
Co-Crystal Structure-Guided Optimization of Dual-Functional Small Molecules for Improving the Peroxygenase Activity of Cytochrome P450BM3

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
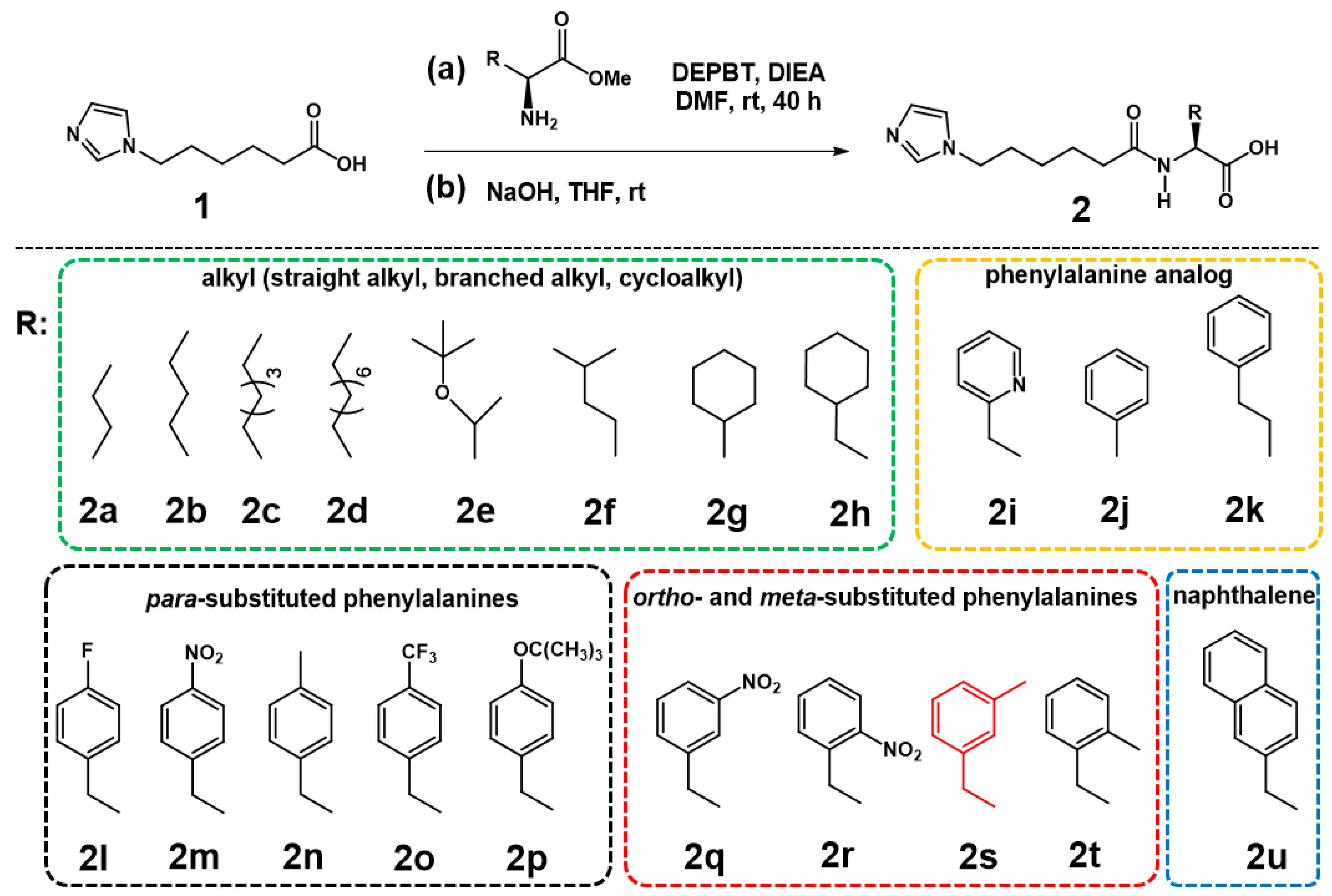
2.1. Design and Synthesis of the DFSMs Equipped with Unnatural Amino Acids
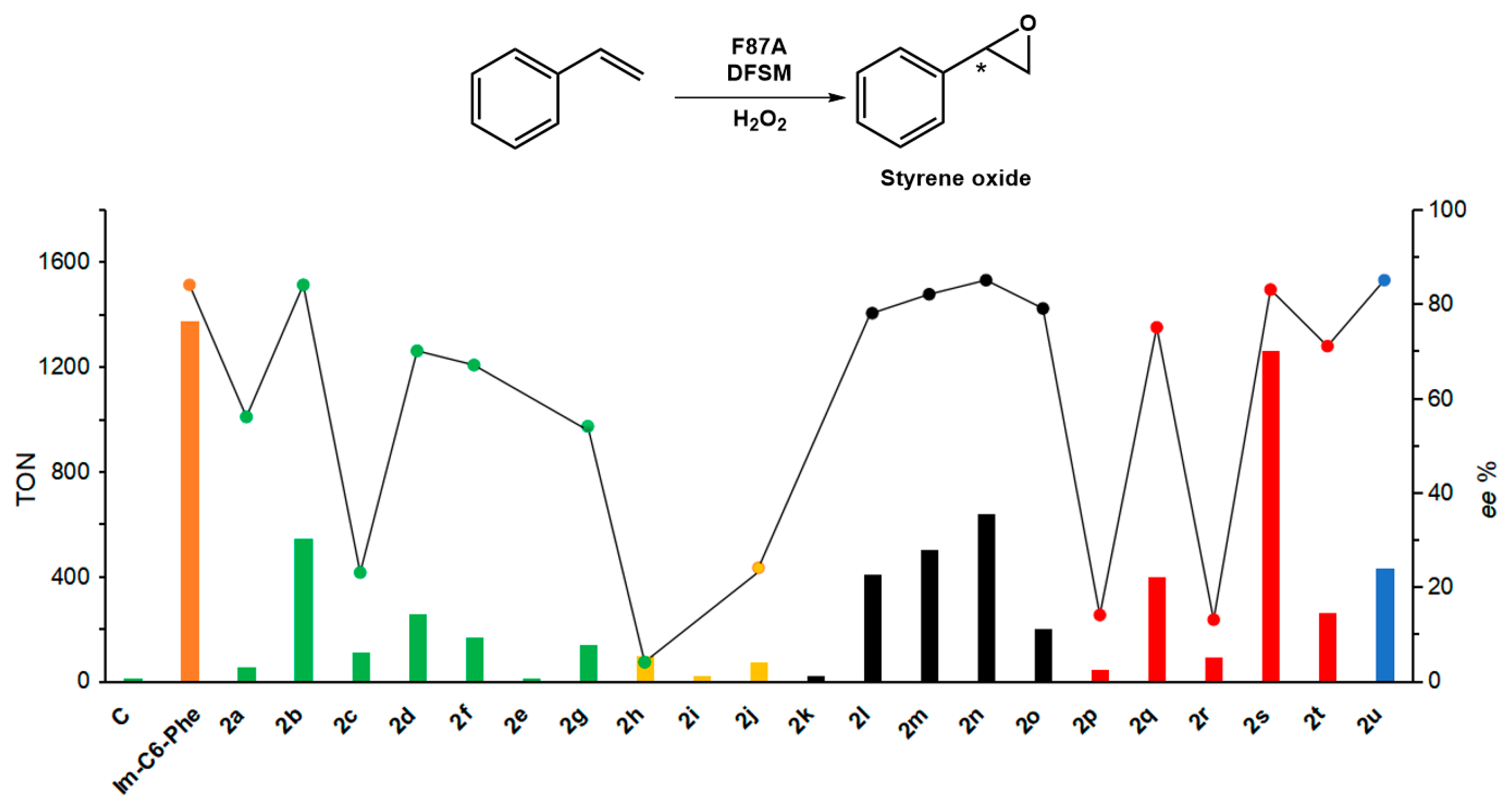
2.2. Effect of the DFSMs on the H2O2-Dependent Epoxidation of P450BM3
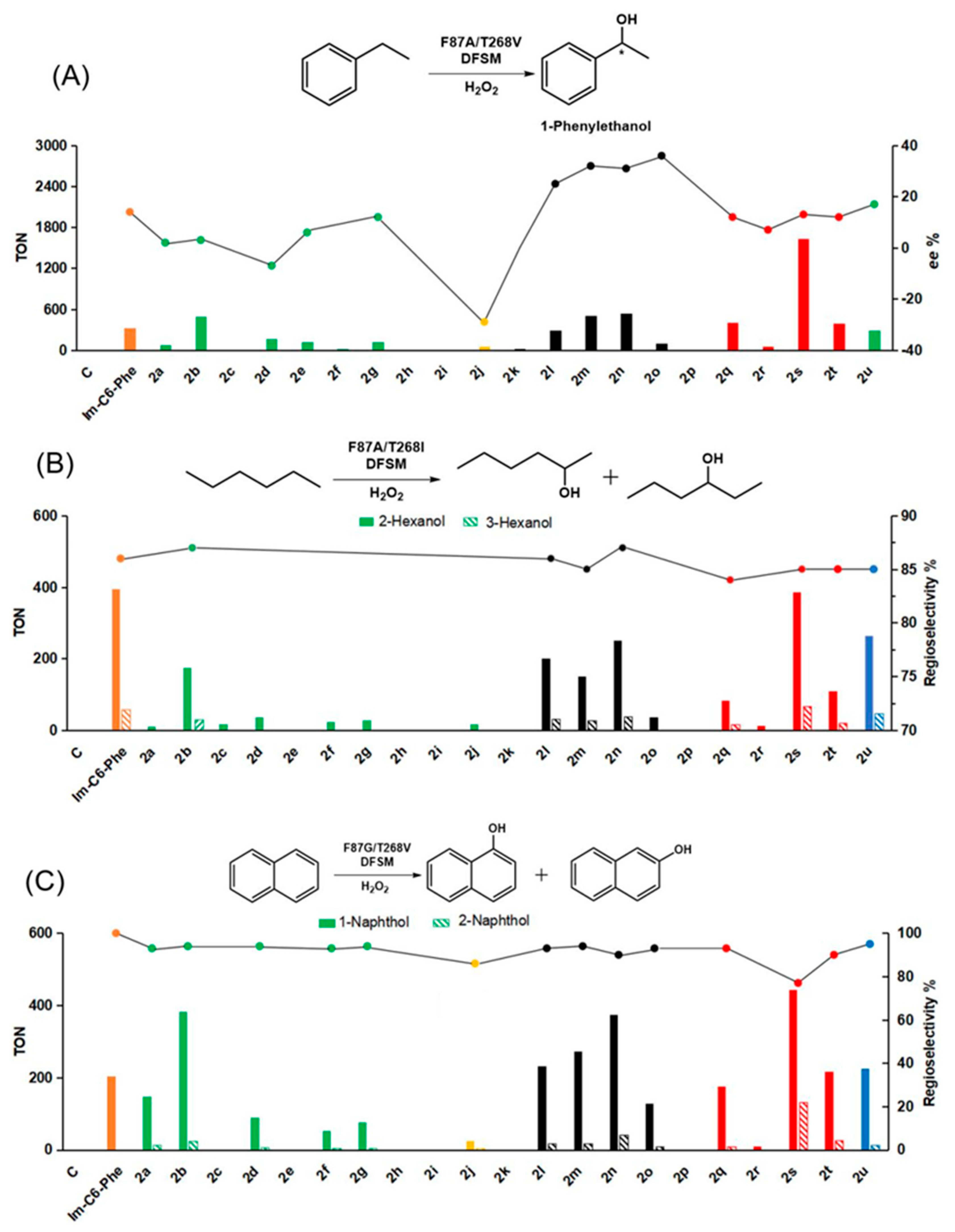
2.3. Effect of the DFSMs on the H2O2-Dependent Hydroxylation of P450BM3
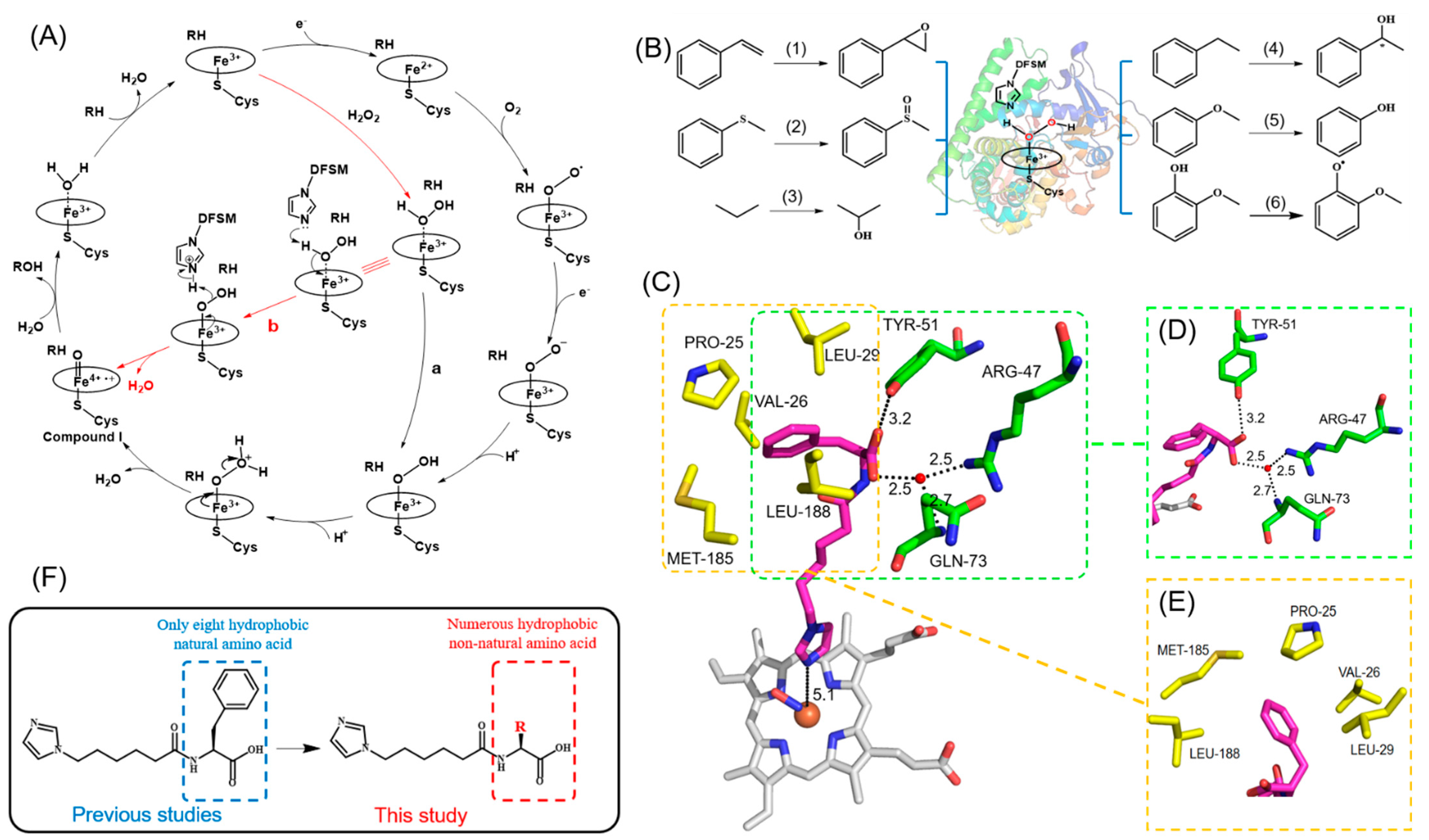
2.4. Molecular Docking Simulation
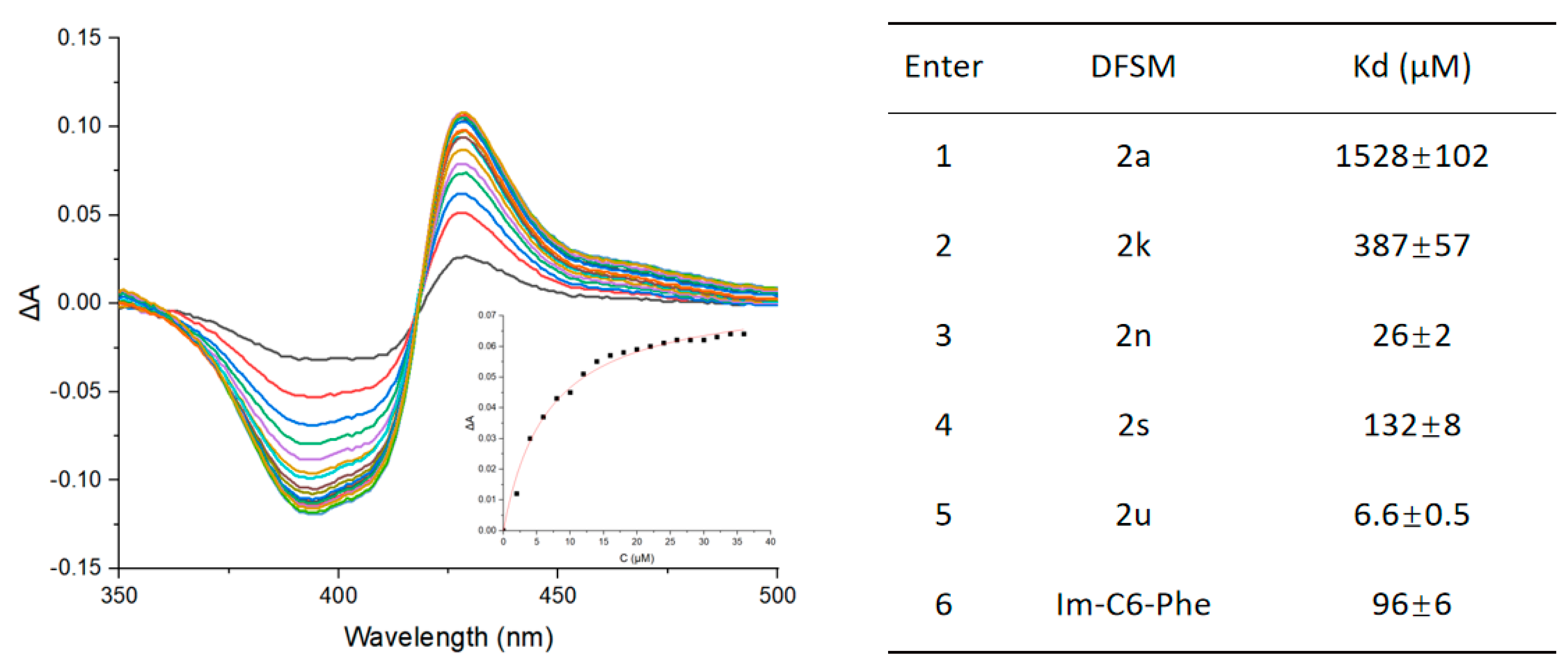
2.5. Dissociation Constant Measurement of DFSMs
3. Materials and Methods
3.1. General Procedure for the Synthesis of DFSMs (2s as an Example)
3.2. General Procedure for Epoxidation of Styrene
3.3. General Procedure for Hydroxylation of Ethylbenzene
3.4. General Procedure for Hydroxylation of n-Hexane
3.5. General Procedure for Hydroxylation of Naphthalene
3.6. Product Analysis
3.6.1. Gas Chromatography (GC)
3.6.2. Chiral Gas Chromatography
3.7. Docking Simulation of the DFSMs
3.8. Determination of Dissociation Constants of DFSMs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valikhani, D.; Bolivar, J.M.; Pelletier, J.N. An overview of cytochrome P450 immobilization strategies for drug metabolism studies, biosensing, and biocatalytic applications: Challenges and opportunities. ACS Catal. 2021, 11, 9418–9434. [Google Scholar] [CrossRef]
- Newmister, S.A.; Srivastava, K.R.; Espinoza, R.V.; Haatveit, K.C.; Khatri, Y.; Martini, R.M.; Garcia-Borràs, M.; Podust, L.M.; Houk, K.N.; Sherman, D.H. Molecular basis of iterative C─H oxidation by TamI, a multifunctional P450 monooxygenase from the tirandamycin biosynthetic pathway. ACS Catal. 2020, 10, 13445–13454. [Google Scholar] [CrossRef] [PubMed]
- Behrendorff, J.B.Y.H.; Gillam, E.M.J. Prospects for Applying Synthetic Biology to Toxicology: Future Opportunities and Current Limitations for the Repurposing of Cytochrome P450 Systems. Chem. Res. Toxicol. 2017, 30, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lei, T.; Shen, C.; Wang, Z.; Cao, D.; Hou, T. ADMET Evaluation in Drug Discovery. 19. Reliable Prediction of Human Cytochrome P450 Inhibition Using Artificial Intelligence Approaches. J. Chem. Inf. Model. 2019, 59, 4587–4601. [Google Scholar] [CrossRef]
- Steck, V.; Kolev, J.N.; Ren, X.; Fasan, R. Mechanism-Guided Design and Discovery of Efficient Cytochrome P450-Derived C-H Amination Biocatalysts. J. Am. Chem. Soc. 2020, 142, 10343–10357. [Google Scholar] [CrossRef]
- Arya, C.K.; Yadav, S.; Fine, J.; Casanal, A.; Chopra, G.; Ramanathan, G.; Vinothkumar, K.R.; Subramanian, R. A 2-Tyr-1-carboxylate Mononuclear Iron Center Forms the Active Site of a Paracoccus Dimethylformamidase. Angew. Chem. Int. Ed. 2020, 59, 16961–16966. [Google Scholar] [CrossRef]
- Mukherjee, G.; Satpathy, J.K.; Bagha, U.K.; Mubarak, M.Q.E.; Sastri, C.V.; de Visser, S.P. Inspiration from Nature: Influence of Engineered Ligand Scaffolds and Auxiliary Factors on the Reactivity of Biomimetic Oxidants. ACS Catal. 2021, 11, 9761–9797. [Google Scholar] [CrossRef]
- Peng, Y.; Gao, C.; Zhang, Z.; Wu, S.; Zhao, J.; Li, A. A Chemoenzymatic Strategy for the Synthesis of Steroid Drugs Enabled by P450 Monooxygenase-Mediated Steroidal Core Modification. ACS Catal. 2022, 12, 2907–29149. [Google Scholar] [CrossRef]
- Chen, H.; Huang, M.; Yan, W.; Bai, W.-J.; Wang, X. Enzymatic Regio- and Enantioselective C–H Oxyfunctionalization of Fatty Acids. ACS Catal. 2021, 11, 10625–10630. [Google Scholar] [CrossRef]
- Espinoza, R.V.; Haatveit, K.C.; Grossman, S.W.; Tan, J.Y.; McGlade, C.A.; Khatri, Y.; Newmister, S.A.; Schmidt, J.J.; Garcia-Borràs, M.; Montgomery, J.; et al. Engineering P450 TamI as an Iterative Biocatalyst for Selective Late-Stage C-H Functionalization and Epoxidation of Tirandamycin Antibiotics. ACS Catal. 2021, 11, 8304–8316. [Google Scholar] [CrossRef]
- Münch, J.; Püllmann, P.; Zhang, W.; Weissenborn, M.J. Enzymatic Hydroxylations of Sp3-Carbons. ACS Catal. 2021, 11, 9168–9203. [Google Scholar] [CrossRef]
- Li, F.; Renata, H. A Chiral-Pool-Based Strategy to Access Trans-Syn-Fused Drimane Meroterpenoids: Chemoenzymatic Total Syntheses of Polysin, N-Acetyl-Polyveoline and the Chrodrimanins. J. Am. Chem. Soc. 2021, 143, 18280–18286. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.C.; Lal, R.G.; Marchetti, L.A.; Arnold, F.H. Biocatalytic One-Carbon Ring Expansion of Aziridines to Azetidines via a Highly Enantioselective [1,2]-Stevens Rearrangement. J. Am. Chem. Soc. 2022, 144, 4739–4745. [Google Scholar] [CrossRef]
- Miller, D.C.; Athavale, S.V.; Arnold, F.H. Combining Chemistry and Protein Engineering for New-to-Nature Biocatalysis. Nat. Synth. 2022, 1, 18–23. [Google Scholar] [CrossRef]
- Nam, D.; Tinoco, A.; Shen, Z.; Adukure, R.D.; Sreenilayam, G.; Khare, S.D.; Fasan, R. Enantioselective Synthesis of α-Trifluoromethyl Amines via Biocatalytic N-H Bond Insertion with Acceptor-Acceptor Carbene Donors. J. Am. Chem. Soc. 2022, 144, 2590–2602. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, M.; Yonemura, K.; Stanfield, J.K.; Suzuki, K.; Shoji, O. Ein Designeraußenmembranprotein Fördert Die Aufnahme von Täuschmolekülen in Einen Auf Zytochrom P450BM3 Beruhenden Ganzzellbiokatalysator. Angew. Chem. Int. Ed. 2022, 61, e202111612. [Google Scholar] [CrossRef]
- Yang, Y.; Cho, I.; Qi, X.; Liu, P.; Arnold, F.H. An Enzymatic Platform for the Asymmetric Amination of Primary, Secondary and Tertiary C(Sp3)-H Bonds. Nat. Chem. 2019, 11, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Peng, W.; Li, Z.; You, C.; Zhao, Y.; Tang, D.; Wang, B.; Li, S. Unexpected Reactions of α,β-Unsaturated Fatty Acids Provide Insight into the Mechanisms of CYP152 Peroxygenases. Angew. Chem. Int. Ed. 2021, 60, 24694–24701. [Google Scholar] [CrossRef]
- Knorrscheidt, A.; Soler, J.; Hünecke, N.; Püllmann, P.; Garcia-Borràs, M.; Weissenborn, M.J. Accessing Chemo- and Regioselective Benzylic and Aromatic Oxidations by Protein Engineering of an Unspecific Peroxygenase. ACS Catal. 2021, 11, 7327–7338. [Google Scholar] [CrossRef]
- Pickl, M.; Kurakin, S.; Cantú Reinhard, F.G.; Schmid, P.; Pöcheim, A.; Winkler, C.K.; Kroutil, W.; de Visser, S.P.; Faber, K. Mechanistic Studies of Fatty Acid Activation by CYP152 Peroxygenases Reveal Unexpected Desaturase Activity. ACS Catal. 2019, 9, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, Y. Mechanistic Investigation of Isonitrile Formation Catalyzed by the Nonheme Iron/α-KG-Dependent Decarboxylase (ScoE). ACS Catal. 2020, 10, 2942–2957. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, S.; Chen, H.; Bai, W.J.; Wang, X. Directed Evolution of a Hydroxylase into a Decarboxylase for Synthesis of 1-Alkenes from Fatty Acids. ACS Catal. 2020, 10, 14375–14379. [Google Scholar] [CrossRef]
- Olmedo, A.; Aranda, C.; del Río, J.C.; Kiebist, J.; Scheibner, K.; Martínez, A.T.; Gutiérrez, A. From Alkanes to Carboxylic Acids: Terminal Oxygenation by a Fungal Peroxygenase. Angew. Chem. Int. Ed. 2016, 128, 12436–12439. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Peroxygenases En Route to Becoming Dream Catalysts. What Are the Opportunities and Challenges? Curr. Opin. Chem. Biol. 2017, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Michele, C.; Thangavelu, S.; Kim, M.K.; Gerrit, J.P. Enantiocomplementary Epoxidation Reactions Catalyzed by an Engineered Cofactor-Independent Non-natural Peroxygenase. Angew. Chem. Int. Ed. 2020, 132, 10460–10464. [Google Scholar] [CrossRef]
- Sigmund, M.-C.; Poelarends, G.J. Current State and Future Perspectives of Engineered and Artificial Peroxygenases for the Oxyfunctionalization of Organic Molecules. Nat. Catal. 2020, 3, 690–702. [Google Scholar] [CrossRef]
- Gomez de Santos, P.; Lazaro, S.; Viña-Gonzalez, J.; Hoang, M.D.; Sánchez-Moreno, I.; Glieder, A.; Hollmann, F.; Alcalde, M. Evolved Peroxygenase–Aryl Alcohol Oxidase Fusions for Self-Sufficient Oxyfunctionalization Reactions. ACS Catal. 2020, 10, 13524–13534. [Google Scholar] [CrossRef]
- Grogan, G. Hemoprotein Catalyzed Oxygenations: P450s, UPOs, and Progress toward Scalable Reactions. JACS Au 2021, 1, 1312–1329. [Google Scholar] [CrossRef]
- Shoji, O.; Watanabe, Y. Peroxygenase Reactions Catalyzed by Cytochromes P450. J. Biol. Inorg. Chem. 2014, 19, 529–539. [Google Scholar] [CrossRef]
- Yu, D.; Wang, J.-B.; Reetz, M.T. Exploiting Designed Oxidase-Peroxygenase Mutual Benefit System for Asymmetric Cascade Reactions. J. Am. Chem. Soc. 2019, 141, 5655–5658. [Google Scholar] [CrossRef] [Green Version]
- Coleman, T.; Kirk, A.M.; Lee, J.H.Z.; Doherty, D.Z.; Bruning, J.B.; Krenske, E.H.; De Voss, J.J.; Bell, S.G. Different Geometric Requirements for Cytochrome P450-Catalyzed Aliphatic versus Aromatic Hydroxylation Results in Chemoselective Oxidation. ACS Catal. 2022, 12, 1258–1267. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.-L. P450(BM3) (CYP102A1): Connecting the Dots. Chem. Soc. Rev. 2012, 41, 1218–1260. [Google Scholar] [CrossRef]
- Ma, N.; Chen, Z.; Chen, J.; Chen, J.; Wang, C.; Zhou, H.; Yao, L.; Shoji, O.; Watanabe, Y.; Cong, Z. Dual-Functional Small Molecules for Generating an Efficient Cytochrome P450BM3 Peroxygenase. Angew. Chem. Int. Ed. 2018, 130, 7754–7759. [Google Scholar] [CrossRef]
- Xu, J.; Wang, C.; Cong, Z. Strategies for Substrate-regulated P450 Catalysis: From Substrate Engineering to Co-catalysis. Chem. Eur. J. 2019, 25, 6853–6863. [Google Scholar] [CrossRef] [PubMed]
- Di, S.; Fan, S.; Jiang, F.; Cong, Z. A Unique P450 Peroxygenase System Facilitated by a Dual-Functional Small Molecule: Concept, Application, and Perspective. Antioxidants 2022, 11, 529. [Google Scholar] [CrossRef]
- Zhao, P.; Chen, J.; Ma, N.; Chen, J.; Qin, X.; Liu, C.; Yao, F.; Yao, L.; Jin, L.; Cong, Z. Enabling Highly (R)-Enantioselective Epoxidation of Styrene by Engineering Unique Non-Natural P450 Peroxygenases. Chem. Sci. 2021, 12, 6307–6314. [Google Scholar] [CrossRef]
- Chen, J.; Kong, F.; Ma, N.; Zhao, P.; Liu, C.; Wang, X.; Cong, Z. Peroxide-Driven Hydroxylation of Small Alkanes Catalyzed by an Artificial P450BM3 Peroxygenase System. ACS Catal. 2019, 9, 7350–7355. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, C.; Ma, N.; Chen, J.; Liu, C.; Wang, F.; Xu, J.; Cong, Z. Regioselective Aromatic O-Demethylation with an Artificial P450BM3 Peroxygenase System. Catal. Sci. Technol. 2020, 10, 1219–1223. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Ma, N.; Zhou, H.; Cong, Z. Selective Hydroxylation of Naphthalene Using the H2O2-Dependent Engineered P450BM3 Driven by Dual-Functional Small Molecules. J. Porphyr. Phthalocyanines 2018, 22, 831–883. [Google Scholar] [CrossRef]
- Ma, N.; Fang, W.; Liu, C.; Qin, X.; Wang, X.; Jin, L.; Wang, B.; Cong, Z. Switching an Artificial P450 Peroxygenase into Peroxidase via Mechanism-Guided Protein Engineering. ACS Catal. 2021, 11, 8449–8455. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, Y.; Chen, Q.; Dong, S.; Feng, Y.; Cong, Z.; Shaik, S.; Wang, B. H-Bonding Networks Dictate the Molecular Mechanism of H2O2 Activation by P450. ACS Catal. 2021, 11, 8774–8785. [Google Scholar] [CrossRef]
- Cong, Z.; Shoji, O.; Kasai, C.; Kawakami, N.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. Activation of Wild-Type Cytochrome P450BM3 by the next Generation of Decoy Molecules: Enhanced Hydroxylation of Gaseous Alkanes and Crystallographic Evidence. ACS Catal. 2015, 5, 150–156. [Google Scholar] [CrossRef]
- Yonemura, K.; Ariyasu, S.; Stanfield, J.K.; Suzuki, K.; Onoda, H.; Kasai, C.; Sugimoto, H.; Aiba, Y.; Watanabe, Y.; Shoji, O. Systematic Evolution of Decoy Molecules for the Highly Efficient Hydroxylation of Benzene and Small Alkanes Catalyzed by Wild-Type Cytochrome P450BM3. ACS Catal. 2020, 10, 9136–9144. [Google Scholar] [CrossRef]
- Karasawa, M.; Stanfield, J.K.; Yanagisawa, S.; Shoji, O.; Watanabe, Y. Whole-Cell Biotransformation of Benzene to Phenol Catalysed by Intracellular Cytochrome P450BM3 Activated by External Additives. Angew. Chem. Int. Ed. 2018, 57, 12264–12269. [Google Scholar] [CrossRef] [PubMed]
- Willot, S.J.P.; Tieves, F.; Girhard, M.; Urlacher, V.B.; Hollmann, F.; de Gonzalo, G. P450BM3-Catalyzed Oxidations Employing Dual Functional Small Molecules. Catalysts. 2019, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.; Zhang, L.; Kryza, T.; Neodo, A.; Bock, N.; De Vita, E.; Williams, E.D.; Engelsberger, E.; Xu, C.; Bakker, A.T.; et al. A Suite of Activity-Based Probes to Dissect the KLK Activome in Drug-Resistant Prostate Cancer. J. Am. Chem. Soc. 2021, 143, 8911–8924. [Google Scholar] [CrossRef] [PubMed]
- Adaligil, E.; Song, A.; Cunningham, C.N.; Fairbrother, W.J. Ribosomal Synthesis of Macrocyclic Peptides with Linear γ4- and β-Hydroxy-γ4-Amino Acids. ACS Chem. Biol. 2021, 16, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, T.M.; Rouget-Virbel, C.; Rincón, J.A.; Barberis, M.; Mateos, C.; García-Cerrada, S.; Agejas, J.; de Frutos, O.; MacMillan, D.W.C. Synthesis of Enantiopure Unnatural Amino Acids by Metallaphotoredox Catalysis. Org. Process Res. Dev. 2021, 25, 1966–1973. [Google Scholar] [CrossRef]
- Hu, L.; Xu, S.; Zhao, Z.; Yang, Y.; Peng, Z.; Yang, M.; Wang, C.; Zhao, J. Ynamides as Racemization-Free Coupling Reagents for Amide and Peptide Synthesis. J. Am. Chem. Soc. 2016, 138, 13135–13138. [Google Scholar] [CrossRef]
- Oleg, T.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Effificient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DFSM | Score (kcal/mol) | L (Fe-N) (Å) | DFSM | Score (kcal/mol) | L (Fe-N) (Å) |
|---|---|---|---|---|---|
| 2a | −5.9 | 5.7 | 2l | −7.2 | 5.6 |
| 2b | −6.6 | 5.1 | 2m | −7.1 | 5.0 |
| 2c | −6.1 | 5.6 | 2n | −7.3 | 5.0 |
| 2d | −6.4 | 5.6 | 2o | −6.6 | 5.6 |
| 2e | −6.8 | 5.9 | 2p | −6.3 | 5.5 |
| 2f | −6.4 | 5.6 | 2q | −7.4 | 5.1 |
| 2g | −6.9 | 5.6 | 2r | −6.9 | 5.7 |
| 2h | −6.9 | 5.8 | 2s | −7.7 | 4.9 |
| 2i | −6.5 | 6.2 | 2t | −7.0 | 5.7 |
| 2j | −6.7 | 5.6 | 2u | −7.4 | 5.1 |
| 2k | −6.9 | 7.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, X.; Jiang, Y.; Chen, J.; Yao, F.; Zhao, P.; Jin, L.; Cong, Z. Co-Crystal Structure-Guided Optimization of Dual-Functional Small Molecules for Improving the Peroxygenase Activity of Cytochrome P450BM3. Int. J. Mol. Sci. 2022, 23, 7901. https://doi.org/10.3390/ijms23147901
Qin X, Jiang Y, Chen J, Yao F, Zhao P, Jin L, Cong Z. Co-Crystal Structure-Guided Optimization of Dual-Functional Small Molecules for Improving the Peroxygenase Activity of Cytochrome P450BM3. International Journal of Molecular Sciences. 2022; 23(14):7901. https://doi.org/10.3390/ijms23147901
Chicago/Turabian StyleQin, Xiangquan, Yiping Jiang, Jie Chen, Fuquan Yao, Panxia Zhao, Longyi Jin, and Zhiqi Cong. 2022. "Co-Crystal Structure-Guided Optimization of Dual-Functional Small Molecules for Improving the Peroxygenase Activity of Cytochrome P450BM3" International Journal of Molecular Sciences 23, no. 14: 7901. https://doi.org/10.3390/ijms23147901
APA StyleQin, X., Jiang, Y., Chen, J., Yao, F., Zhao, P., Jin, L., & Cong, Z. (2022). Co-Crystal Structure-Guided Optimization of Dual-Functional Small Molecules for Improving the Peroxygenase Activity of Cytochrome P450BM3. International Journal of Molecular Sciences, 23(14), 7901. https://doi.org/10.3390/ijms23147901