
Vitamin D–VDR Novel Anti-Inflammatory Molecules—New Insights into Their Effects on Liver Diseases

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Selection of Studies
3. Hepatocytes and Liver Damage
4. Physiology and Metabolism of Vitamin D
5. Vitamin D Receptor (VDR)
6. Vitamin D, VDR, and Liver Diseases
7. Possible Mechanisms of the Association between Vitamin D–VDR and Liver Disease
7.1. Vitamin D–VDR and Hepatitis B Virus (HBV)
Vitamin D Supplementation and HBV
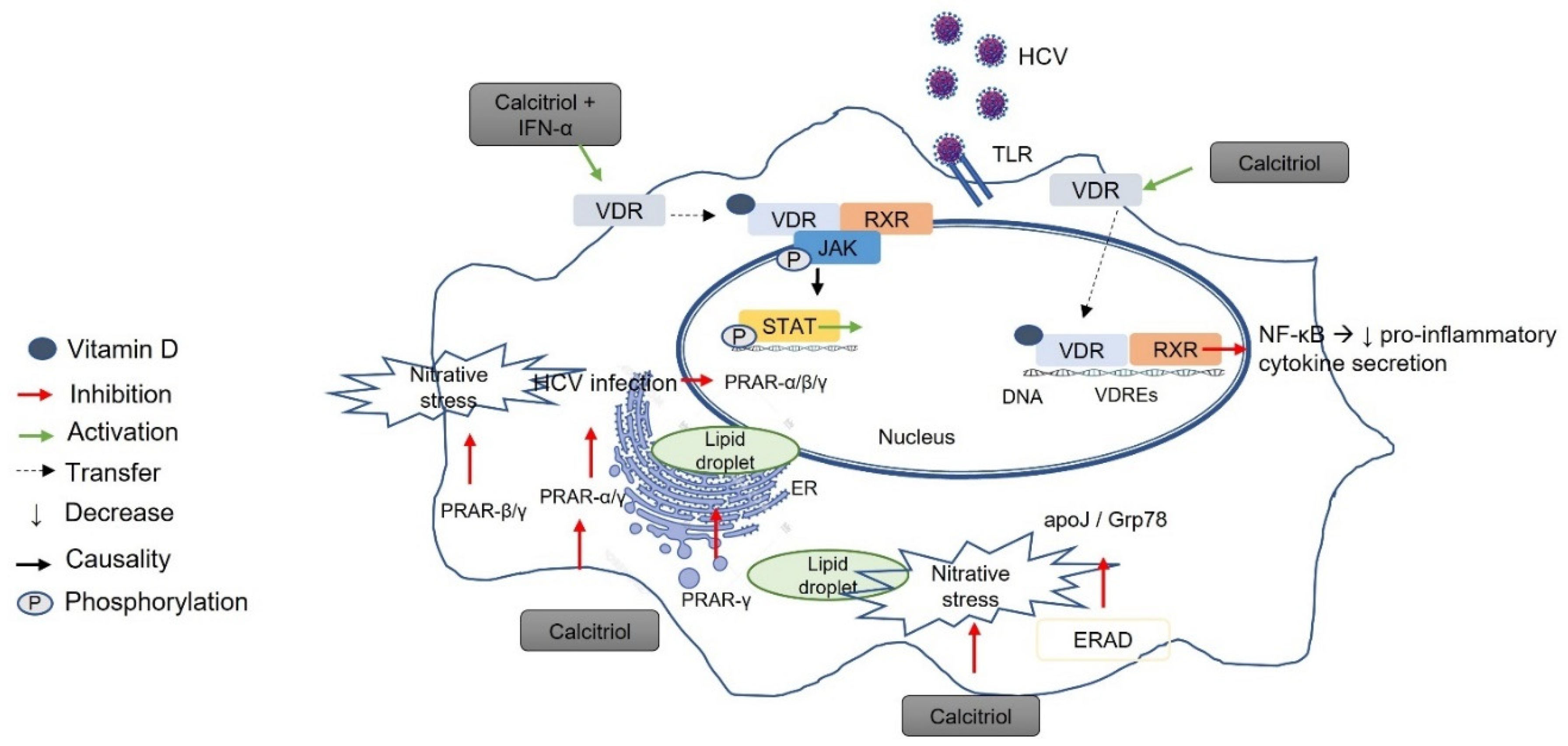
7.2. Vitamin D–VDR and Hepatitis C Virus (HCV)
Vitamin D Supplementation and HCV
7.3. Vitamin D–VDR and Autoimmune Hepatitis
Vitamin D Supplementation and AIH
7.4. Vitamin D–VDR and Primary Biliary Cholangitis (PBC)
Vitamin D Supplementation and PBC
7.5. Vitamin D–VDR and NAFLD
Vitamin D Supplementation and NAFLD
8. Vitamin D–VDR-Related Genetic Polymorphisms in Liver Disease
9. Vitamin D Signaling and Gut Microbiota in Liver Disease
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Tarantino, G.; Scalera, A.; Finelli, C. Liver-spleen axis: Intersection between immunity, infections and metabolism. World J. Gastroenterol. 2013, 19, 3534–3542. [Google Scholar] [CrossRef]
- Barrea, L.; Di Somma, C. Nutrition, inflammation and liver-spleen axis. Crit. Rev. Food Sci. Nutr. 2018, 58, 3141–3158. [Google Scholar] [CrossRef]
- van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Rowe, I.A. Lessons from Epidemiology: The Burden of Liver Disease. Dig. Dis. 2017, 35, 304–309. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, 27. [Google Scholar] [CrossRef]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 24. [Google Scholar] [CrossRef]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef]
- Mouli, V.P.; Ananthakrishnan, A.N. Review article: Vitamin D and inflammatory bowel diseases. Aliment. Pharm. 2014, 39, 125–136. [Google Scholar] [CrossRef]
- Triantos, C.; Aggeletopoulou, I.; Thomopoulos, K.; Mouzaki, A. Vitamin D–liver disease association: Biological basis and mechanisms of action. Hepatology 2021, 74, 1065–1073. [Google Scholar] [CrossRef]
- Hossein-nezhad, A.; Holick, M.F. Vitamin D for health: A global perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef]
- Battault, S.; Whiting, S.J.; Peltier, S.L.; Sadrin, S.; Gerber, G.; Maixent, J.M. Vitamin D metabolism, functions and needs: From science to health claims. Eur. J. Nutr. 2013, 52, 429–441. [Google Scholar] [CrossRef]
- Ronti, T.; Lupattelli, G.; Mannarino, E. The endocrine function of adipose tissue: An update. Clin. Endocrinol. 2006, 64, 355–365. [Google Scholar] [CrossRef]
- Hewison, M.; Burke, F.; Evans, K.N.; Lammas, D.A.; Sansom, D.M.; Liu, P.; Modlin, R.L.; Adams, J.S. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J. Steroid Biochem. Mol. Biol. 2007, 103, 316–321. [Google Scholar] [CrossRef]
- Kongsbak, M.; von Essen, M.R.; Boding, L.; Levring, T.B.; Schjerling, P.; Lauritsen, J.P.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Vitamin D up-regulates the vitamin D receptor by protecting it from proteasomal degradation in human CD4+ T cells. PLoS ONE 2014, 9, e96695. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Ooi, J.H.; Li, Y.; Rogers, C.J.; Cantorna, M.T. Vitamin D Regulates the Gut Microbiome and Protects Mice from Dextran Sodium Sulfate–Induced Colitis. J. Nutr. 2013, 143, 1679–1686. [Google Scholar] [CrossRef]
- Abramovitch, S.; Dahan-Bachar, L.; Sharvit, E.; Weisman, Y.; Ben Tov, A.; Brazowski, E.; Reif, S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut 2011, 60, 1728–1737. [Google Scholar] [CrossRef]
- Petta, S.; Camma, C.; Scazzone, C.; Tripodo, C.; Di Marco, V.; Bono, A.; Cabibi, D.; Licata, G.; Porcasi, R.; Marchesini, G.; et al. Low vitamin D serum level is related to severe fibrosis and low responsiveness to interferon-based therapy in genotype 1 chronic hepatitis C. Hepatology 2010, 51, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Putz-Bankuti, C.; Pilz, S.; Stojakovic, T.; Scharnagl, H.; Pieber, T.R.; Trauner, M.; Obermayer-Pietsch, B.; Stauber, R.E. Association of 25-hydroxyvitamin D levels with liver dysfunction and mortality in chronic liver disease. Liver Int. 2012, 32, 845–851. [Google Scholar] [CrossRef]
- Stokes, C.S.; Krawczyk, M.; Reichel, C.; Lammert, F.; Grunhage, F. Vitamin D deficiency is associated with mortality in patients with advanced liver cirrhosis. Eur. J. Clin. Investig. 2014, 44, 176–183. [Google Scholar] [CrossRef]
- Luo, Y.Q.; Wu, X.X.; Ling, Z.X.; Cheng, Y.W.; Yuan, L.; Xiang, C. Association between serum vitamin D and severity of liver fibrosis in chronic hepatitis C patients: A systematic meta-analysis. J. Zhejiang Univ. Sci. B 2014, 15, 900–906. [Google Scholar] [CrossRef]
- Trepo, E.; Ouziel, R.; Pradat, P.; Momozawa, Y.; Quertinmont, E.; Gervy, C.; Gustot, T.; Degre, D.; Vercruysse, V.; Deltenre, P.; et al. Marked 25-hydroxyvitamin D deficiency is associated with poor prognosis in patients with alcoholic liver disease. J. Hepatol. 2013, 59, 344–350. [Google Scholar] [CrossRef]
- Guo, G.Y.; Shi, Y.Q.; Wang, L.; Ren, X.; Han, Z.Y.; Guo, C.C.; Cui, L.N.; Wang, J.B.; Zhu, J.; Wang, N.; et al. Serum vitamin D level is associated with disease severity and response to ursodeoxycholic acid in primary biliary cirrhosis. Aliment. Pharm. 2015, 42, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.; Fisher, A. Vitamin D and parathyroid hormone in outpatients with noncholestatic chronic liver disease. Clin. Gastroenterol. Hepatol. 2007, 5, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Malham, M.; Jorgensen, S.P.; Ott, P.; Agnholt, J.; Vilstrup, H.; Borre, M.; Dahlerup, J.F. Vitamin D deficiency in cirrhosis relates to liver dysfunction rather than aetiology. World J. Gastroenterol. 2011, 17, 922–925. [Google Scholar] [CrossRef]
- Triantos, C.; Kalafateli, M.; Aggeletopoulou, I.; Diamantopoulou, G.; Spantidea, P.I.; Michalaki, M.; Vourli, G.; Konstantakis, C.; Assimakopoulos, S.F.; Manolakopoulos, S.; et al. Vitamin D-related immunomodulation in patients with liver cirrhosis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alvarez, M.; Pineda-Tenor, D.; Jimenez-Sousa, M.A.; Fernandez-Rodriguez, A.; Guzman-Fulgencio, M.; Resino, S. Relationship of vitamin D status with advanced liver fibrosis and response to hepatitis C virus therapy: A meta-analysis. Hepatology 2014, 60, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Carrat, F.; Geri, G.; Pol, S.; Piroth, L.; Halfon, P.; Poynard, T.; Souberbielle, J.C.; Cacoub, P. Low 25-OH vitamin D serum levels correlate with severe fibrosis in HIV-HCV co-infected patients with chronic hepatitis. J. Hepatol. 2011, 55, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Haussler, C.A.; Bartik, L.; Whitfield, G.K.; Hsieh, J.C.; Slater, S.; Jurutka, P.W. Vitamin D receptor: Molecular signaling and actions of nutritional ligands in disease prevention. Nutr. Rev. 2008, 66, 1753–4887. [Google Scholar] [CrossRef]
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Vitamin D genomics: From in vitro to in vivo. Front. Endocrinol. 2018, 9, 250. [Google Scholar] [CrossRef]
- Campbell, M.J. Vitamin D and the RNA transcriptome: More than mRNA regulation. Front. Physiol. 2014, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Udomsinprasert, W.; Jittikoon, J. Vitamin D and liver fibrosis: Molecular mechanisms and clinical studies. Biomed. Pharm. 2019, 109, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, H.; Chahal, S.; Gunton, J.E. Vitamin D in liver disease: Current evidence and potential directions. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 4, 907–916. [Google Scholar] [CrossRef]
- Norman, A.W. Minireview: Vitamin D receptor: New assignments for an already busy receptor. Endocrinology 2006, 147, 5542–5548. [Google Scholar] [CrossRef]
- Gascon-Barre, M.; Demers, C.; Mirshahi, A.; Neron, S.; Zalzal, S.; Nanci, A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology 2003, 37, 1034–1042. [Google Scholar] [CrossRef]
- Veldman, C.M.; Cantorna, M.T.; DeLuca, H.F. Expression of 1,25-dihydroxyvitamin D(3) receptor in the immune system. Arch. Biochem. Biophys. 2000, 374, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Kamen, D.L.; Tangpricha, V. Vitamin D and molecular actions on the immune system: Modulation of innate and autoimmunity. J. Mol. Med. 2010, 88, 441–450. [Google Scholar] [CrossRef]
- Griffin, M.D.; Lutz, W.; Phan, V.A.; Bachman, L.A.; McKean, D.J.; Kumar, R. Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: A vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 6800–6805. [Google Scholar] [CrossRef]
- von Essen, M.R.; Kongsbak, M.; Schjerling, P.; Olgaard, K.; Odum, N.; Geisler, C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010, 11, 344–349. [Google Scholar] [CrossRef]
- Nagpal, S.; Na, S.; Rathnachalam, R. Noncalcemic actions of vitamin D receptor ligands. Endocr. Rev. 2005, 26, 662–687. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Konstantakis, C.; Tselekouni, P.; Kalafateli, M.; Triantos, C. Vitamin D deficiency in patients with liver cirrhosis. Ann. Gastroenterol. 2016, 29, 297–306. [Google Scholar] [CrossRef]
- Chan, H.L.; Elkhashab, M.; Trinh, H.; Tak, W.Y.; Ma, X.; Chuang, W.L.; Kim, Y.J.; Martins, E.B.; Lin, L.; Dinh, P.; et al. Association of baseline vitamin D levels with clinical parameters and treatment outcomes in chronic hepatitis B. J. Hepatol. 2015, 63, 1086–1092. [Google Scholar] [CrossRef]
- Wong, G.L.; Chan, H.L.; Chan, H.Y.; Tse, C.H.; Chim, A.M.; Lo, A.O.; Wong, V.W. Adverse effects of vitamin D deficiency on outcomes of patients with chronic hepatitis B. Clin. Gastroenterol. Hepatol. 2015, 13, 783–790. [Google Scholar] [CrossRef]
- Chen, E.Q.; Bai, L.; Zhou, T.Y.; Fe, M.; Zhang, D.M.; Tang, H. Sustained suppression of viral replication in improving vitamin D serum concentrations in patients with chronic hepatitis B. Sci. Rep. 2015, 5, 15441. [Google Scholar] [CrossRef]
- Farnik, H.; Bojunga, J.; Berger, A.; Allwinn, R.; Waidmann, O.; Kronenberger, B.; Keppler, O.T.; Zeuzem, S.; Sarrazin, C.; Lange, C.M. Low vitamin D serum concentration is associated with high levels of hepatitis B virus replication in chronically infected patients. Hepatology 2013, 58, 1270–1276. [Google Scholar] [CrossRef]
- Hoan, N.X.; Khuyen, N.; Binh, M.T.; Giang, D.P.; Van Tong, H.; Hoan, P.Q.; Trung, N.T.; Anh, D.T.; Toan, N.L.; Meyer, C.G.; et al. Association of vitamin D deficiency with hepatitis B virus—Related liver diseases. BMC Infect. Dis. 2016, 16, 016–1836. [Google Scholar] [CrossRef]
- Gotlieb, N.; Tachlytski, I.; Lapidot, Y.; Sultan, M.; Safran, M.; Ben-Ari, Z. Hepatitis B virus downregulates vitamin D receptor levels in hepatoma cell lines, thereby preventing vitamin D-dependent inhibition of viral transcription and production. Mol. Med. 2018, 24, 018–0055. [Google Scholar] [CrossRef]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil. Med. Res. 2021, 8, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.; Mushtaq, S.; Ahmed, S.Z.; Shahid, M.A. The Role of Vitamin D in HBV infection. Eur. J. Biotechnol. Biosci. 2015, 3, 35–41. [Google Scholar]
- Cantorna, M.T.; Waddell, A. The vitamin D receptor turns off chronically activated T cells. Ann. N. Y. Acad. Sci. 2014, 1317, 70–75. [Google Scholar] [CrossRef]
- Khoo, A.L.; Joosten, I.; Michels, M.; Woestenenk, R.; Preijers, F.; He, X.H.; Netea, M.G.; van der Ven, A.J.; Koenen, H.J. 1,25-Dihydroxyvitamin D3 inhibits proliferation but not the suppressive function of regulatory T cells in the absence of antigen-presenting cells. Immunology 2011, 134, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Ojaimi, S.; Skinner, N.A.; Strauss, B.J.G.; Sundararajan, V.; Woolley, I.; Visvanathan, K. Vitamin d deficiency impacts on expression of toll-like receptor-2 and cytokine profile: A pilot study. J. Transl. Med. 2013, 11, 176. [Google Scholar] [CrossRef]
- He, Q.; Huang, Y.; Zhang, L.; Yan, Y.; Liu, J.; Song, X.; Chen, W. Association between vitamin D receptor polymorphisms and hepatitis B virus infection susceptibility: A meta-analysis study. Gene 2018, 645, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Youssry, S.; Shalaby, T.; Maher, A.S.; Ghoneim, H. Association of hepatitis B vaccine response to vitamin D supplementation and ultraviolet B (UVB) exposure during different time intervals in experimental animals. Immunol. Res. 2022, 70, 537–545. [Google Scholar] [CrossRef]
- Kashi, D.S.; Oliver, S.J. Vitamin D and the hepatitis B vaccine response: A prospective cohort study and a randomized, placebo-controlled oral vitamin D(3) and simulated sunlight supplementation trial in healthy adults. Eur. J. Nutr. 2021, 60, 475–491. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Xu, H.-J.; Li, Y.; Hu, C.-M.; Yang, J.-Y.; Sun, M.-Y. The Anti-Inflammatory Effects of Vitamin D in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2736. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-M.; Sun, H.-Y.; Chiu, W.-T.; Su, H.-C.; Chien, Y.-C.; Chong, L.-W.; Chang, H.-C.; Bai, C.-H.; Young, K.-C.; Tsao, C.-W. Calcitriol inhibits HCV infection via blockade of activation of PPAR and interference with endoplasmic reticulum-associated degradation. Viruses 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Gal-Tanamy, M.; Bachmetov, L.; Ravid, A.; Koren, R.; Erman, A.; Tur-Kaspa, R.; Zemel, R. Vitamin D: An innate antiviral agent suppressing hepatitis C virus in human hepatocytes. Hepatology 2011, 54, 1570–1579. [Google Scholar] [CrossRef]
- Murayama, A.; Saitoh, H.; Takeuchi, A.; Yamada, N.; Matsumura, T.; Shiina, M.; Muramatsu, M.; Wakita, T.; Imawari, M.; Kato, T. Vitamin D derivatives inhibit hepatitis C virus production through the suppression of apolipoprotein. Antivir. Res. 2018, 160, 55–63. [Google Scholar] [CrossRef]
- Ravid, A.; Rapaport, N.; Issachar, A.; Erman, A.; Bachmetov, L.; Tur-Kaspa, R.; Zemel, R. 25-Hydroxyvitamin D Inhibits Hepatitis C Virus Production in Hepatocellular Carcinoma Cell Line by a Vitamin D Receptor-Independent Mechanism. Int. J. Mol. Sci. 2019, 20, 2367. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Kato, T.; Sugiyama, N.; Tasaka-Fujita, M.; Murayama, A.; Masaki, T.; Wakita, T.; Imawari, M. 25-hydroxyvitamin D3 suppresses hepatitis C virus production. Hepatology 2012, 56, 1231–1239. [Google Scholar] [CrossRef]
- Gutierrez, J.A.; Jones, K.A.; Flores, R.; Singhania, A.; Woelk, C.H.; Schooley, R.T.; Wyles, D.L. Vitamin D metabolites inhibit hepatitis C virus and modulate cellular gene expression. J. Virol. Antivir. Res. 2014, 3. [Google Scholar] [CrossRef]
- Saleh, M.; Welsch, C.; Cai, C.; Döring, C.; Gouttenoire, J.; Friedrich, J.; Haselow, K.; Sarrazin, C.; Badenhoop, K.; Moradpour, D.; et al. Differential modulation of hepatitis C virus replication and innate immune pathways by synthetic calcitriol-analogs. J. Steroid Biochem. Mol. Biol. 2018, 183, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Gouttenoire, J.; Duong, F.H.; Morikawa, K.; Heim, M.H.; Moradpour, D. Vitamin D receptor and Jak–STAT signaling crosstalk results in calcitriol-mediated increase of hepatocellular response to IFN-α. J. Immunol. 2014, 192, 6037–6044. [Google Scholar] [CrossRef]
- Hii, C.S.; Ferrante, A. The Non-Genomic Actions of Vitamin D. Nutrients 2016, 8, 135. [Google Scholar] [CrossRef]
- Kitson, M.T.; Sarrazin, C.; Toniutto, P.; Eslick, G.D.; Roberts, S.K. Vitamin D level and sustained virologic response to interferon-based antiviral therapy in chronic hepatitis C: A systematic review and meta-analysis. J. Hepatol. 2014, 61, 1247–1252. [Google Scholar] [CrossRef]
- Udomsinprasert, W.; Jittikoon, J.; Sukkho, S.; Pojarassangkul, N.; Sangroongruangsri, S.; Chaikledkaew, U. Decreased circulating vitamin D reflects adverse outcomes of hepatitis C virus infection: A systematic review and meta-analysis. J. Infect. 2020, 15, 30402–30407. [Google Scholar] [CrossRef] [PubMed]
- de Azevedo, L.A.; Matte, U.; da Silveira, T.R.; Álvares-da-Silva, M.R. Genetic variants underlying vitamin D metabolism and VDR-TGFβ-1-SMAD3 interaction may impact on HCV progression: A study based on dbGaP data from the HALT-C study. J. Hum. Genet. 2017, 62, 969–977. [Google Scholar] [CrossRef]
- Murayama, A.; Kato, T. Chapter Nine—Inhibition of hepatitis C virus by vitamin D. In Vitamins and Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 117, pp. 227–238. [Google Scholar]
- Sriphoosanaphan, S.; Thanapirom, K.; Kerr, S.J.; Suksawatamnuay, S.; Thaimai, P.; Sittisomwong, S.; Sonsiri, K.; Srisoonthorn, N.; Teeratorn, N.; Tanpowpong, N.; et al. Effect of vitamin D supplementation in patients with chronic hepatitis C after direct-acting antiviral treatment: A randomized, double-blind, placebo-controlled trial. PeerJ 2021, 9, e10709. [Google Scholar] [CrossRef]
- Abu-Mouch, S.; Fireman, Z.; Jarchovsky, J.; Zeina, A.R.; Assy, N. Vitamin D supplementation improves sustained virologic response in chronic hepatitis C (genotype 1)-naïve patients. World J. Gastroenterol. 2011, 17, 5184–5190. [Google Scholar] [CrossRef] [PubMed]
- Bitetto, D.; Fabris, C.; Fornasiere, E.; Pipan, C.; Fumolo, E.; Cussigh, A.; Bignulin, S.; Cmet, S.; Fontanini, E.; Falleti, E.; et al. Vitamin D supplementation improves response to antiviral treatment for recurrent hepatitis C. Transpl. Int. 2011, 24, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Nimer, A.; Mouch, A. Vitamin D improves viral response in hepatitis C genotype 2-3 naïve patients. World J. Gastroenterol. 2012, 18, 800–805. [Google Scholar] [CrossRef]
- Vosoghinia, H.; Esmaeilzadeh, A.; Ganji, A.; Hosseini, S.M.-R.; Jamehdar, S.A.; Salehi, M.; Bahari, A.; Ghanaei, O.; Sahebari, M.; Rajabzadeh, F. Vitamin D in standard HCV regimen (PEG-interferon plus ribavirin), its effect on the early virologic response rate: A clinical trial. Razavi Int. J. Med. 2016, 4. [Google Scholar] [CrossRef]
- Villar, L.M.; Del Campo, J.A.; Ranchal, I.; Lampe, E.; Romero-Gomez, M. Association between vitamin D and hepatitis C virus infection: A meta-analysis. World J. Gastroenterol. 2013, 19, 5917–5924. [Google Scholar] [CrossRef]
- Kim, H.B.; Myung, S.K.; Lee, Y.J.; Park, B.J.; Group, K.M.A.S. Efficacy of vitamin D supplementation in combination with conventional antiviral therapy in patients with chronic hepatitis C infection: A meta—Analysis of randomised controlled trials. J. Hum. Nutr. Diet. 2018, 31, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Behera, M.K.; Shukla, S.K.; Dixit, V.K.; Nath, P.; Abhilash, V.B.; Asati, P.K.; Jain, A.K. Effect of vitamin D supplementation on sustained virological response in genotype 1/4 chronic hepatitis C treatment-naïve patients from India. Indian J. Med. Res. 2018, 148, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Ladero, J.M.; Torrejón, M.J.; Sánchez-Pobre, P.; Suárez, A.; Cuenca, F.; de la Orden, V.; Devesa, M.J.; Rodrigo, M.; Estrada, V.; López-Alonso, G.; et al. Vitamin D deficiency and vitamin D therapy in chronic hepatitis C. Ann. Hepatol. 2013, 12, 199–204. [Google Scholar] [CrossRef]
- Esmat, G.; El Raziky, M.; Elsharkawy, A.; Sabry, D.; Hassany, M.; Ahmed, A.; Assem, N.; El Kassas, M.; Doss, W. Impact of vitamin D supplementation on sustained virological response in chronic hepatitis C genotype 4 patients treated by pegylated interferon/ribavirin. J. Interferon Cytokine Res. 2015, 35, 49–54. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Jun, D.W.; Park, S.J.; Sohn, J.H.; Kim, S.G.; Lee, S.W.; Jeong, S.W.; Kim, M.Y.; Kim, W.; Shim, J.J.; et al. Effects of vitamin D supplements in patients with chronic hepatitis C: A randomized, multi-center, open label study. Korean J. Intern. Med. 2020, 35, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, M.; Nikolova, D.; Bjelakovic, G.; Gluud, C. Vitamin D supplementation for chronic liver diseases in adults. Cochrane Database Syst. Rev. 2021, 8, Cd011564. [Google Scholar] [CrossRef]
- Kondo, Y.; Kato, T.; Kimura, O.; Iwata, T.; Ninomiya, M.; Kakazu, E.; Miura, M.; Akahane, T.; Miyazaki, Y.; Kobayashi, T. 1 (OH) vitamin D3 supplementation improves the sensitivity of the immune-response during Peg-IFN/RBV therapy in chronic hepatitis C patients-case controlled trial. PLoS ONE 2013, 8, e63672. [Google Scholar] [CrossRef] [PubMed]
- Omori-Mizuno, Y.; Nakayama, N.; Inao, M.; Funyu, J.; Asabe, S.; Tomita, K.; Nishikawa, K.; Hosoda, Y.; Tanaka, M.; Hashimoto, Y.; et al. Randomized study comparing vitamin D3 and 1α-Hydroxyvitamin D3 in combination with pegylated interferon/ribavirin therapy for chronic hepatitis C. J. Gastroenterol. Hepatol. 2015, 30, 1384–1390. [Google Scholar] [CrossRef]
- Chen, H.W.; Chiu, Y.L. Relationships Between Vitamin D Status and Cytokine: Results from Interferon-Based Therapy in Non-Cirrhotic, Treatment-Naïve Patients with Chronic Hepatitis C Infection. J. Inflamm. Res. 2020, 13, 1207–1218. [Google Scholar] [CrossRef]
- Sergi, C.M. Vitamin D supplementation for autoimmune hepatitis: A need for further investigation. World J. Hepatol. 2022, 14, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zhang, H.; Zhao, Q.; Bu, H.; Wang, H.; Guo, H. Correlation of vitamin D with inflammatory factors, oxidative stress and T cell subsets in patients with autoimmune hepatitis. Exp. Med. 2020, 19, 3419–3424. [Google Scholar] [CrossRef]
- Umeda, N.; Endo-Umeda, K.; Nakashima, H.; Kato, S.; Seki, S.; Makishima, M. Frontline Science: Concanavalin A-induced acute hepatitis is attenuated in vitamin D receptor knockout mice with decreased immune cell function. J. Leukoc. Biol. 2019, 106, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Efe, C.; Kav, T.; Aydin, C.; Cengiz, M.; Imga, N.N.; Purnak, T.; Smyk, D.S.; Torgutalp, M.; Turhan, T.; Ozenirler, S.; et al. Low serum vitamin D levels are associated with severe histological features and poor response to therapy in patients with autoimmune hepatitis. Dig. Dis. Sci. 2014, 59, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Montano-Loza, A.J. Evolving Role of Vitamin D in Immune-Mediated Disease and Its Implications in Autoimmune Hepatitis. Dig. Dis. Sci. 2019, 64, 324–344. [Google Scholar] [CrossRef] [PubMed]
- Luong, K.V.Q.; Nguyễn, L.T.H. The role of vitamin d in primary biliary cirrhosis: Possible genetic and cell signaling mechanisms. Gastroenterol. Res. Pract. 2013, 2013, 602321. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Liu, W.; Sun, T.; Huang, Y.; Wang, Y.; Deb, D.K.; Yoon, D.; Kong, J.; Thadhani, R.; Li, Y.C. 1,25-Dihydroxyvitamin D promotes negative feedback regulation of TLR signaling via targeting microRNA-155-SOCS1 in macrophages. J. Immunol. 2013, 190, 3687–3695. [Google Scholar] [CrossRef] [PubMed]
- Kempinska-Podhorodecka, A.; Milkiewicz, M.; Wasik, U.; Ligocka, J.; Zawadzki, M.; Krawczyk, M.; Milkiewicz, P. Decreased Expression of Vitamin D Receptor Affects an Immune Response in Primary Biliary Cholangitis via the VDR-miRNA155-SOCS1 Pathway. Int. J. Mol. Sci. 2017, 18, 289. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Wang, Z.; Peng, C.; Wang, P.; Sui, J.; Wang, Y.; Sun, G.; Liu, M. Serum vitamin D level is related to disease progression in primary biliary cholangitis. Scand. J. Gastroenterol. 2020, 55, 1333–1340. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Vitamin D up-regulates glucose transporter 4 (GLUT4) translocation and glucose utilization mediated by cystathionine-γ-lyase (CSE) activation and H2S formation in 3T3L1 adipocytes. J. Biol. Chem. 2012, 287, 42324–42332. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and Toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Q.; Chai, Y.; Liu, Y.; Li, F.; Wang, B.; Zhu, C.; Cui, J.; Qu, H.; Zhu, M. 1,25(OH)2D3 downregulates the Toll-like receptor 4-mediated inflammatory pathway and ameliorates liver injury in diabetic rats. J. Endocrinol. Investig. 2015, 38, 1083–1091. [Google Scholar] [CrossRef]
- Savastano, S.; Barrea, L.; Savanelli, M.C.; Nappi, F.; Di Somma, C.; Orio, F.; Colao, A. Low vitamin D status and obesity: Role of nutritionist. Rev. Endocr. Metab. Disord. 2017, 18, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Savastano, S.; Capone, D.; Colao, A. Spleen: A new role for an old player? World J. Gastroenterol. 2011, 17, 3776–3784. [Google Scholar] [CrossRef]
- Mohamed, H.K. Effect of vitamin D on the spleen of adult male rats fed on diet with high fat: A histological and immunohistochemical study. Egypt. J. Histol. 2019, 42, 1001–1017. [Google Scholar] [CrossRef]
- Zeng, Y.; Luo, M.; Pan, L.; Chen, Y.; Guo, S.; Luo, D.; Zhu, L.; Liu, Y.; Xu, S.; Zhang, R.; et al. Vitamin D signaling maintains intestinal innate immunity and gut microbiota: Potential intervention for metabolic syndrome and NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G542–G553. [Google Scholar] [CrossRef] [PubMed]
- Eliades, M.; Spyrou, E. Vitamin D: A new player in non-alcoholic fatty liver disease? World J. Gastroenterol. 2015, 21, 1718–1727. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Z.; Lin, Y.; Zhang, J.; Zhang, Y.; Liu, P.; Zeng, H.; Yu, M.; Chen, X.; Ning, L.; et al. Vitamin D receptor targets hepatocyte nuclear factor 4α and mediates protective effects of vitamin D in nonalcoholic fatty liver disease. J. Biol. Chem. 2020, 295, 3891–3905. [Google Scholar] [CrossRef]
- Shin, D.-J.; Osborne, T.F. FGF15/FGFR4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J. Biol. Chem. 2009, 284, 11110–11120. [Google Scholar] [CrossRef] [PubMed]
- Bozic, M.; Guzmán, C.; Benet, M.; Sánchez-Campos, S.; García-Monzón, C.; Gari, E.; Gatius, S.; Valdivielso, J.M.; Jover, R. Hepatocyte vitamin D receptor regulates lipid metabolism and mediates experimental diet-induced steatosis. J. Hepatol. 2016, 65, 748–757. [Google Scholar] [CrossRef] [PubMed]
- García-Monzón, C.; Petrov, P.D.; Rey, E.; Marañón, P.; Del Pozo-Maroto, E.; Guzmán, C.; Rodríguez de Cía, J.; Casado-Collado, A.J.; Vargas-Castrillón, J.; Saez, A.; et al. Angiopoietin-Like Protein 8 Is a Novel Vitamin D Receptor Target Gene Involved in Nonalcoholic Fatty Liver Pathogenesis. Am. J. Pathol. 2018, 188, 2800–2810. [Google Scholar] [CrossRef]
- Arai, T.; Atsukawa, M.; Tsubota, A.; Koeda, M.; Yoshida, Y.; Okubo, T.; Nakagawa, A.; Itokawa, N.; Kondo, C.; Nakatsuka, K.; et al. Association of vitamin D levels and vitamin D-related gene polymorphisms with liver fibrosis in patients with biopsy-proven nonalcoholic fatty liver disease. Dig. Liver Dis. 2019, 51, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients 2017, 9, 1015. [Google Scholar] [CrossRef]
- Kitson, M.T.; Roberts, S.K. D-livering the message: The importance of vitamin D status in chronic liver disease. J. Hepatol. 2012, 57, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Del Ben, M.; Angelico, F.; Di Martino, M.; Fraioli, A.; La Torre, G.; Saulle, R.; Perri, L.; Morini, S.; Tiberti, C.; et al. No effects of oral vitamin D supplementation on non-alcoholic fatty liver disease in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled trial. BMC Med. 2016, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Amani, R.; Hajiani, E.; Cheraghian, B. Does vitamin D improve liver enzymes, oxidative stress, and inflammatory biomarkers in adults with non-alcoholic fatty liver disease? A randomized clinical trial. Endocrine 2014, 47, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, S.; Meng, Y.; Yu, Q.; Wang, Q.; Xu, H.; Yuan, H.; Li, X.; Chen, L. Effects of Vitamin D Supplementation in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. Metab. 2020, 18, e97205. [Google Scholar] [CrossRef] [PubMed]
- Gad, A.I.; Elmedames, M.R.; Abdelhai, A.R.; Marei, A.M.; Abdel-Ghani, H.A. Efficacy of vitamin D supplementation on adult patients with non-alcoholic fatty liver disease: A single-center experience. Gastroenterol. Hepatol. Bed Bench 2021, 14, 44–52. [Google Scholar] [PubMed]
- Rezaei, S.; Tabrizi, R.; Nowrouzi-Sohrabi, P.; Jalali, M.; Shabani-Borujeni, M.; Modaresi, S.; Gholamalizadeh, M.; Doaei, S. The Effects of Vitamin D Supplementation on Anthropometric and Biochemical Indices in Patients With Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. Front. Pharmacol. 2021, 12, 732496. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, L.; Asadi, S.; Al-Mousavi, Z.; Niknam, R. A randomized controlled clinical trial comparing calcitriol versus cholecalciferol supplementation to reduce insulin resistance in patients with non-alcoholic fatty liver disease. Clin. Nutr. 2021, 40, 2999–3005. [Google Scholar] [CrossRef]
- Sindhughosa, D.A.; Wibawa, I.D.N.; Mariadi, I.K.; Somayana, G. Additional treatment of vitamin D for improvement of insulin resistance in non-alcoholic fatty liver disease patients: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 7716. [Google Scholar] [CrossRef]
- Hamzehzadeh Alamdari, A.; Ahrabi, S.; Khoshbaten, M.; Roustaei, S.; Araqchin Ahrabi, S.; Asghari Jafarabadi, M. Effect of Oral and parenteral routes of vitamin D supplementation on serum 25(OH) vitamin D levels in patients with non-alcoholic fatty liver disease. Casp. J. Intern. Med. 2022, 13, 23–28. [Google Scholar] [CrossRef]
- Guo, X.F.; Wang, C.; Yang, T.; Ma, W.J.; Zhai, J.; Zhao, T.; Xu, T.C.; Li, J.; Liu, H.; Sinclair, A.J.; et al. The effects of fish oil plus vitamin D(3) intervention on non-alcoholic fatty liver disease: A randomized controlled trial. Eur. J. Nutr. 2022, 61, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.S.; Quaglia, A.; Dhawan, A.; Wu, H.; Lanham-New, S.; Hart, K.H.; Fitzpatrick, E.; Moore, J.B. Vitamin D status and associated genetic polymorphisms in a cohort of UK children with non-alcoholic fatty liver disease. Pediatr. Obes. 2018, 13, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; White, S.W.; Marsh, J.A.; Lye, S.J.; Connor, K.L.; Maganga, R.; Ayonrinde, O.T.; Olynyk, J.K.; Mori, T.A.; Beilin, L.J.; et al. Association between liver-specific gene polymorphisms and their expression levels with nonalcoholic fatty liver disease. Hepatology 2013, 57, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Sadeghnia, H.R.; Tabatabaeizadeh, S.A.; Bahrami-Taghanaki, H.; Behboodi, N.; Esmaeili, H.; Ferns, G.A.; Mobarhan, M.G.; Avan, A. Genetic and epigenetic factors influencing vitamin D status. J. Cell. Physiol. 2018, 233, 4033–4043. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Strassburg, C.P.; Manns, M.P. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology 2002, 35, 126–131. [Google Scholar] [CrossRef]
- Kempinska-Podhorecka, A.; Wunsch, E.; Jarowicz, T.; Raszeja-Wyszomirska, J.; Loniewska, B.; Kaczmarczyk, M.; Milkiewicz, M.; Milkiewicz, P. Vitamin d receptor polymorphisms predispose to primary biliary cirrhosis and severity of the disease in polish population. Gastroenterol. Res. Pract. 2012, 408723, 29. [Google Scholar] [CrossRef]
- Halmos, B.; Szalay, F.; Cserniczky, T.; Nemesanszky, E.; Lakatos, P.; Barlage, S.; Schmitz, G.; Romics, L.; Csaszar, A. Association of primary biliary cirrhosis with vitamin D receptor BsmI genotype polymorphism in a Hungarian population. Dig. Dis. Sci. 2000, 45, 1091–1095. [Google Scholar] [CrossRef]
- Lakatos, L.P.; Bajnok, E.; Hegedus, D.; Toth, T.; Lakatos, P.; Szalay, F. Vitamin D receptor, oestrogen receptor-alpha gene and interleukin-1 receptor antagonist gene polymorphisms in Hungarian patients with primary biliary cirrhosis. Eur. J. Gastroenterol. Hepatol. 2002, 14, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nezu, S.; Uegaki, S.; Kikuchi, K.; Shibuya, A.; Miyakawa, H.; Takahashi, S.; Bianchi, I.; Zermiani, P.; Podda, M.; et al. Vitamin D receptor polymorphisms are associated with increased susceptibility to primary biliary cirrhosis in Japanese and Italian populations. J. Hepatol. 2009, 50, 1202–1209. [Google Scholar] [CrossRef]
- Falleti, E.; Bitetto, D.; Fabris, C.; Cussigh, A.; Fontanini, E.; Fornasiere, E.; Fumolo, E.; Bignulin, S.; Cmet, S.; Minisini, R.; et al. Vitamin D receptor gene polymorphisms and hepatocellular carcinoma in alcoholic cirrhosis. World J. Gastroenterol. 2010, 16, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zeng, H.; Zhang, G.; Zhou, W.; Yan, Q.; Dai, L.; Wang, X. The associated ion between the VDR gene polymorphisms and susceptibility to hepatocellular carcinoma and the clinicopathological features in subjects infected with HBV. Biomed. Res. Int. 2013, 953974, 23. [Google Scholar] [CrossRef] [PubMed]
- Suneetha, P.V.; Sarin, S.K.; Goyal, A.; Kumar, G.T.; Shukla, D.K.; Hissar, S. Association between vitamin D receptor, CCR5, TNF-alpha and TNF-beta gene polymorphisms and HBV infection and severity of liver disease. J. Hepatol. 2006, 44, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Tu, X.; Zhu, Y.; Zhou, L.; Pfeiffer, T.; Feltens, R.; Stoecker, W.; Zhong, R. Genetic association of vitamin D receptor polymorphisms with autoimmune hepatitis and primary biliary cirrhosis in the Chinese. J. Gastroenterol. Hepatol. 2005, 20, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Tsounis, E.P.; Tourkochristou, E.; Sapsani, A.; Aggeletopoulou, I.; Lourida, T.; Ζisimopoulos, Κ.; Tzikopoulos, T.; Diamantopoulou, G.; Tsintoni, A.; Thomopoulos, K.; et al. The role of vitamin D receptor polymorphisms in the course of chronic hepatitis C infection. Ann. Gastroenterol. 2022, 35, 203–212. [Google Scholar] [CrossRef]
- Baur, K.; Mertens, J.C.; Schmitt, J.; Iwata, R.; Stieger, B.; Eloranta, J.J.; Frei, P.; Stickel, F.; Dill, M.T.; Seifert, B.; et al. Combined effect of 25-OH vitamin D plasma levels and genetic vitamin D receptor (NR 1I1) variants on fibrosis progression rate in HCV patients. Liver Int. 2012, 32, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Triantos, C.; Aggeletopoulou, I.; Kalafateli, M.; Spantidea, P.I.; Vourli, G.; Diamantopoulou, G.; Tapratzi, D.; Michalaki, M.; Manolakopoulos, S.; Gogos, C.; et al. Prognostic significance of vitamin D receptor (VDR) gene polymorphisms in liver cirrhosis. Sci. Rep. 2018, 8, 14065. [Google Scholar] [CrossRef]
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D counteracts fibrogenic TGF-beta signalling in human hepatic stellate cells both receptor-dependently and independently. Gut 2015, 64, 791–799. [Google Scholar] [CrossRef]
- Petta, S.; Grimaudo, S.; Marco, V.D.; Scazzone, C.; Macaluso, F.S.; Camma, C.; Cabibi, D.; Pipitone, R.; Craxi, A. Association of vitamin D serum levels and its common genetic determinants, with severity of liver fibrosis in genotype 1 chronic hepatitis C patients. J. Viral Hepat. 2013, 20, 486–493. [Google Scholar] [CrossRef]
- Grunhage, F.; Hochrath, K.; Krawczyk, M.; Hoblinger, A.; Obermayer-Pietsch, B.; Geisel, J.; Trauner, M.; Sauerbruch, T.; Lammert, F. Common genetic variation in vitamin D metabolism is associated with liver stiffness. Hepatology 2012, 56, 1883–1891. [Google Scholar] [CrossRef]
- Gombart, A.F.; Pierre, A.; Maggini, S. A Review of Micronutrients and the Immune System-Working in Harmony to Reduce the Risk of Infection. Nutrients 2020, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Vernia, F.; Valvano, M.; Longo, S.; Cesaro, N.; Viscido, A.; Latella, G. Vitamin D in Inflammatory Bowel Diseases. Mechanisms of Action and Therapeutic Implications. Nutrients 2022, 14, 269. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Bouillon, R. Extra-Skeletal Effects of Vitamin D. Front. Horm. Res. 2018, 50, 72–88. [Google Scholar] [PubMed]
- Damoiseaux, J.; Smolders, J. The Engagement Between Vitamin D and the Immune System: Is Consolidation by a Marriage to Be Expected? EBioMedicine 2018, 31, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Triantos, C.; Aggeletopoulou, I.; Mantzaris, G.J.; Mouzaki, A. Molecular basis of vitamin D action in inflammatory bowel disease. Autoimmun. Rev. 2022, 21, 103136. [Google Scholar] [CrossRef]
- Barrea, L.; Muscogiuri, G. Nutrition and immune system: From the Mediterranean diet to dietary supplementary through the microbiota. Crit. Rev. Food Sci. Nutr. 2021, 61, 3066–3090. [Google Scholar] [CrossRef] [PubMed]
- Barbáchano, A.; Fernández-Barral, A.; Ferrer-Mayorga, G.; Costales-Carrera, A.; Larriba, M.J.; Muñoz, A. The endocrine vitamin D system in the gut. Mol. Cell. Endocrinol. 2017, 453, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Anty, R.; Tonohouan, M.; Ferrari-Panaia, P.; Piche, T.; Pariente, A.; Anstee, Q.M.; Gual, P.; Tran, A. Low Levels of 25-Hydroxy Vitamin D are Independently Associated with the Risk of Bacterial Infection in Cirrhotic Patients. Clin. Transl. Gastroenterol. 2014, 5, e56. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Hsieh, Y.C.; Huo, T.I.; Yang, U.C.; Lin, C.H.; Li, C.P.; Huang, Y.H.; Hou, M.C.; Lin, H.C.; Lee, K.C. Active Vitamin D(3) Treatment Attenuated Bacterial Translocation via Improving Intestinal Barriers in Cirrhotic Rats. Mol. Nutr. Food Res. 2021, 65, e2000937. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhang, R.; Luo, M.; Zhang, T.; Pan, L.; Xu, S.; Pan, L.; Ren, F.; Ji, C.; Hu, R. Impaired 25-hydroxylation of vitamin D in liver injury suppresses intestinal Paneth cell defensins, leading to gut dysbiosis and liver fibrogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G685–G695. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shang, X.; Jin, S.; Ma, Z.; Wang, H.; Ao, N.; Yang, J.; Du, J. Vitamin D ameliorates high-fat-diet-induced hepatic injury via inhibiting pyroptosis and alters gut microbiota in rats. Arch. Biochem. Biophys. 2021, 705, 108894. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggeletopoulou, I.; Thomopoulos, K.; Mouzaki, A.; Triantos, C. Vitamin D–VDR Novel Anti-Inflammatory Molecules—New Insights into Their Effects on Liver Diseases. Int. J. Mol. Sci. 2022, 23, 8465. https://doi.org/10.3390/ijms23158465
Aggeletopoulou I, Thomopoulos K, Mouzaki A, Triantos C. Vitamin D–VDR Novel Anti-Inflammatory Molecules—New Insights into Their Effects on Liver Diseases. International Journal of Molecular Sciences. 2022; 23(15):8465. https://doi.org/10.3390/ijms23158465
Chicago/Turabian StyleAggeletopoulou, Ioanna, Konstantinos Thomopoulos, Athanasia Mouzaki, and Christos Triantos. 2022. "Vitamin D–VDR Novel Anti-Inflammatory Molecules—New Insights into Their Effects on Liver Diseases" International Journal of Molecular Sciences 23, no. 15: 8465. https://doi.org/10.3390/ijms23158465
APA StyleAggeletopoulou, I., Thomopoulos, K., Mouzaki, A., & Triantos, C. (2022). Vitamin D–VDR Novel Anti-Inflammatory Molecules—New Insights into Their Effects on Liver Diseases. International Journal of Molecular Sciences, 23(15), 8465. https://doi.org/10.3390/ijms23158465