
Comparative Transcriptomic Analysis of Root and Leaf Transcript Profiles Reveals the Coordinated Mechanisms in Response to Salinity Stress in Common Vetch




Abstract
:1. Introduction
2. Results
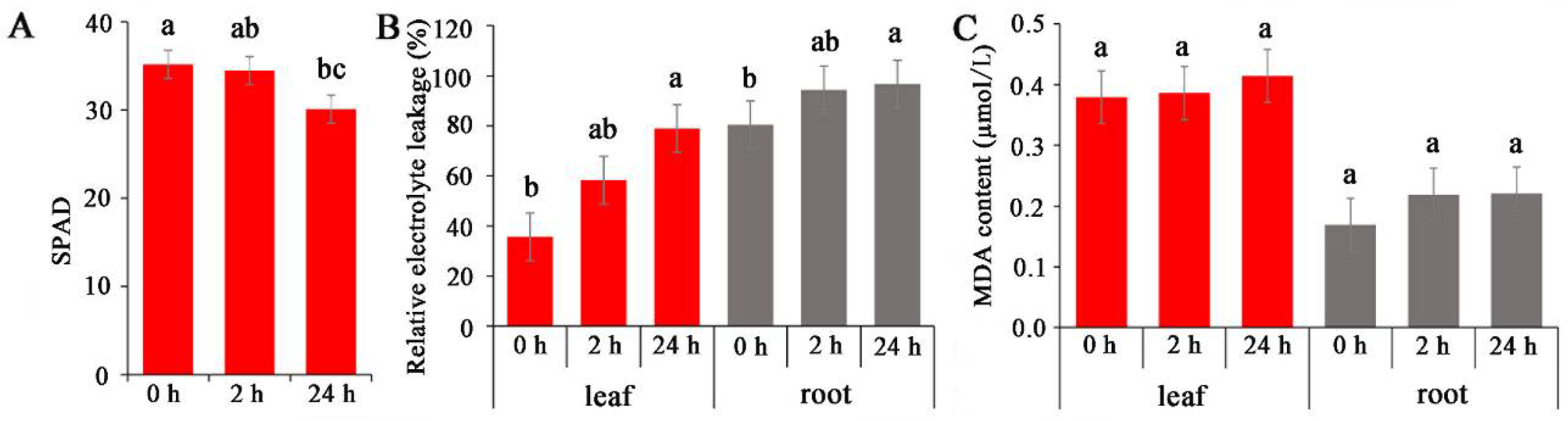
2.1. Physiological Characteristics of Common Vetch under Salinity Stress
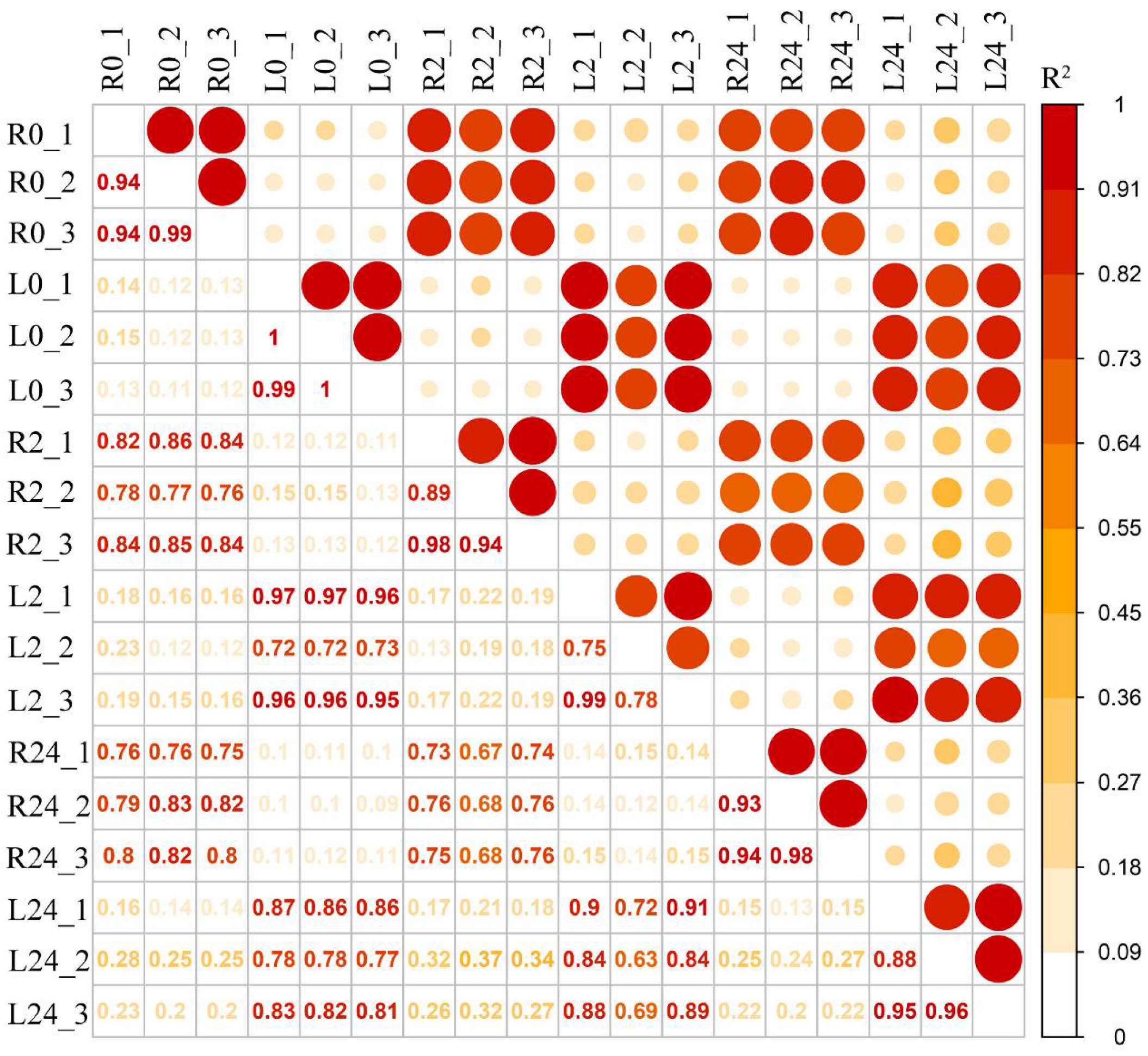
2.2. Transcriptome Assembly and Annotation of Transcripts under Salinity Stress
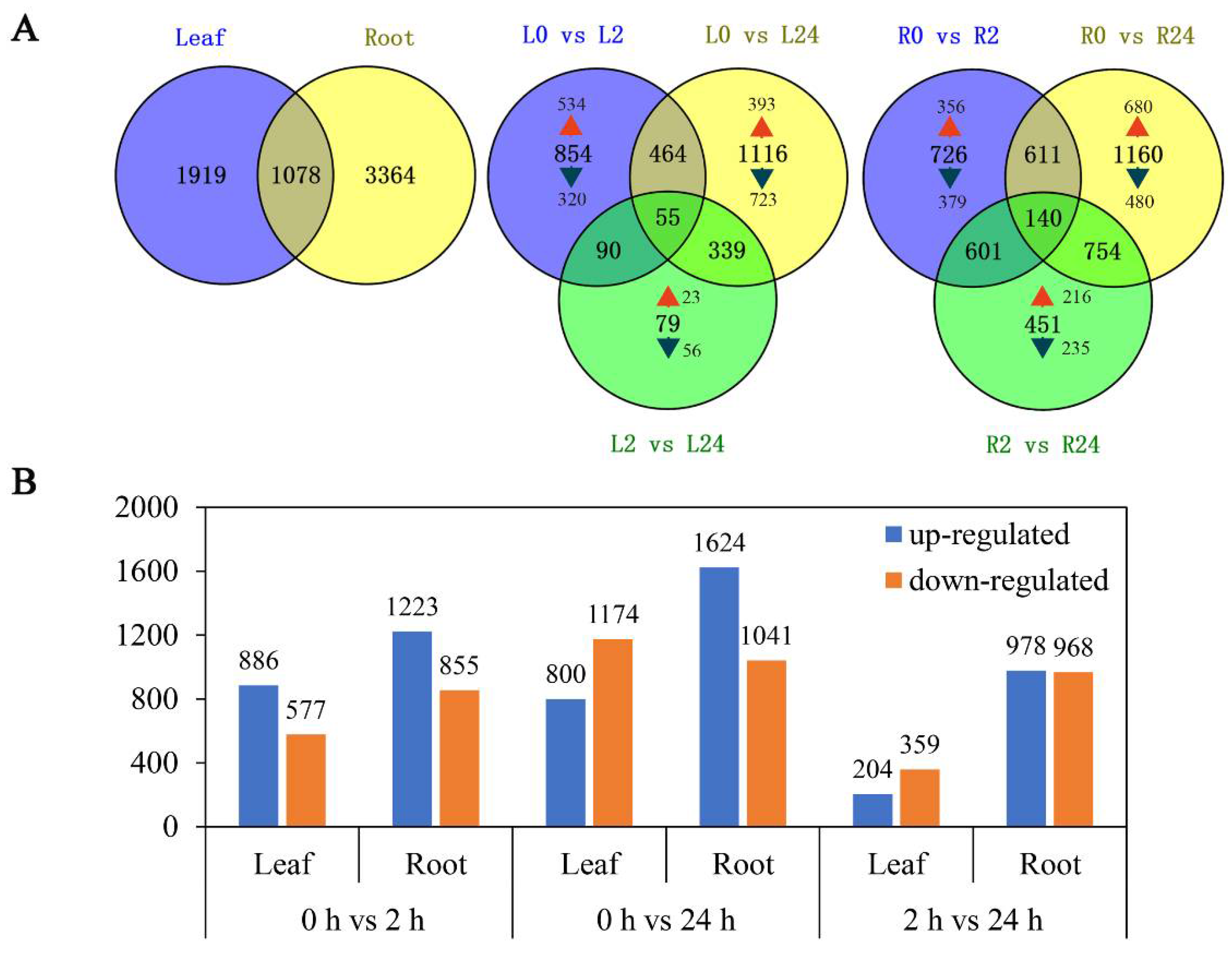
2.3. Identification and Analysis of Genes Differentially Expressed in Response to Salinity Stress
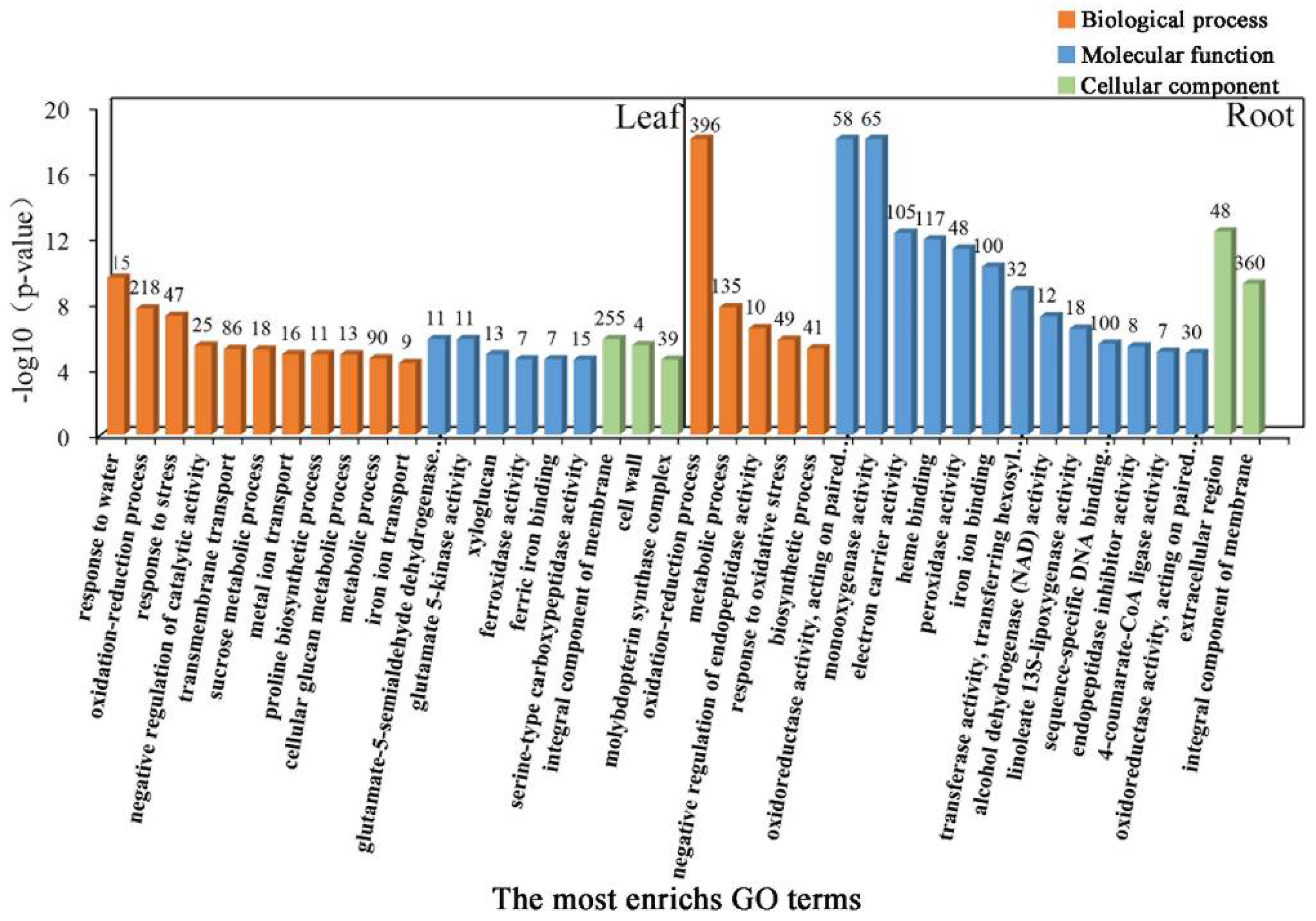
2.4. GO Enrichment Analysis
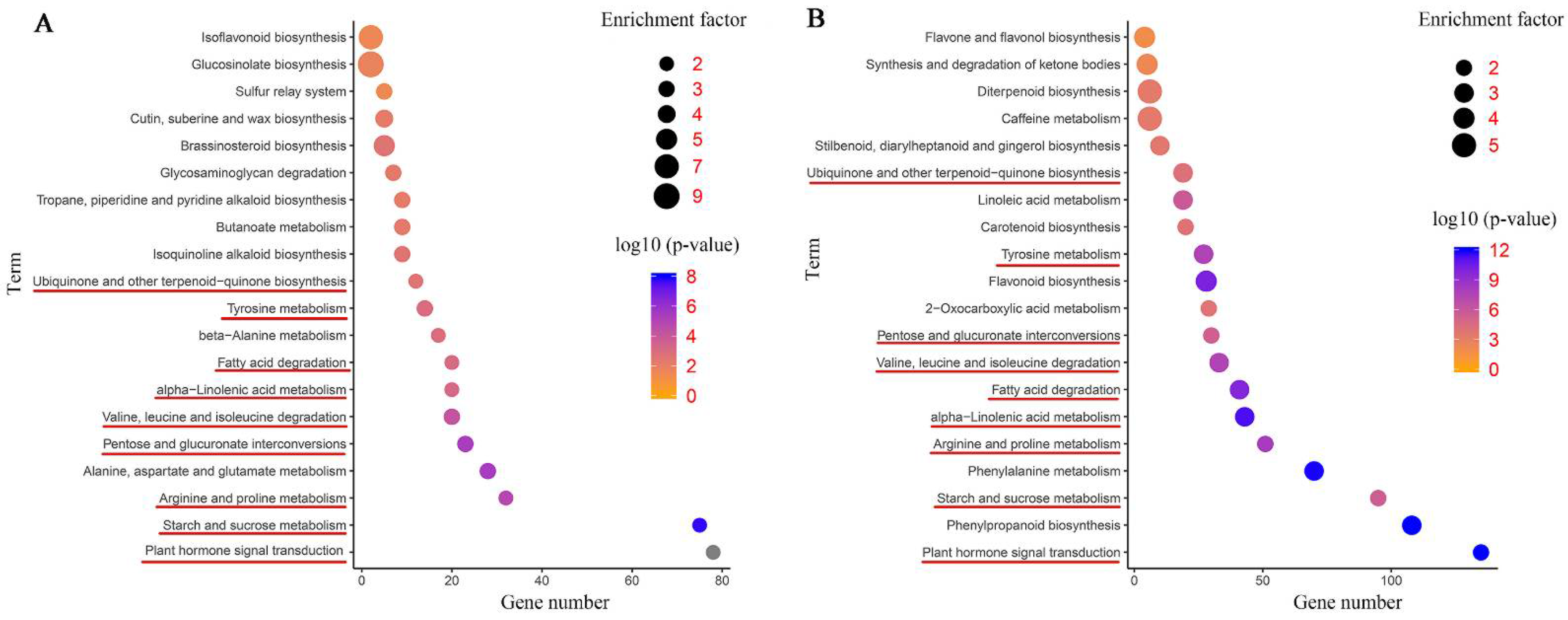
2.5. KEGG Pathway Enrichment Analysis
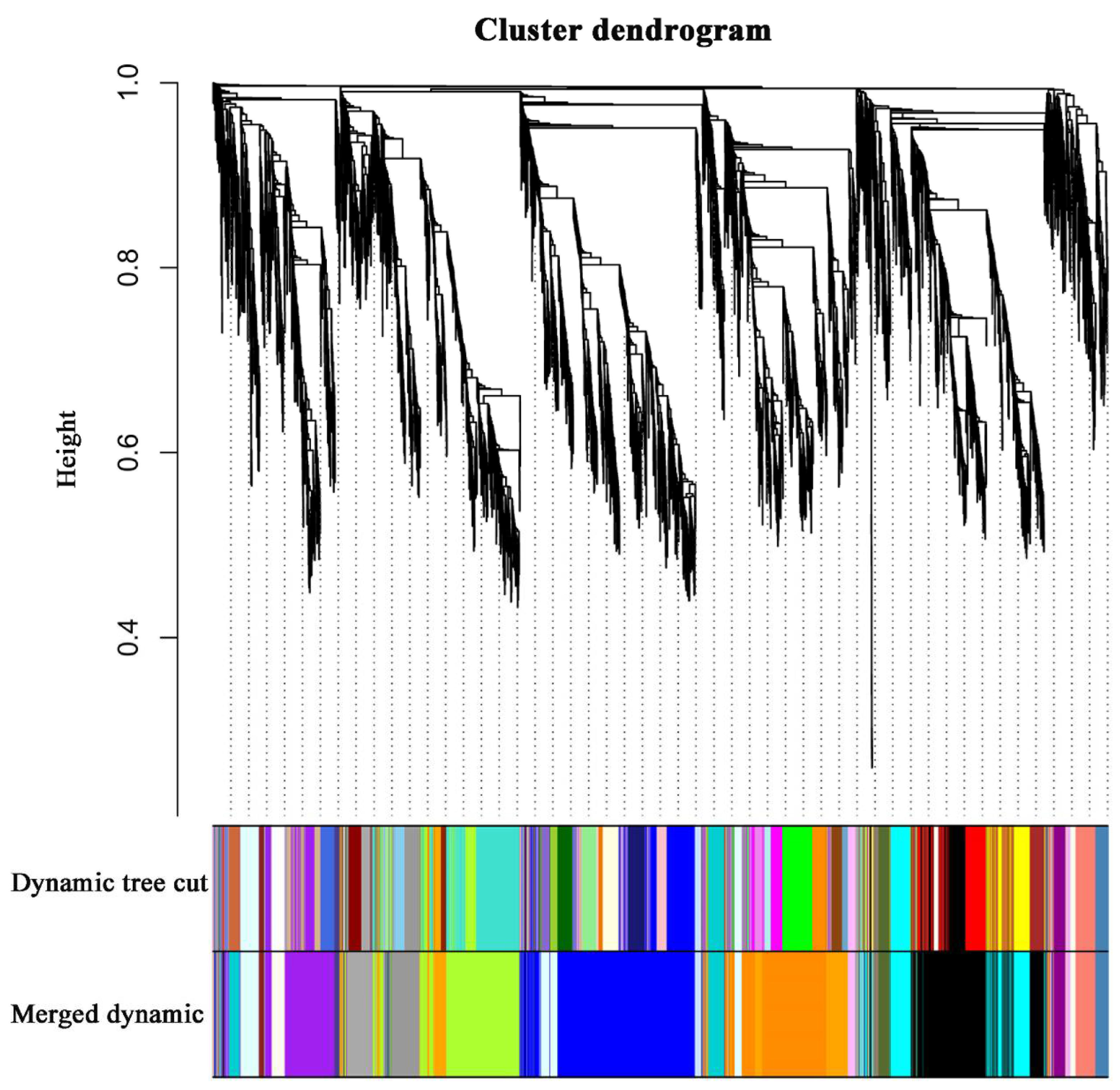
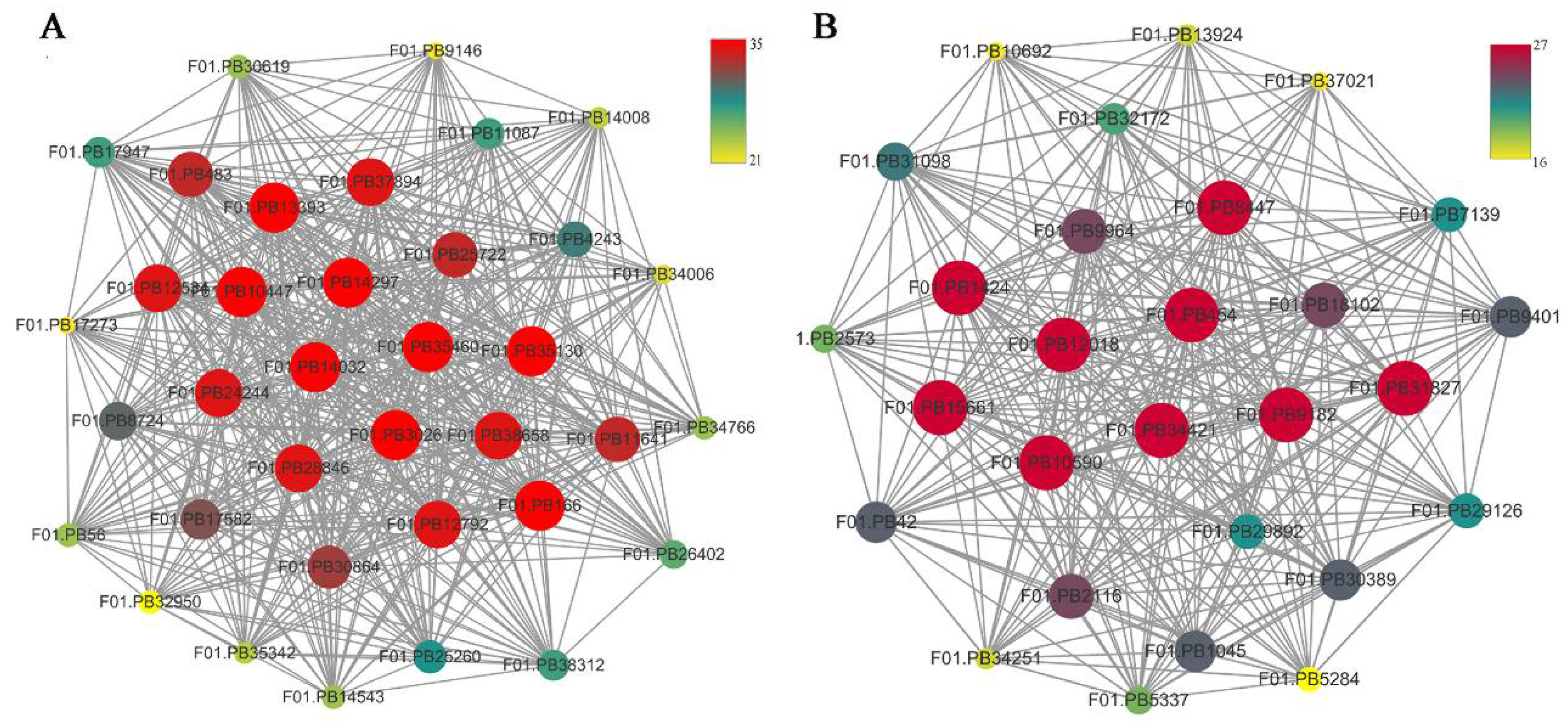
2.6. WGCNA and Identification of Key Genes
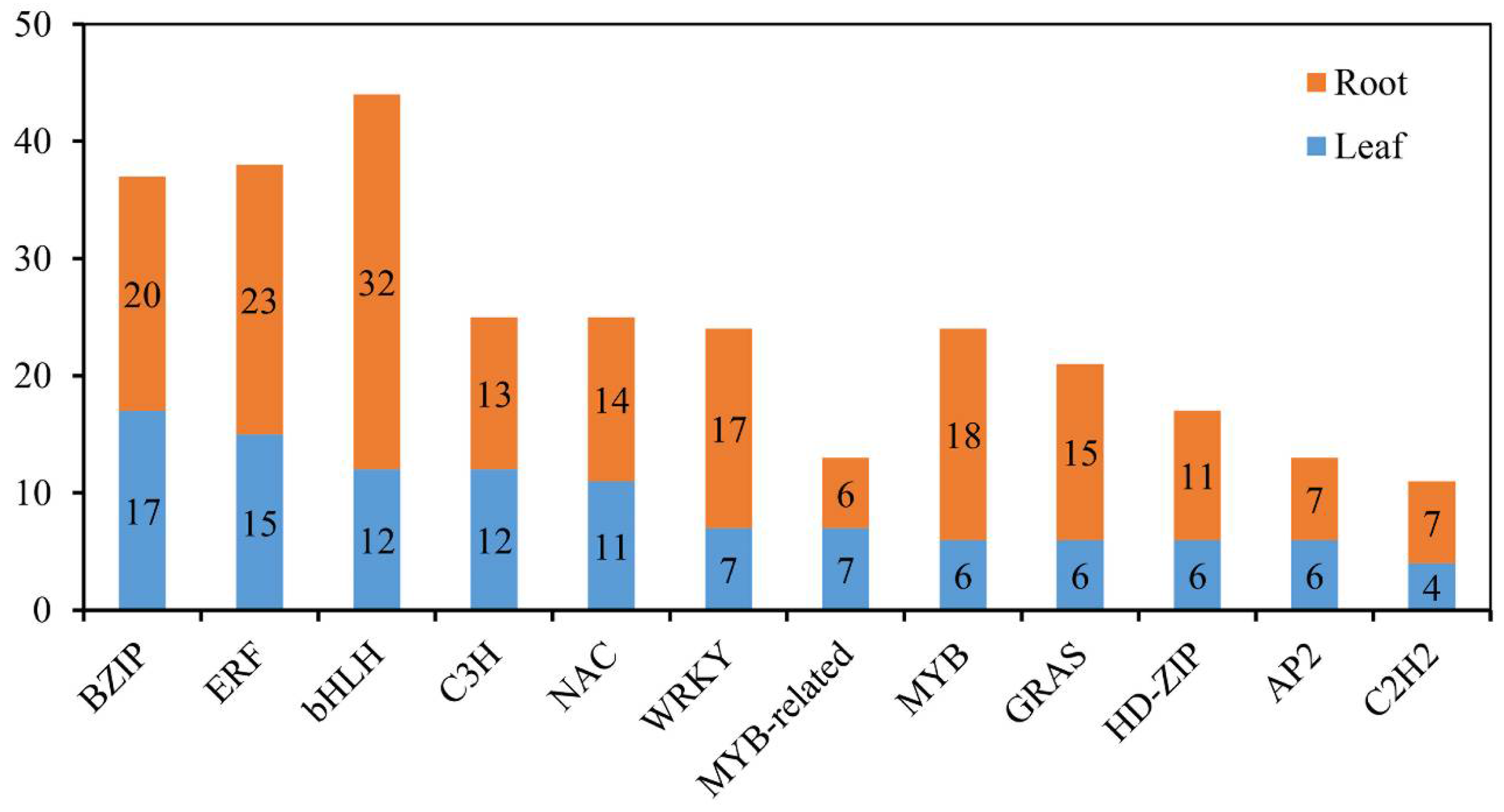
2.7. Identification of TFs in Response to Salinity Stress
2.8. Validation of Gene Expression by qRT-PCR
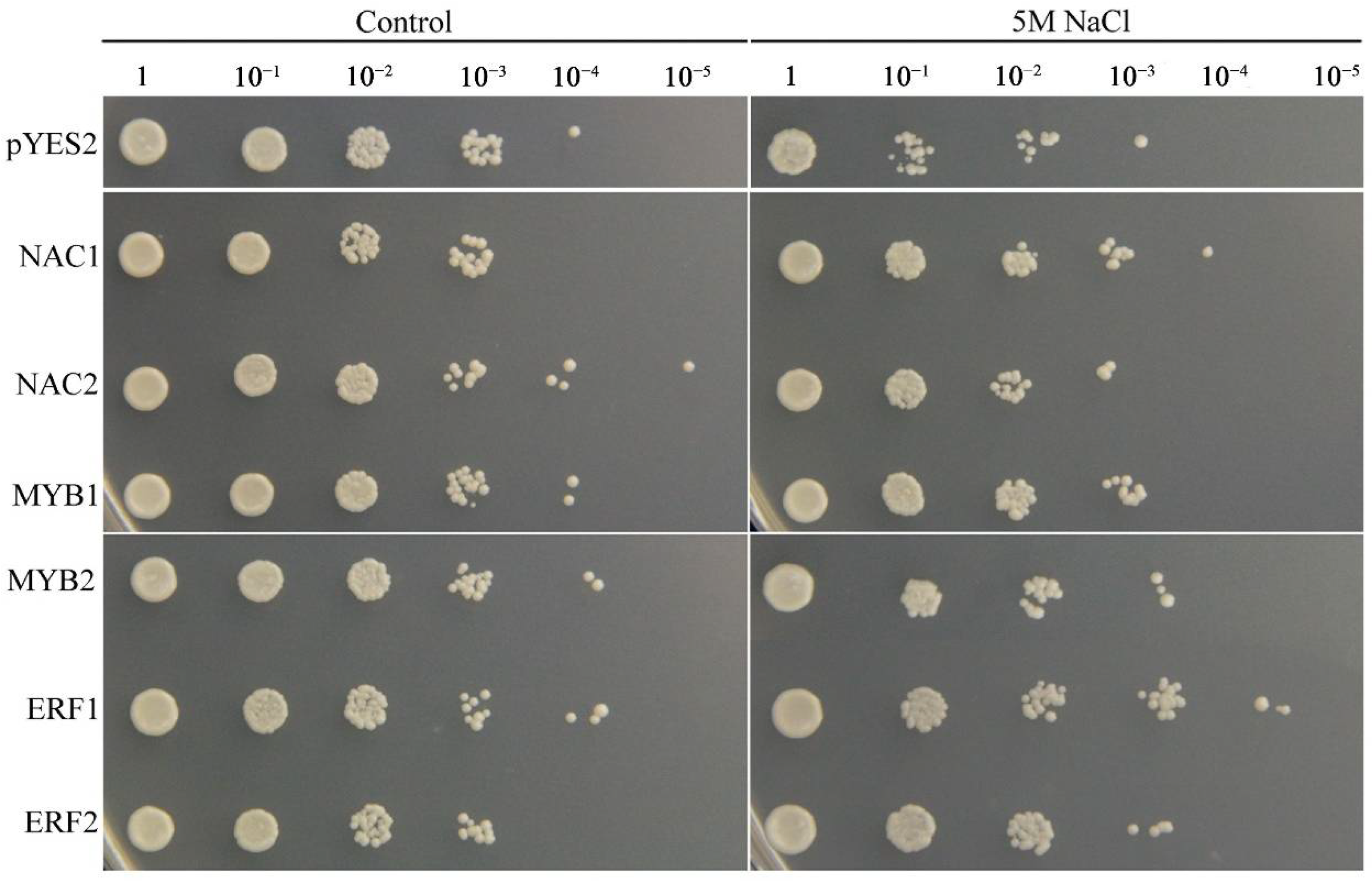
2.9. Functional Confirmation of TFs in Transgenic Yeast
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Physiological Measurements
4.3. RNA Isolation, Library Construction, and Sequencing
4.3.1. RNA Isolation
4.3.2. PacBio Library Construction and Sequencing
4.3.3. Illumina HiSeq Library Construction and Sequencing
4.3.4. Mapping Reads from the PacBio Library and Annotation Analysis
4.4. Identification of Differentially Expressed Genes (DEGs)
4.5. qRT-PCR Analysis
4.6. Salinity Tolerance Tests of Transgenic Yeast
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, P.; Abdel Latef, A.A.; Hashem, A.; Abd Allah, E.F.; Gucel, S.; Tran, L.S. Nitric Oxide Mitigates Salt Stress by Regulating Levels of Osmolytes and Antioxidant Enzymes in Chickpea. Front. Plant Sci. 2016, 7, 347. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Deinlein, U.; Stephan, A.B.; Horie, T.; Luo, W.; Xu, G.; Schroeder, J.I. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014, 19, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Steinhorst, L.; Kudla, J. Calcium and Reactive Oxygen Species Rule the Waves of Signaling. Plant Physiol. 2013, 163, 471–485. [Google Scholar] [CrossRef] [Green Version]
- Kurusu, T.; Kuchitsu, K.; Tada, Y. Plant signaling networks involving Ca2+ and Rboh/Nox-mediated ROS production under salinity stress. Front. Plant Sci. 2015, 6, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ma, M.; Feng, Y.; Li, H.; Wang, Y.; Ma, Y.; Li, M.; An, F.; Guo, H. EIN2-Directed Translational Regulation of Ethylene Signaling in Arabidopsis. Cell 2015, 163, 670–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Song, L.; Li, K.; Zhang, X.; Liu, S.; Qin, Y.; Li, P. Polysaccharides from Grateloupia filicina enhance tolerance of rice seeds (Oryza sativa L.) under salt stress. Int. J. Biol. Macromol. 2018, 124, 1197–1204. [Google Scholar] [CrossRef]
- Makavitskaya, M.; Svistunenko, D.; Navaselsky, I.; Hryvusevich, P.; Mackievic, V.; Rabadanova, C.; Tyutereva, E.; Samokhina, V.; Straltsova, D.; Sokolik, A.; et al. Novel roles of ascorbate in plants: Induction of cytosolic Ca2+ signals and efflux from cells via anion channels. J. Exp. Bot. 2018, 69, 3477–3489. [Google Scholar] [CrossRef]
- Abdelrahman, M.; Jogaiah, S.; Burritt, D.J.; Tran, L.-S.P. Legume genetic resources and transcriptome dynamics under abiotic stress conditions. Plant Cell Environ. 2018, 41, 1972–1983. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Zheng, Z.; Grumet, R.; Loescher, W.; Zhu, J.-K.; Yang, P.; Hu, Y.; Chan, Z. Transcriptomic and Physiological Variations of Three Arabidopsis Ecotypes in Response to Salt Stress. PLoS ONE 2013, 8, e69036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhu, J.; Zhang, Y.; Fan, F.; Li, W.; Wang, F.; Zhong, W.; Wang, C.; Yang, J. Comparative transcriptome analysis reveals molecular response to salinity stress of salt-tolerant and sensitive genotypes of indica rice at seedling stage. Sci. Rep. 2018, 8, 2085. [Google Scholar] [CrossRef] [Green Version]
- Villarino, G.H.; Bombarely, A.; Giovannoni, J.J.; Scanlon, M.J.; Mattson, N.S. Transcriptomic Analysis of Petunia hybrida in Response to Salt Stress Using High Throughput RNA Sequencing. PLoS ONE 2014, 9, e94651. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, Y.; Luo, C.; Xiang, Y.; An, L. De Novo Transcriptome Sequencing of Desert Herbaceous Achnatherum splendens (Achnatherum) Seedlings and Identification of Salt Tolerance Genes. Genes 2016, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jiang, D.; Liu, B.; Luo, W.; Lu, J.; Ma, T.; Wan, D. Transcriptome dynamics of a desert poplar (Populus pruinosa) in response to continuous salinity stress. Plant Cell Rep. 2014, 33, 1565–1579. [Google Scholar] [CrossRef]
- Khadka, V.S.; Vaughn, K.; Xie, J.; Swaminathan, P.; Ma, Q.; Cramer, G.R.; Fennell, A.Y. Transcriptomic response is more sensitive to water deficit in shoots than roots of Vitis riparia (Michx.). BMC Plant Biol. 2019, 19, 72. [Google Scholar] [CrossRef]
- Tian, X.; Wang, Z.; Zhang, Q.; Ci, H.; Wang, P.; Yu, L.; Jia, G. Genome-wide transcriptome analysis of the salt stress tolerance mechanism in Rosa chinensis. PLoS ONE 2018, 13, e0200938. [Google Scholar] [CrossRef]
- Yaish, M.W.; Patankar, H.V.; Assaha, D.V.M.; Zheng, Y.; Al-Yahyai, R.; Sunkar, R. Genome-wide expression profiling in leaves and roots of date palm (Phoenix dactylifera L.) exposed to salinity. BMC Genom. 2017, 18, 246. [Google Scholar] [CrossRef] [Green Version]
- Rihawi, S.; Iñiguez, L.; Knaus, W.; Zaklouta, M.; Wurzinger, M.; Soelkner, J.; Larbi, A.; Bomfim, M. Fattening performance of lambs of different Awassi genotypes, fed under cost-reducing diets and contrasting housing conditions. Small Rumin. Res. 2010, 94, 38–44. [Google Scholar] [CrossRef]
- Garduño-Castro, Y.; Espinoza-Ortega, A.; Gonzalez-Esquivel, C.; Mateo-Salazar, B.; Arriaga-Jordán, C.M. Intercropped oats (Avena sativa)—Common vetch (Vicia sativa) silage in the dry season for small-scale dairy systems in the Highlands of Central Mexico. Trop. Anim. Health Prod. 2008, 41, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Henman, D.; King, R.; Dunshea, F. Common vetch (Vicia sativa cv Morava) is an alternative protein source in pig diets. Asia Pac. J. Clin. Nutr. 2002, 11, S249. [Google Scholar]
- Seymour, M.; Siddique, K.; Brandon, N.; Martin, L.; Jackson, E. Response of vetch (Vicia spp.) to plant density in south-western Australia. Aust. J. Exp. Agric. 2002, 42, 1043–1051. [Google Scholar] [CrossRef]
- Min, X.; Liu, Z.; Wang, Y.; Liu, W. Products, comparative transcriptomic analysis provides insights into the coordinated mechanisms of leaves and roots response to cold stress in Common Vetch. Ind. Crops Prod. 2020, 158, 112949. [Google Scholar] [CrossRef]
- Orak, A.; Ates, E. Resistance to salinity stress and available water levels at the seedling stage of the common vetch (Vicia sativa L.). Plant Soil Environ. 2011, 51, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Bilgili, U.; Carpici, E.B.; Aşik, B.B.; Celik, N. Root and shoot response of common vetch (Vicia sativa L.), forage pea (Pisum sativum L.) and canola (Brassica napus L.) to salt stress during early se. Turk. J. Field Crops 2011, 16, 33–38. [Google Scholar]
- Aydinoğlu, B.; Shabani, A.; Safavi, S.M. Impact of priming on seed germination, seedling growth and gene expression in common vetch under salinity stress. Cell. Mol. Biol. 2019, 65, 18–24. [Google Scholar] [CrossRef]
- Duarte, B.; Santos, D.; Marques, J.C.; Caçador, I. Ecophysiological adaptations of two halophytes to salt stress: Photosynthesis, PS II photochemistry and anti-oxidant feedback—Implications for resilience in climate change. Plant Physiol. Biochem. 2013, 67, 178–188. [Google Scholar] [CrossRef]
- Panda, S.; Upadhyay, R. Salt Stress Injury Induces Oxidative Alterations and Antioxidative Defence in the Roots of Lemna minor. Biol. Plant. 2004, 48, 249–253. [Google Scholar] [CrossRef]
- Du, X.; Wang, G.; Ji, J.; Shi, L.; Guan, C.; Jin, C. Comparative transcriptome analysis of transcription factors in different maize varieties under salt stress conditions. Plant Growth Regul. 2016, 81, 183–195. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Q.; Xu, W.; Zhang, J.; Wang, X.; Nie, G.; Yao, L.; Wang, H.; Lin, C. De Novo Assembly and Discovery of Genes That Involved in Drought Tolerance in the Common Vetch. Int. J. Mol. Sci. 2019, 20, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-León, E.; López-Moreno, F.J.; Atero-Calvo, S.; Albacete, A.; Ruiz, J.M.; Blasco, B. CAX1a TILLING Mutations Modify the Hormonal Balance Controlling Growth and Ion Homeostasis in Brassica rapa Plants Subjected to Salinity. Agronomy 2020, 10, 1699. [Google Scholar] [CrossRef]
- Dong, L.; Lu, Y.; Liu, S. Genome-wide member identification, phylogeny and expression analysis of PEBP gene family in wheat and its progenitors. PeerJ 2020, 8, e10483. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lan, H.; Huang, P.; Zhang, Y.; Yuan, X.; Huang, X.; Huang, J.; Zhang, H. Characterization of OsPM19L1 encoding an AWPM-19-like family protein that is dramatically induced by osmotic stress in rice. Genet. Mol. Res. 2015, 14, 11994–12005. [Google Scholar] [CrossRef]
- Seok, H.-Y.; Nguyen, L.V.; Van Nguyen, D.; Lee, S.-Y.; Moon, Y.-H. Investigation of a Novel Salt Stress-Responsive Pathway Mediated by Arabidopsis DEAD-Box RNA Helicase Gene AtRH17 Using RNA-Seq Analysis. Int. J. Mol. Sci. 2020, 21, 1595. [Google Scholar] [CrossRef] [Green Version]
- Fini, A.; Brunetti, C.; Di Ferdinando, M.; Ferrini, F.; Tattini, M. Stress-induced flavonoid biosynthesis and the antioxidant machinery of plants. Plant Signal. Behav. 2011, 6, 709–711. [Google Scholar] [CrossRef]
- Ghasemzadeh, A.; Ghasemzadeh, N. Flavonoids and phenolic acids: Role and biochemical activity in plants and human. J. Med. Plants Res. 2011, 5, 6697–6703. [Google Scholar] [CrossRef]
- Yu, L.; Ma, J.; Niu, Z.; Bai, X.; Lei, W.; Shao, X.; Chen, N.; Zhou, F.; Wan, D. Tissue-Specific Transcriptome Analysis Reveals Multiple Responses to Salt Stress in Populus euphratica Seedlings. Genes 2017, 8, 372. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Han, H.; Yan, J.; Chen, F.; Wei, W. A New AP2/ERF Transcription Factor from the Oil Plant Jatropha curcas Confers Salt and Drought Tolerance to Transgenic Tobacco. Appl. Biochem. Biotechnol. 2015, 176, 582–597. [Google Scholar] [CrossRef]
- Liu, L.; Tang, X.; Wu, Y.; Huang, Y.; Tang, Y. The roles of MYB transcription factors on plant defense responses and its molecular mechanism. Yi Chuan/Hereditas 2008, 30, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Brankova, L.; Ivanov, S.; Alexieva, V. The induction of microsomal NADPH: Cytochrome P450 and NADH: Cytochrome b5 reductases by long-term salt treatment of cotton (Gossypium hirsutum L.) and bean (Phaseolus vulgaris L.) plants. Plant Physiol. Biochem. 2007, 45, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Deleu, C.; Coustaut, M.; Niogret, M.; Larher, F. Three new osmotic stress-regulated cDNAs identified by differential display polymerase chain reaction in rapeseed leaf discs. Plant Cell Environ. 1999, 22, 979–988. [Google Scholar] [CrossRef]
- Sivakumar, P.; Sharmila, P.; Saradhi, P.P. Proline Alleviates Salt-Stress-Induced Enhancement in Ribulose-1,5-Bisphosphate Oxygenase Activity. Biochem. Biophys. Res. Commun. 2000, 279, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Baskar, V.; Venkatesh, R.; Ramalingam, S. Flavonoids (Antioxidants Systems) in Higher Plants and Their Response to Stresses. In Antioxidants and Antioxidant Enzymes in Higher Plants; Springer: Berlin/Heidelberg, Germany, 2018; pp. 253–268. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, S.; Li, C.; Zhang, P.; Zhang, P. Transcriptome sequencing of Antarctic moss under salt stress emphasizes the important roles of the ROS-scavenging system. Gene 2019, 696, 122–134. [Google Scholar] [CrossRef]
- Springob, K.; Nakajima, J.-I.; Yamazaki, M.; Saito, K. Recent advances in the biosynthesis and accumulation of anthocyanins. Nat. Prod. Rep. 2003, 20, 288–303. [Google Scholar] [CrossRef]
- Geng, X.; Chen, Y.; Zhang, S.; Gao, Z.; Liu, S.; Yang, Q.; Wu, J.; Chen, X. Genome-Wide Analysis of Type-III Polyketide Synthases in Wheat and Possible Roles in Wheat Sheath-Blight Resistance. Int. J. Mol. Sci. 2022, 23, 7187. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Wang, T.; Liu, H.; Xu, X.; Chen, K.; Zhang, P. The moss flavone synthase I positively regulates the tolerance of plants to drought stress and UV-B radiation. Plant Sci. 2020, 298, 110591. [Google Scholar] [CrossRef]
- Přerostová, S.; Dobrev, P.I.; Gaudinova, A.; Hosek, P.; Soudek, P.; Knirsch, V.; Vankova, R. Hormonal dynamics during salt stress responses of salt-sensitive Arabidopsis thaliana and salt-tolerant Thellungiella salsuginea. Plant Sci. 2017, 264, 188–198. [Google Scholar] [CrossRef]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Khurana, J.P. Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 2009, 276, 3148–3162. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Jain, M. Genome-wide survey and comprehensive expression profiling of Aux/IAA gene family in chickpea and soybean. Front. Plant Sci. 2015, 6, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, M.J.; Terrile, M.C.; Windels, D.; Lombardo, M.C.; Bartoli, C.G.; Vazquez, F.; Estelle, M.; Casalongué, C.A. MiR393 Regulation of Auxin Signaling and Redox-Related Components during Acclimation to Salinity in Arabidopsis. PLoS ONE 2014, 9, e107678. [Google Scholar] [CrossRef] [Green Version]
- Amin, I.; Rasool, S.; Mir, M.A.; Wani, W.; Masoodi, K.Z.; Ahmad, P. Ion homeostasis for salinity tolerance in plants: A molecular approach. Physiol. Plant. 2020, 171, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Liu, Z. Basic Helix-Loop-Helix (bHLH) transcription factor family in Yellow horn (Xanthoceras sorbifolia Bunge): Genome-wide characterization, chromosome location, phylogeny, structures and expression patterns. Int. J. Biol. Macromol. 2020, 160, 711–723. [Google Scholar] [CrossRef]
- Wang, J.Y.; Wang, J.P.; He, Y. A Populus euphratica NAC protein regulating Na+/K+ homeostasis improves salt tolerance in Arabidopsis thaliana. Gene 2013, 521, 265–273. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Plumb-Dhindsa, P.; Thorpe, T.A. Leaf Senescence: Correlated with Increased Levels of Membrane Permeability and Lipid Peroxidation, and Decreased Levels of Superoxide Dismutase and Catalase. J. Exp. Bot. 1981, 32, 93–101. [Google Scholar] [CrossRef]
- Hatem, A.; Bozdağ, D.; Toland, A.E.; Çatalyürek, Ü.V. Benchmarking short sequence mapping tools. BMC Bioinform. 2013, 14, 184. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, R.; Wang, R.; Chen, S. Transcriptomics analysis revealing candidate networks and genes for the body size sexual dimorphism of Chinese tongue sole (Cynoglossus semilaevis). Funct. Integr. Genom. 2018, 18, 327–339. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Zhou, Q.; Wu, Y.; Chai, X.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Jin, X.; Ndayambaza, B.; Min, X.; Zhang, Z.; Wang, Y.; Liu, W. Transcriptome-Wide Characterization and Functional Identification of the Aquaporin Gene Family During Drought Stress in Common Vetch. DNA Cell Biol. 2019, 38, 374–384. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | ReadSum | BaseSum | GC (%) | Q30 (%) | Total Mapped | Uniquely Mapped | Multiple Mapped |
|---|---|---|---|---|---|---|---|
| CL-0 h | 25,651,280 | 7,672,360,004 | 42.65 | 92.32 | 79.42% | 26.51% | 73.49% |
| CL-0 h | 27,015,618 | 8,079,601,436 | 42.69 | 92.62 | 79.72% | 26.62% | 73.38% |
| CL-0 h | 28,011,278 | 8,374,751,502 | 42.77 | 91.78 | 79.45% | 26.01% | 73.99% |
| CR-0 h | 26,924,164 | 8,045,472,396 | 42.93 | 90.68 | 76.02% | 30.23% | 69.77% |
| CR-0 h | 26,568,062 | 7,930,048,480 | 42.69 | 90.77 | 76.75% | 30.79% | 69.21% |
| CR-0 h | 25,307,189 | 7,551,301,734 | 42.79 | 90.35 | 76.09% | 30.89% | 69.11% |
| TL-2 h | 25,859,885 | 7,713,984,870 | 42.81 | 92.18 | 80.22% | 26.04% | 73.96% |
| TL-2 h | 23,973,183 | 7,167,050,540 | 45.33 | 91.2 | 75.45% | 19.69% | 80.31% |
| TL-2 h | 28,540,428 | 8,532,717,458 | 42.88 | 91.19 | 79.23% | 25.98% | 74.02% |
| TR-2 h | 34,472,027 | 1,030,8164,942 | 42.42 | 92.87 | 76.41% | 31.41% | 68.59% |
| TR-2 h | 21,406,284 | 6,411,072,890 | 42.43 | 92.69 | 72.95% | 32.61% | 67.39% |
| TR-2 h | 30,183,602 | 9,029,440,866 | 42.65 | 93.16 | 76.47% | 30.74% | 69.26% |
| TL-24 h | 23,924,641 | 7,160,307,252 | 42.17 | 91.49 | 81.99% | 26.76% | 73.24% |
| TL-24 h | 30,612,059 | 9,143,959,668 | 42.98 | 92.74 | 77.55% | 28.48% | 71.52% |
| TL-24 h | 34,902,231 | 10,444,591,882 | 42.57 | 92.8 | 79.06% | 28.08% | 71.92% |
| TR-24 h | 28,348,083 | 8,475,035,384 | 43.11 | 91.61 | 74.99% | 30.10% | 69.90% |
| TR-24 h | 28,097,797 | 8,358,094,914 | 43.07 | 92.41 | 76.49% | 29.67% | 70.33% |
| TR-24 h | 30,363,513 | 9,045,723,502 | 42.72 | 92.04 | 76.94% | 29.67% | 70.33% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Wang, Q.; Min, X.; Liu, W.; Liu, Z. Comparative Transcriptomic Analysis of Root and Leaf Transcript Profiles Reveals the Coordinated Mechanisms in Response to Salinity Stress in Common Vetch. Int. J. Mol. Sci. 2022, 23, 8477. https://doi.org/10.3390/ijms23158477
Lin X, Wang Q, Min X, Liu W, Liu Z. Comparative Transcriptomic Analysis of Root and Leaf Transcript Profiles Reveals the Coordinated Mechanisms in Response to Salinity Stress in Common Vetch. International Journal of Molecular Sciences. 2022; 23(15):8477. https://doi.org/10.3390/ijms23158477
Chicago/Turabian StyleLin, Xiaoshan, Qiuxia Wang, Xueyang Min, Wenxian Liu, and Zhipeng Liu. 2022. "Comparative Transcriptomic Analysis of Root and Leaf Transcript Profiles Reveals the Coordinated Mechanisms in Response to Salinity Stress in Common Vetch" International Journal of Molecular Sciences 23, no. 15: 8477. https://doi.org/10.3390/ijms23158477
APA StyleLin, X., Wang, Q., Min, X., Liu, W., & Liu, Z. (2022). Comparative Transcriptomic Analysis of Root and Leaf Transcript Profiles Reveals the Coordinated Mechanisms in Response to Salinity Stress in Common Vetch. International Journal of Molecular Sciences, 23(15), 8477. https://doi.org/10.3390/ijms23158477