M.J.; Baba, T.; Kim, B.R.; Ohmori, S.; Guengerich, F.P. Expression of Modified Human Cytochrome P450 3A4 in Escherichia Coli and Purification and Reconstitution of the Enzyme. Arch. Biochem. Biophys. 1993, 305, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Grinkova, Y.V.; Denisov, I.G.; McLean, M.A.; Sligar, S.G. Oxidase Uncoupling in Heme Monooxygenases: Human Cytochrome P450 CYP3A4 in Nanodiscs. Biochem. Biophys. Res. Commun. 2013, 430, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Holmstock, N.; Gonzalez, F.J.; Baes, M.; Annaert, P.; Augustijns, P. PXR/CYP3A4-Humanized Mice for Studying Drug–Drug Interactions Involving Intestinal P-Glycoprotein. Mol. Pharm. 2013, 10, 1056–1062. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Regev-Shoshani, G.; Shoseyov, O.; Kerem, Z. Influence of Lipophilicity on the Interactions of Hydroxy Stilbenes with Cytochrome P450 3A4. Biochem. Biophys. Res. Commun. 2004, 323, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Schroer, K.; Kittelmann, M.; Lütz, S. Recombinant Human Cytochrome P450 Monooxygenases for Drug Metabolite Synthesis. Biotechnol. Bioeng. 2010, 106, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinković, D.; Jhoti, H. Crystal Structure of Human Cytochrome P450 2C9 with Bound Warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Vinković, D.M.; Ward, A.; Angove, H.C.; Day, P.J.; Vonrhein, C.; Tickle, I.J.; Jhoti, H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science 2004, 305, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The Structure of Human Microsomal Cytochrome P450 3A4 Determined by X-ray Crystallography to 2.05-A Resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [PubMed]
- Gay, S.C.; Shah, M.B.; Talakad, J.C.; Maekawa, K.; Roberts, A.G.; Wilderman, P.R.; Sun, L.; Yang, J.Y.; Huelga, S.C.; Hong, W.-X.; et al. Crystal Structure of a Cytochrome P450 2B6 Genetic Variant in Complex with the Inhibitor 4-(4-Chlorophenyl)Imidazole at 2.0-Å Resolution. Mol. Pharmacol. 2010, 77, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Rowland, P.; Blaney, F.E.; Smyth, M.G.; Jones, J.J.; Leydon, V.R.; Oxbrow, A.K.; Lewis, C.J.; Tennant, M.G.; Modi, S.; Eggleston, D.S.; et al. Crystal Structure of Human Cytochrome P450 2D6. J. Biol. Chem. 2006, 281, 7614–7622. [Google Scholar] [CrossRef] [PubMed]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the Oxidation of Polycyclic Aromatic Hydrocarbons Exhibited by the Structure of Human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef]
- Yano, J.K.; Hsu, M.-H.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structures of Human Microsomal Cytochrome P450 2A6 Complexed with Coumarin and Methoxsalen. Nat. Struct. Mol. Biol. 2005, 12, 822–823. [Google Scholar] [CrossRef]
- da Fonseca, R.; Menziani, M.C.; Melo, A.; Ramos, M.J. Modelling the Metabolic Action of Human and Rat CYP1A2 and Its Relationship with the Carcinogenicity of Heterocyclic Amines. Mol. Phys. 2003, 101, 2731–2741. [Google Scholar] [CrossRef]
- Ekins, S.; de Groot, M.J.; Jones, J.P. Pharmacophore and Three-Dimensional Quantitative Structure Activity Relationship Methods for Modeling Cytochrome P450 Active Sites. Drug Metab. Dispos. 2001, 29, 936–944. [Google Scholar] [PubMed]
- Brändén, G.; Sjögren, T.; Schnecke, V.; Xue, Y. Structure-Based Ligand Design to Overcome CYP Inhibition in Drug Discovery Projects. Drug Discovery Today 2014, 19, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Colthart, A.M.; Tietz, D.R.; Ni, Y.; Friedman, J.L.; Dang, M.; Pochapsky, T.C. Detection of Substrate-Dependent Conformational Changes in the P450 Fold by Nuclear Magnetic Resonance. Sci. Rep. 2016, 6, 22035. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Halpert, J.R. Structure–Function Analysis of Cytochromes P450 2B. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.J.; Lill, M.A. Substrate Tunnels in Enzymes: Structure–Function Relationships and Computational Methodology. Proteins Struct. Funct. Bioinf. 2015, 83, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Smieško, M. Spontaneous Ligand Access Events to Membrane-Bound Cytochrome P450 2D6 Sampled at Atomic Resolution. Sci. Rep. 2019, 9, 16411. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G.; Nandekar, P.P.; Bruce, N.J.; Wade, R.C. Differing Membrane Interactions of Two Highly Similar Drug-Metabolizing Cytochrome P450 Isoforms: CYP 2C9 and CYP 2C19. Int. J. Mol. Sci. 2019, 20, 4328. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting Drug Metabolism: Experiment and/or Computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O. Cytochrome P450 Structure–Function: Insights from Molecular Dynamics Simulations. Drug Metab. Rev. 2016, 48, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.L.; Wienkers, L.C. Covalent Alteration of the CYP3A4 Active Site: Evidence for Multiple Substrate Binding Domains. Arch. Biochem. Biophys. 2001, 391, 49–55. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Multiple Sequential Steps Involved in the Binding of Inhibitors to Cytochrome P450 3A4. J. Biol. Chem. 2007, 282, 6863–6874. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, M.; Sjogren, T. Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef]
- Bren, U.; Oostenbrink, C. Cytochrome P450 3A4 Inhibition by Ketoconazole: Tackling the Problem of Ligand Cooperativity Using Molecular Dynamics Simulations and Free-Energy Calculations. J. Chem. Inf. Model. 2012, 52, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of Mutation Pathogenicity Prediction Methods on Missense Variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef]
- Radloff, R.; Gras, A.; Zanger, U.M.; Masquelier, C.; Arumugam, K.; Karasi, J.-C.; Arendt, V.; Seguin-Devaux, C.; Klein, K. Novel CYP2B6 Enzyme Variants in a Rwandese Population: Functional Characterization and Assessment of in Silico Prediction Tools. Hum. Mutat. 2013, 34, 725–734. [Google Scholar] [CrossRef]
- Martiny, V.Y.; Miteva, M.A. Advances in Molecular Modeling of Human Cytochrome P450 Polymorphism. J. Mol. Biol. 2013, 425, 3978–3992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miteva, M.A.; Wang, L.; Alexov, E. Analyzing Effects of Naturally Occurring Missense Mutations. Comput. Math. Methods Med. 2012, 2012, 805827. [Google Scholar] [CrossRef] [PubMed]
- Guttman, Y.; Nudel, A.; Kerem, Z. Polymorphism in Cytochrome P450 3A4 Is Ethnicity Related. Front. Genet. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Ramsland, P.A. Latest Developments in Molecular Docking: 2010–2011 in Review. J. Mol. Recognit. 2013, 26, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Huff, H.C.; Vasan, A.; Roy, P.; Kaul, A.; Tajkhorshid, E.; Das, A. Differential Interactions of Selected Phytocannabinoids with Human CYP2D6 Polymorphisms. Biochemistry 2021, 60, 2749–2760. [Google Scholar] [CrossRef]
- de Waal, P.W.; Sunden, K.F.; Furge, L.L. Molecular Dynamics of CYP2D6 Polymorphisms in the Absence and Presence of a Mechanism-Based Inactivator Reveals Changes in Local Flexibility and Dominant Substrate Access Channels. PLoS ONE 2014, 9, e108607. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, O.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Watanabe, Y.; Oda, A. Evaluation of Influence of Single Nucleotide Polymorphisms in Cytochrome P450 2B6 on Substrate Recognition Using Computational Docking and Molecular Dynamics Simulation. PLoS ONE 2014, 9, e96789. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, L.; Kuriakose, B.B.; Mushfiq, S.; Muthusamy, K. Mechanistic Insights on NsSNPs on Binding Site of Renin and Cytochrome P450 Proteins: A Computational Perceptual Study for Pharmacogenomics Evaluation. J. Cell. Biochem. 2021, 122, 1460–1474. [Google Scholar] [CrossRef]
- Brown, D.K.; Tastan Bishop, Ö. The Role of Structural Bioinformatics in Drug Discovery via Computational SNP Analysis – A Proposed Protocol for Analyzing Variation at the Protein Level. Glob. Heart 2017, 12, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Peng, L.; Zhou, Y.; Zhang, Y.; Zhang, J.Z.H. Computational Alanine Scanning with Interaction Entropy for Protein–Ligand Binding Free Energies. J. Chem. Theory Comput. 2018, 14, 1772–1780. [Google Scholar] [CrossRef]
- Basheer, L.; Schultz, K.; Fichman, M.; Kerem, Z. Use of in Vitro and Predictive in Silico Models to Study the Inhibition of Cytochrome P4503A by Stilbenes. PLoS ONE 2015, 10, e0141061. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.P. Mechanism-Based Inhibition of CYP3A4 and Other Cytochromes P450. In Annual Reports in Medicinal Chemistry; Macor, J.E., Ed.; Academic Press: Cambridge, MA, USA, 2009; Volume 44, pp. 535–553. [Google Scholar]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and Induction of Human Cytochrome P450 Enzymes: Current Status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar] [CrossRef] [PubMed]
- Conner, K.P.; Woods, C.M.; Atkins, W.M. Interactions of Cytochrome P450s with Their Ligands. Arch. Biochem. Biophys. 2011, 507, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tawa, G.J.; Wallqvist, A. Identifying Cytochrome P450 Functional Networks and Their Allosteric Regulatory Elements. PLoS ONE 2013, 8, e81980. [Google Scholar] [CrossRef] [PubMed]
- Rossato, G.; Ernst, B.; Smiesko, M.; Spreafico, M.; Vedani, A. Probing Small-Molecule Binding to Cytochrome P450 2D6 and 2C9: An in Silico Protocol for Generating Toxicity Alerts. ChemMedChem 2010, 5, 2088–2101. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Hu, L.; Li, H.; Su, J.; Cao, Z.; Zhang, W. Novel Natural Inhibitors of CYP1A2 Identified by in Silico and in Vitro Screening. Int. J. Mol. Sci. 2011, 12, 3250–3262. [Google Scholar] [CrossRef]
- Joshi, P.; McCann, G.J.P.; Sonawane, V.R.; Vishwakarma, R.A.; Chaudhuri, B.; Bharate, S.B. Identification of Potent and Selective CYP1A1 Inhibitors via Combined Ligand and Structure-Based Virtual Screening and Their in Vitro Validation in Sacchrosomes and Live Human Cells. J. Chem. Inf. Model. 2017, 57, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Puskás, I.; Roewer, N.; Förster, C.; Broscheit, J. Three-Dimensional Quantitative Structure–Activity Relationship and Docking Studies in a Series of Anthocyanin Derivatives as Cytochrome P450 3A4 Inhibitors. AABC 2014, 7, 11–21. [Google Scholar] [CrossRef]
- Li, Y.; Ning, J.; Wang, Y.; Wang, C.; Sun, C.; Huo, X.; Yu, Z.; Feng, L.; Zhang, B.; Tian, X.; et al. Drug Interaction Study of Flavonoids toward CYP3A4 and Their Quantitative Structure Activity Relationship (QSAR) Analysis for Predicting Potential Effects. Toxicol. Lett. 2018, 294, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kiani, Y.S.; Ranaghan, K.E.; Jabeen, I.; Mulholland, A.J. Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors. Int. J. Mol. Sci. 2019, 20, 4468. [Google Scholar] [CrossRef]
- Basheer, L.; Schultz, K.; Guttman, Y.; Kerem, Z. In Silico and in Vitro Inhibition of Cytochrome P450 3A by Synthetic Stilbenoids. Food Chem. 2017, 237, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Guttman, Y.; Yedidia, I.; Nudel, A.; Zhmykhova, Y.; Kerem, Z.; Carmi, N. New Grapefruit Cultivars Exhibit Low Cytochrome P4503A4-Inhibition Activity. Food Chem. Toxicol. 2020, 137, 111135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hellum, B.H.; Liang, A.; Nilsen, O.G. Inhibitory Mechanisms of Human CYPs by Three Alkaloids Isolated from Traditional Chinese Herbs. Phytother. Res. 2015, 29, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.X.; Zhou, Z.W.; He, Z.X.; Zhang, X.; Zhou, S.F.; Zhu, S. Estimation of the Binding Modes with Important Human Cytochrome P450 Enzymes, Drug Interaction Potential, Pharmacokinetics, and Hepatotoxicity of Ginger Components Using Molecular Docking, Computational, and Pharmacokinetic Modeling Studies. Drug Des. Dev. Ther. 2015, 9, 841–866. [Google Scholar] [CrossRef]
- Saraceno, M.; Coi, A.; Bianucci, A.M. Molecular Modelling of Human CYP2D6 and Molecular Docking of a Series of Ajmalicine- and Quinidine-like Inhibitors. Int. J. Biol. Macromol. 2008, 42, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Ghalehshahi, H.G.; Balalaie, S.; Sohbati, H.R.; Azizian, H.; Alavijeh, M.S. Synthesis, CYP 450 Evaluation, and Docking Simulation of Novel 4-Aminopyridine and Coumarin Derivatives. Arch. Pharm. 2019, 352, 1800247. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. A Mechanism of O-Demethylation of Aristolochic Acid I by Cytochromes P450 and Their Contributions to This Reaction in Human and Rat Livers: Experimental and Theoretical Approaches. Int. J. Mol. Sci. 2015, 16, 27561–27575. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kehinde, I.; Ramharack, P.; Nlooto, M.; Gordon, M. Molecular Dynamic Mechanism(s) of Inhibition of Bioactive Antiviral Phytochemical Compounds Targeting Cytochrome P450 3A4 and P-Glycoprotein. J. Biomol. Struct. Dyn. 2020, 40, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hochleitner, J.; Akram, M.; Ueberall, M.; Davis, R.A.; Waltenberger, B.; Stuppner, H.; Sturm, S.; Ueberall, F.; Gostner, J.M.; Schuster, D. A Combinatorial Approach for the Discovery of Cytochrome P450 2D6 Inhibitors from Nature. Sci. Rep. 2017, 7, 8071. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Yu, Y.; Shen, J.; Yang, L.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. Classification of Cytochrome P450 Inhibitors and Noninhibitors Using Combined Classifiers. J. Chem. Inf. Model. 2011, 51, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Veith, H.; Xia, M.; Austin, C.P.; Huang, R. Predictive Models for Cytochrome P450 Isozymes Based on Quantitative High Throughput Screening Data. J. Chem. Inf. Model. 2011, 51, 2474–2481. [Google Scholar] [CrossRef]
- Lapins, M.; Worachartcheewan, A.; Spjuth, O.; Georgiev, V.; Prachayasittikul, V.; Nantasenamat, C.; Wikberg, J.E.S. A Unified Proteochemometric Model for Prediction of Inhibition of Cytochrome P450 Isoforms. PLoS ONE 2013, 8, e66566. [Google Scholar] [CrossRef]
- Lee, J.H.; Basith, S.; Cui, M.; Kim, B.; Choi, S. In Silico Prediction of Multiple-Category Classification Model for Cytochrome P450 Inhibitors and Non-Inhibitors Using Machine-Learning Method. SAR QSAR Environ. Res. 2017, 28, 863–874. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharmaceutics 2018, 15, 4336–4345. [Google Scholar] [CrossRef] [PubMed]
- Rácz, A.; Bajusz, D.; Héberger, K. Effect of Dataset Size and Train/Test Split Ratios in QSAR/QSPR Multiclass Classification. Molecules 2021, 26, 1111. [Google Scholar] [CrossRef] [PubMed]
- Esaki, T.; Watanabe, R.; Kawashima, H.; Ohashi, R.; Natsume-Kitatani, Y.; Nagao, C.; Mizuguchi, K. Data Curation Can Improve the Prediction Accuracy of Metabolic Intrinsic Clearance. Mol. Inf. 2019, 38, 1800086. [Google Scholar] [CrossRef] [PubMed]
- Guermazi, R.; Chaabane, I.; Hammami, M. AECID: Asymmetric Entropy for Classifying Imbalanced Data. Inf. Sci. 2018, 467, 373–397. [Google Scholar] [CrossRef]
- Pereira, J.; Saraiva, F. Convolutional Neural Network Applied to Detect Electricity Theft: A Comparative Study on Unbalanced Data Handling Techniques. Int. J. Electr. Power Energy Syst. 2021, 131, 107085. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Peach, M.L.; Sitzmann, M.; Nicklaus, M.C. QSAR Modeling of Imbalanced High-Throughput Screening Data in PubChem. J. Chem. Inf. Model. 2014, 54, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Tsai, C.F.; Hu, Y.H.; Jhang, J.S. Clustering-Based Undersampling in Class-Imbalanced Data. Inf. Sci. 2017, 409–410, 17–26. [Google Scholar] [CrossRef]
- Guttman, Y.; Kerem, Z. Dietary Inhibitors of CYP3A4 Are Revealed Using Virtual Screening by Using a New Deep-Learning Classifier. J. Agric. Food Chem. 2022, 70, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Yang, Y.; Huang, S.; Chen, L.; Kuang, Z.; Heng, Y.; Mei, H. Molecular Image-Based Convolutional Neural Network for the Prediction of ADMET Properties. Chemom. Intell. Lab. Syst. 2019, 194, 103853. [Google Scholar] [CrossRef]
- Nembri, S.; Grisoni, F.; Consonni, V.; Todeschini, R. In Silico Prediction of Cytochrome P450-Drug Interaction: QSARs for CYP3A4 and CYP2C9. Int. J. Mol. Sci. 2016, 17, 914. [Google Scholar] [CrossRef] [PubMed]
- Su, B.H.; Tu, Y.S.; Lin, C.; Shao, C.Y.; Lin, O.A.; Tseng, Y.J. Rule-Based Prediction Models of Cytochrome P450 Inhibition. J. Chem. Inf. Model. 2015, 55, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, M.; Steinbeck, C. Review on Natural Products Databases: Where to Find Data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT Online: Collection of Open Natural Products Database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Hirko, K.A.; Rocque, G.; Reasor, E.; Taye, A.; Daly, A.; Cutress, R.I.; Copson, E.R.; Lee, D.W.; Lee, K.H.; Im, S.A.; et al. The Impact of Race and Ethnicity in Breast Cancer—Disparities and Implications for Precision Oncology. BMC Med. 2022, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Yip, V.L.M.; Pertinez, H.; Meng, X.; Maggs, J.L.; Carr, D.F.; Park, B.K.; Marson, A.G.; Pirmohamed, M. Evaluation of Clinical and Genetic Factors in the Population Pharmacokinetics of Carbamazepine. Br. J. Clin. Pharmacol. 2021, 87, 2572–2588. [Google Scholar] [CrossRef] [PubMed]
- Hanser, T.; Barber, C.; Marchaland, J.F.; Werner, S. Applicability Domain: Towards a More Formal Definition. SAR QSAR Environ. Res. 2016, 27, 865–881. [Google Scholar] [CrossRef] [PubMed]


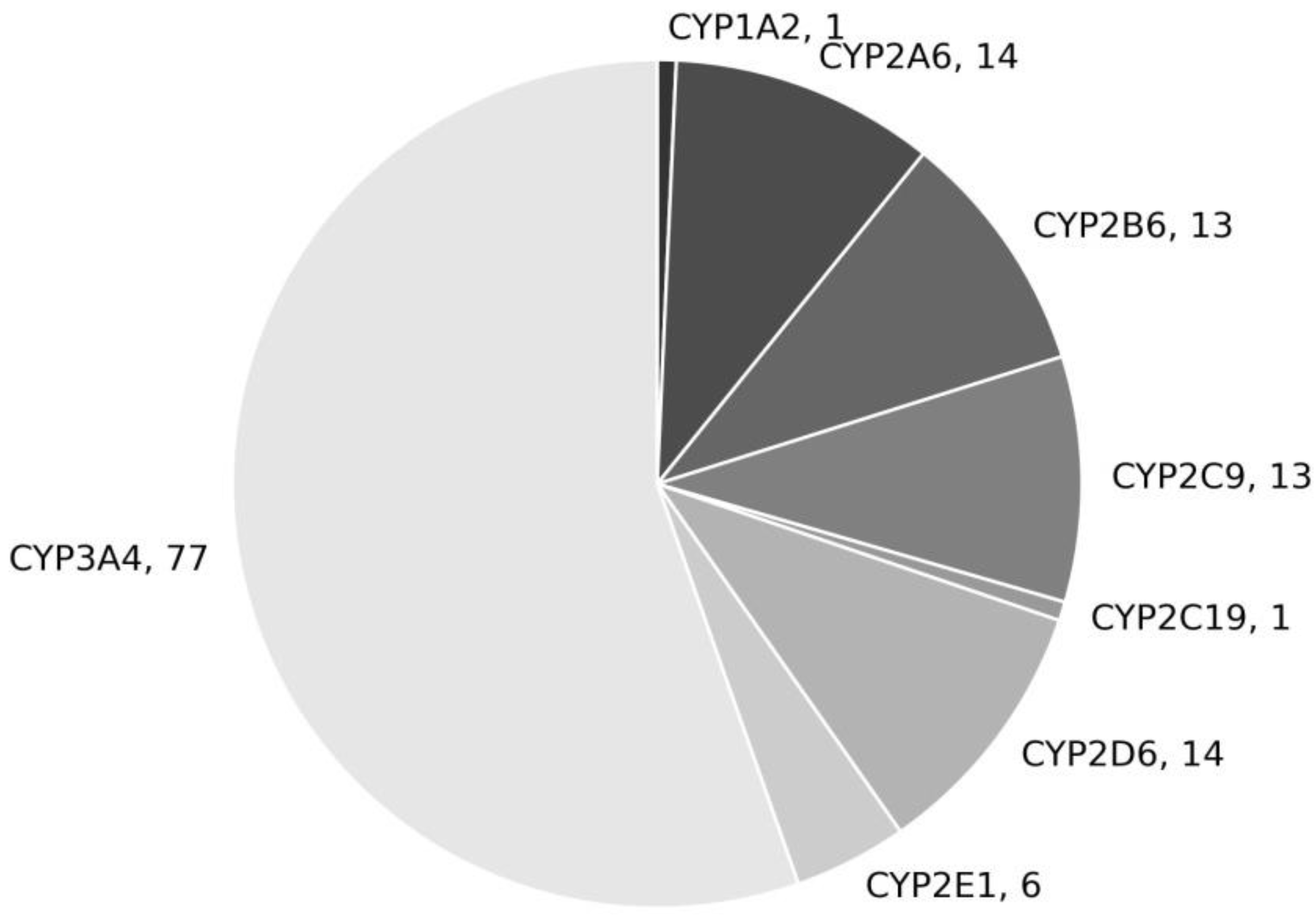{kind=link}
| CYP | Bioassay | Compounds | Substances | References |
|---|---|---|---|---|
| CYP1A2 | 410 | 8354 | 9198 | [82,86,94] |
| CYP2C9 | 883 | 9385 | 10,320 | [82,86] |
| 1,645,842 | 5094 | 5242 | - | |
| CYP2C19 | 899 | 9385 | 10,320 | [82,86] |
| CYP2D6 | 891 | 9385 | 10,320 | [81,82,86] |
| 1,645,840 | 5094 | 5242 | - | |
| CYP3A4 | 884 | 13,076 | 14,155 | [82,86] |
| 885 | 13,076 | 14,155 | [82] | |
| 1,645,841 | 5094 | 5242 | ||
| CYP1A2, CYP2D6, CYP2C9, CYP2C19, CYP3A4 | 1851 | 16,560 | 17,143 | [82,83,85,86,87,94,95,96] |
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 | |
|---|---|---|---|---|---|
| Substances | 9198 | 10,320 | 10,320 | 10,320 | 14,155 |
| Shared substances with AID1581 | 7942 (86%) | 9124 (88%) | 9124 (88%) | 9124 (88%) | 9124 (64%) |
| Pearson’s correlation coefficient of activity scores | 0.937 | 0.996 | 0.995 | 0.999 | 0.994 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guttman, Y.; Kerem, Z. Computer-Aided (In Silico) Modeling of Cytochrome P450-Mediated Food–Drug Interactions (FDI). Int. J. Mol. Sci. 2022, 23, 8498. https://doi.org/10.3390/ijms23158498
Guttman Y, Kerem Z. Computer-Aided (In Silico) Modeling of Cytochrome P450-Mediated Food–Drug Interactions (FDI). International Journal of Molecular Sciences. 2022; 23(15):8498. https://doi.org/10.3390/ijms23158498
Chicago/Turabian StyleGuttman, Yelena, and Zohar Kerem. 2022. "Computer-Aided (In Silico) Modeling of Cytochrome P450-Mediated Food–Drug Interactions (FDI)" International Journal of Molecular Sciences 23, no. 15: 8498. https://doi.org/10.3390/ijms23158498
APA StyleGuttman, Y., & Kerem, Z. (2022). Computer-Aided (In Silico) Modeling of Cytochrome P450-Mediated Food–Drug Interactions (FDI). International Journal of Molecular Sciences, 23(15), 8498. https://doi.org/10.3390/ijms23158498