Correction of Fanconi Anemia Mutations Using Digital Genome Engineering

 , , , ,
, , , ,  ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
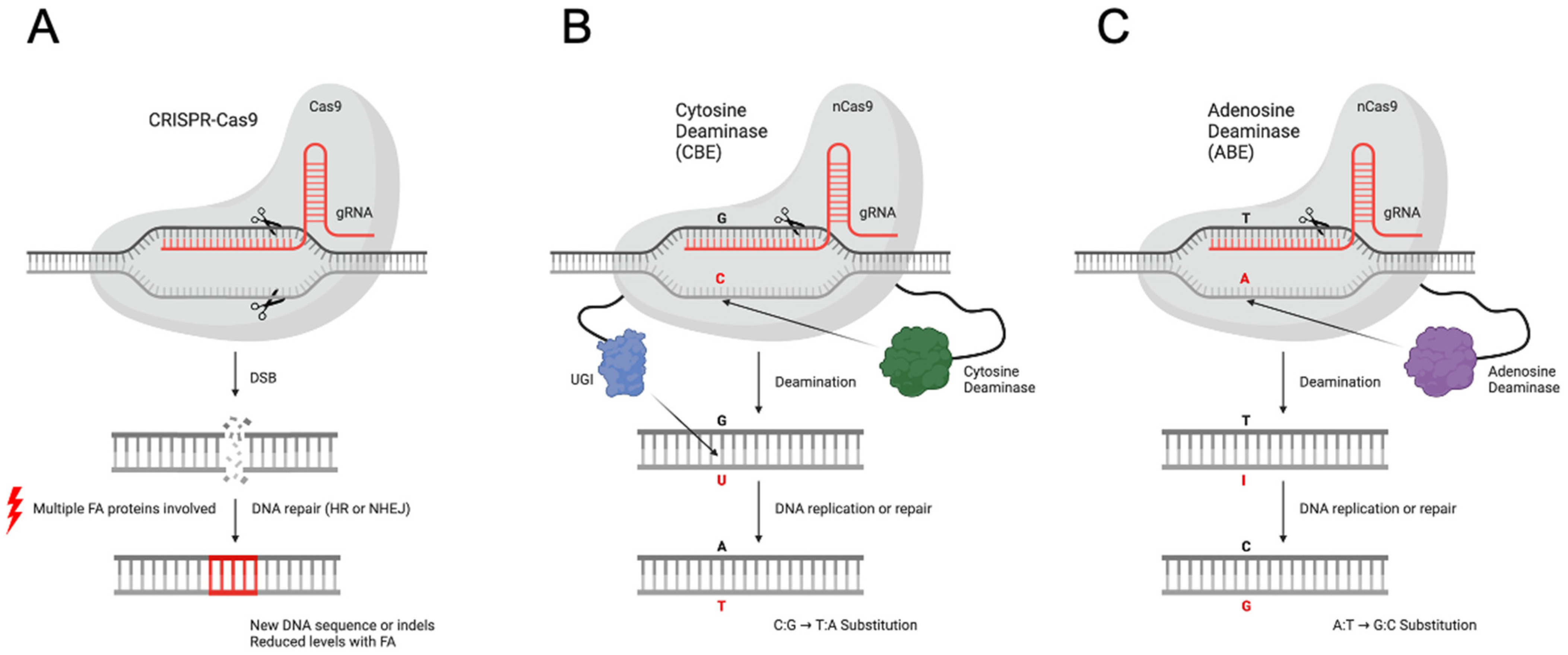
:1. Introduction
2. Results
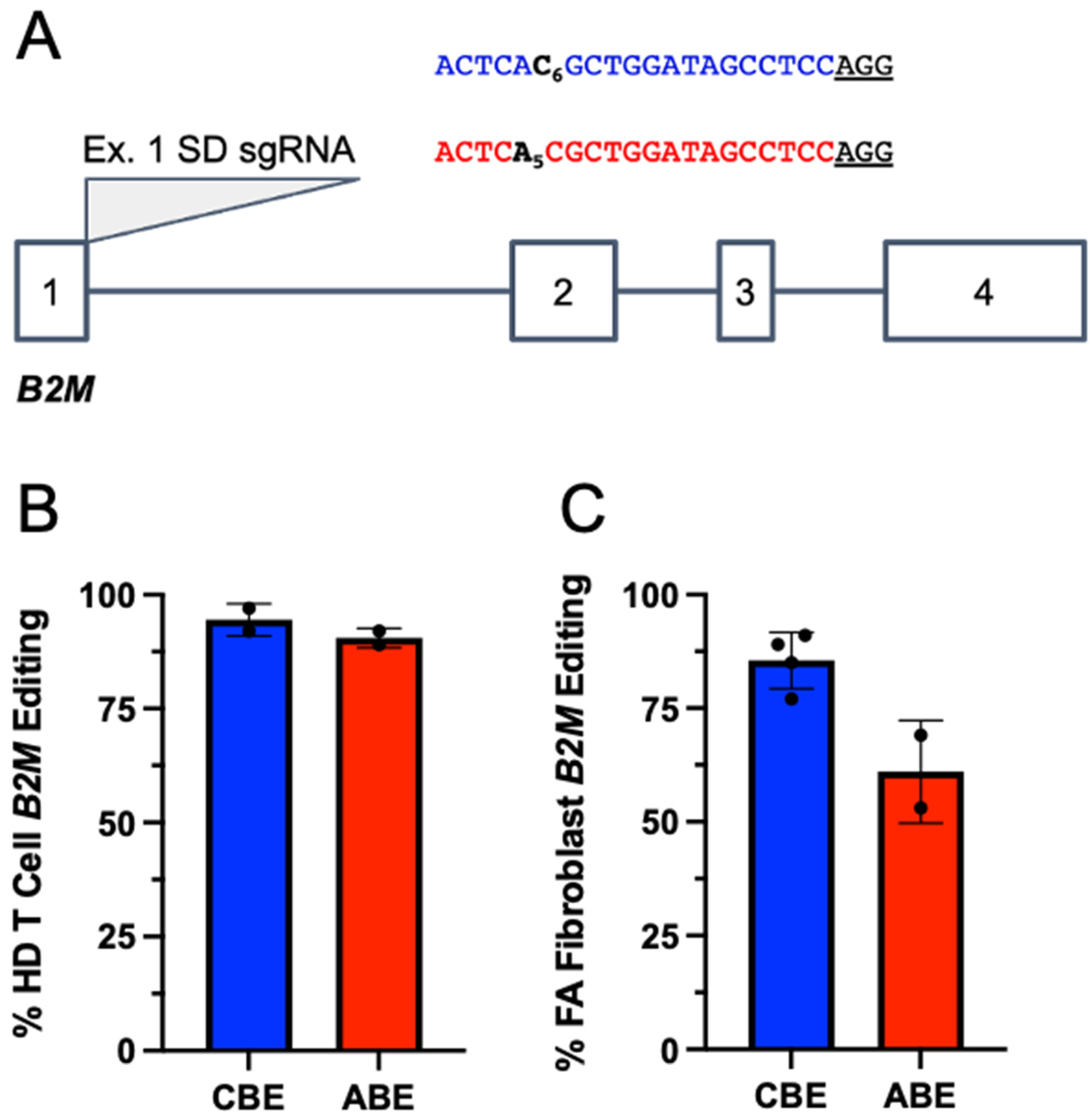
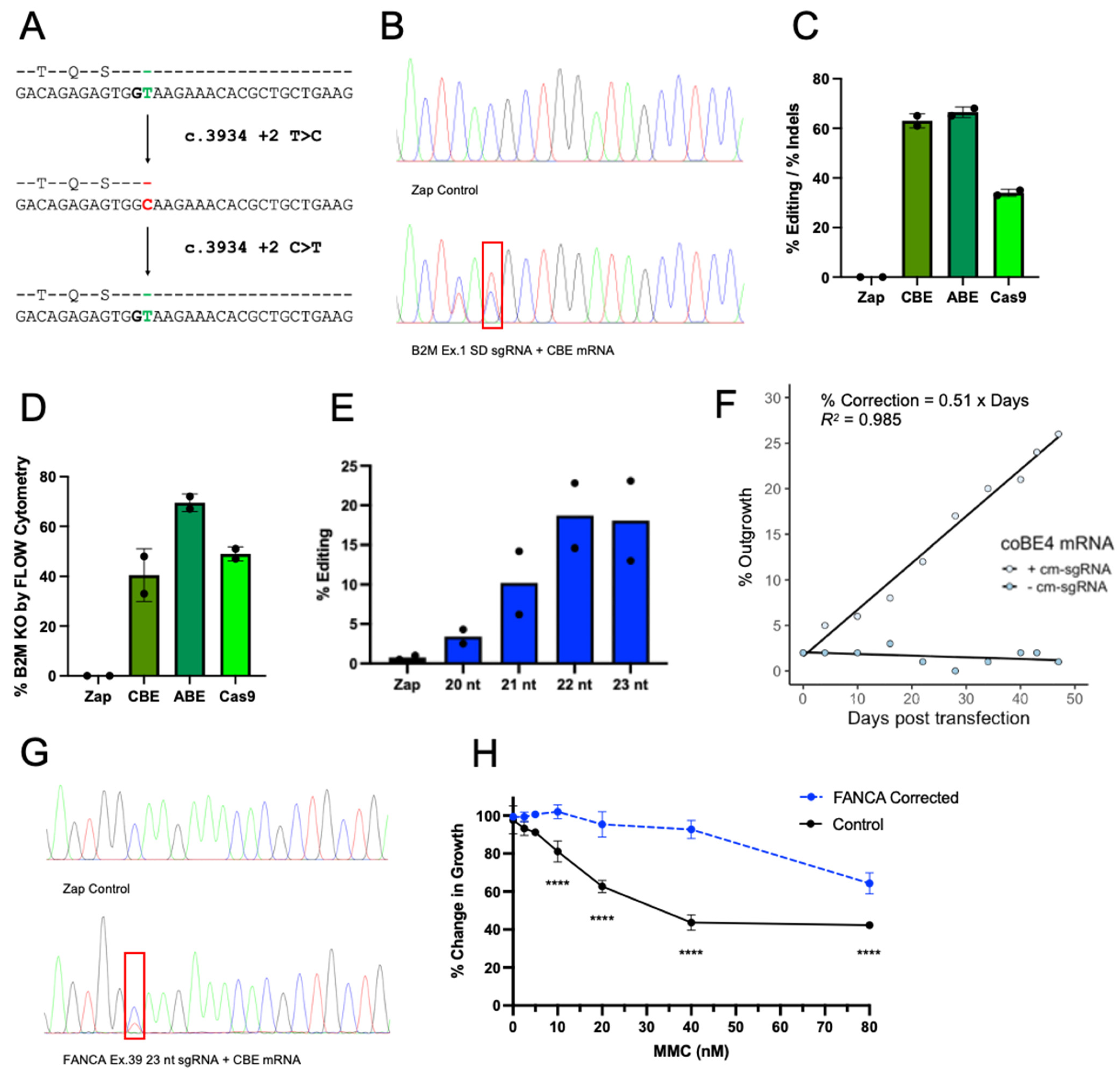
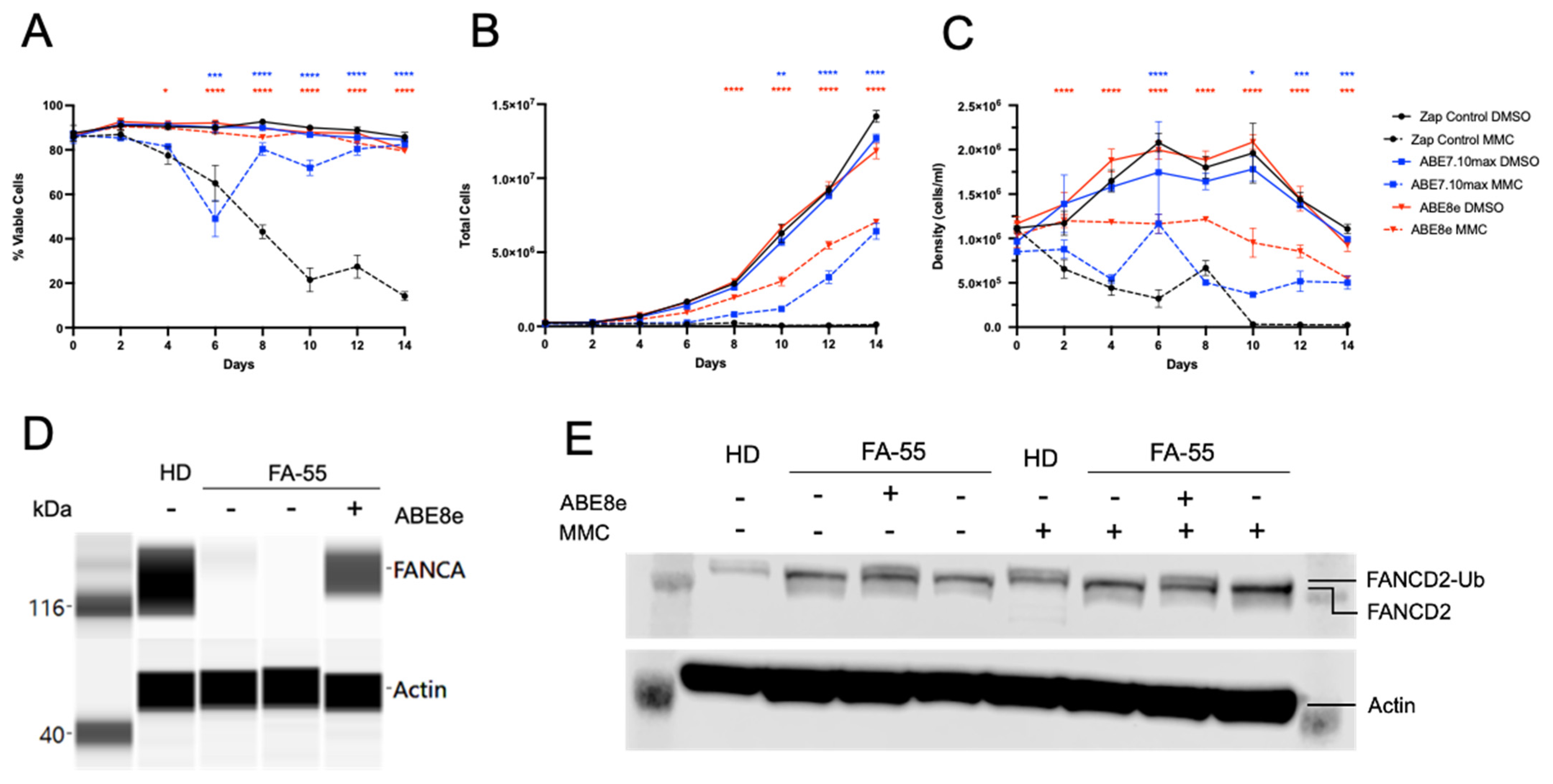
2.1. Base Editors Are Highly Functional in FA Cells
2.2. Genetic Correction with Base Editor Confers Phenotypic Rescue of FA Cells
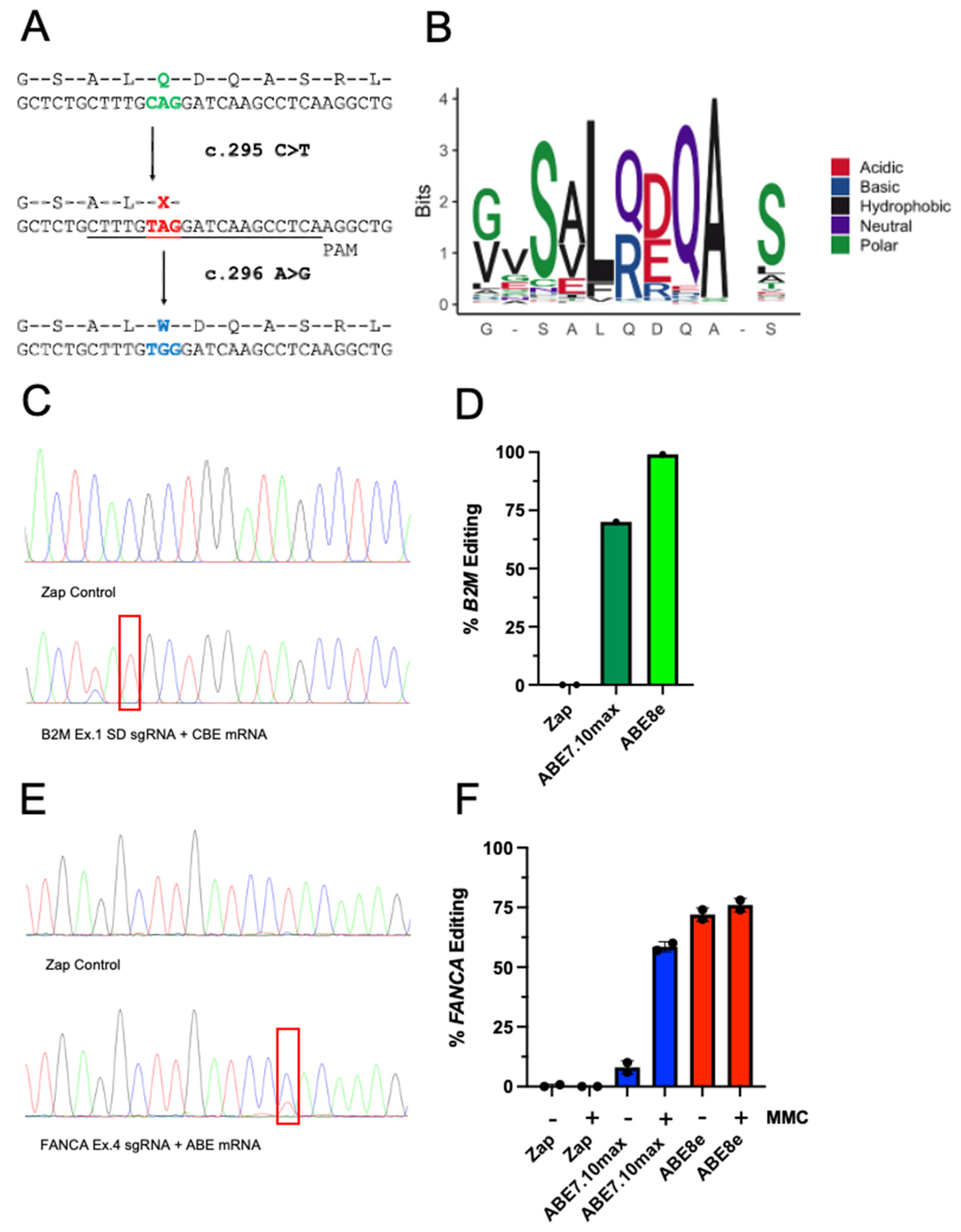
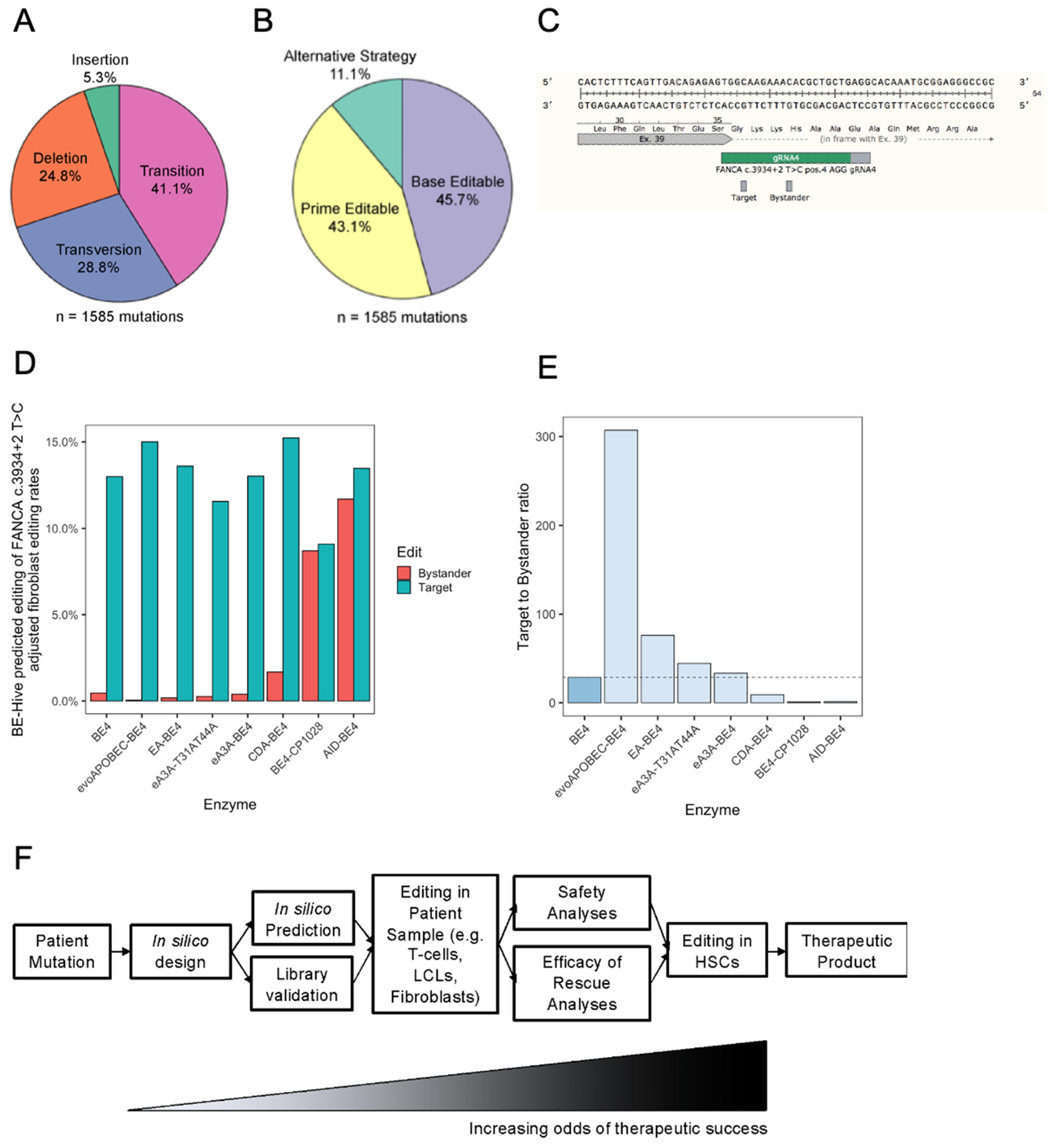
2.3. The Most Common FA Mutation Is Highly Amenable to Base Editing
2.4. Prediction of FA Mutation Outcomes with Base Editors
3. Discussion
4. Materials and Methods
4.1. Fibroblast Culture
4.2. Preparation of LCL Feeder Cells
4.3. LCL Transformation
4.4. LCL Culture
4.5. Fibroblast Base Editing
4.6. LCL Base Editing
4.7. Genomic Analysis
4.8. In Vitro Base Editing Assays
4.9. sgRNAs and PCR Primers
4.10. Flow Cytometry
4.11. MMC Sensitivity Assay
4.12. FANCA Western Blot
4.13. FANCD2 Western Blot
4.14. Statistical Analysis
4.15. Computational Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenberg, P.S.; Tamary, H.; Alter, B.P. How High Are Carrier Frequencies of Rare Recessive Syndromes? Contemporary Estimates for Fanconi Anemia in the United States and Israel. Am. J. Med. Genet. A 2011, 155A, 1877–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malric, A.; Defachelles, A.-S.; Leblanc, T.; Lescoeur, B.; Lacour, B.; Peuchmaur, M.; Maurage, C.-A.; Pierron, G.; Guillemot, D.; d’Enghien, C.D.; et al. Fanconi Anemia and Solid Malignancies in Childhood: A National Retrospective Study. Pediatr. Blood Cancer 2015, 62, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Rosenberg, P.S. VACTERL-H Association and Fanconi Anemia. Mol. Syndromol. 2013, 4, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walden, H.; Deans, A.J. The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef]
- Pang, Q.; Andreassen, P.R. Fanconi Anemia Proteins and Endogenous Stresses. Mutat. Res. 2009, 668, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.-S.; Pondarre, C.; et al. Bone Marrow Failure in Fanconi Anemia Is Triggered by an Exacerbated p53/p21 DNA Damage Response That Impairs Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef] [Green Version]
- MacMillan, M.L.; Hughes, M.R.; Agarwal, S.; Daley, G.Q. Cellular Therapy for Fanconi Anemia: The Past, Present, and Future. Biol. Blood Marrow Transplant. 2011, 17, S109–S114. [Google Scholar] [CrossRef] [Green Version]
- Moreno, O.M.; Paredes, A.C.; Suarez-Obando, F.; Rojas, A. An Update on Fanconi Anemia: Clinical, Cytogenetic and Molecular Approaches (Review). Biomed. Rep. 2021, 15, 74. [Google Scholar] [CrossRef]
- Tan, P.-L.; Wagner, J.E.; Auerbach, A.D.; Defor, T.E.; Slungaard, A.; Macmillan, M.L. Successful Engraftment without Radiation after Fludarabine-Based Regimen in Fanconi Anemia Patients Undergoing Genotypically Identical Donor Hematopoietic Cell Transplantation. Pediatr. Blood Cancer 2006, 46, 630–636. [Google Scholar] [CrossRef]
- Ebens, C.L.; MacMillan, M.L.; Wagner, J.E. Hematopoietic Cell Transplantation in Fanconi Anemia: Current Evidence, Challenges and Recommendations. Expert Rev. Hematol. 2017, 10, 81–97. [Google Scholar] [CrossRef]
- Hayashi, R.J. Considerations in Preparative Regimen Selection to Minimize Rejection in Pediatric Hematopoietic Transplantation in Non-Malignant Diseases. Front. Immunol. 2020, 11, 567423. [Google Scholar] [CrossRef]
- Osborn, M.J.; Gabriel, R.; Webber, B.R.; DeFeo, A.P.; McElroy, A.N.; Jarjour, J.; Starker, C.G.; Wagner, J.E.; Joung, J.K.; Voytas, D.F.; et al. Fanconi Anemia Gene Editing by the CRISPR/Cas9 System. Hum. Gene Ther. 2015, 26, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Skvarova Kramarzova, K.; Osborn, M.J.; Webber, B.R.; DeFeo, A.P.; McElroy, A.N.; Kim, C.J.; Tolar, J. CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells. Int. J. Mol. Sci. 2017, 18, 1269. [Google Scholar] [CrossRef]
- Román-Rodríguez, F.J.; Ugalde, L.; Álvarez, L.; Díez, B.; Ramírez, M.J.; Risueño, C.; Cortón, M.; Bogliolo, M.; Bernal, S.; March, F.; et al. NHEJ-Mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell 2019, 25, 607–621.e7. [Google Scholar] [CrossRef] [Green Version]
- Shafqat, S.; Tariq, E.; Parnes, A.D.; Dasouki, M.J.; Ahmed, S.O.; Hashmi, S.K. Role of Gene Therapy in Fanconi Anemia: A Systematic and Literature Review with Future Directions. Hematol. Oncol. Stem Cell Ther. 2021, 14, 290–301. [Google Scholar] [CrossRef]
- Río, P.; Navarro, S.; Wang, W.; Sánchez-Domínguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful Engraftment of Gene-Corrected Hematopoietic Stem Cells in Non-Conditioned Patients with Fanconi Anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef]
- Sevilla, J.; Rio, P.; Navarro, S.; Sanchez-Dominguez, R.; Segovia, J.C.; Wang, W.; Zubicaray, J.; Yanez, R.; Casado, J.A.; Gimenez, Y.; et al. Follow-Up of a Phase I/II Gene Therapy Trial in Patients with Fanconi Anemia, Subtype A. In Molecular Therapy; Cell Press: Cambridge, MA, USA, 2021; Volume 29, p. 18. [Google Scholar]
- Kelly, P.F.; Radtke, S.; von Kalle, C.; Balcik, B.; Bohn, K.; Mueller, R.; Schuesler, T.; Haren, M.; Reeves, L.; Cancelas, J.A.; et al. Stem Cell Collection and Gene Transfer in Fanconi Anemia. Mol. Ther. 2007, 15, 211–219. [Google Scholar] [CrossRef]
- Rocket Pharmaceuticals Presents Positive Clinical Data from Fanconi Anemia, Leukocyte Adhesion Deficiency-I, and Pyruvate Kinase Deficiency Programs at 24th Annual Meeting of the American Society of Gene and Cell Therapy. Available online: https://ir.rocketpharma.com/news-releases/news-release-details/rocket-pharmaceuticals-presents-positive-clinical-data-fanconi (accessed on 26 April 2022).
- Webber, B.R.; Lonetree, C.-L.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.A.; et al. Highly Efficient Multiplex Human T Cell Engineering without Double-Strand Breaks Using Cas9 Base Editors. Nat. Commun. 2019, 10, 5222. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Hölz, K.; Pavlic, A.; Lietard, J.; Somoza, M.M. Specificity and Efficiency of the Uracil DNA Glycosylase-Mediated Strand Cleavage Surveyed on Large Sequence Libraries. Sci. Rep. 2019, 9, 17822. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 Genome Editing Induces a p53-Mediated DNA Damage Response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J. Chromothripsis as an on-Target Consequence of CRISPR-Cas9 Genome Editing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Adikusuma, F.; Piltz, S.; Corbett, M.A.; Turvey, M.; McColl, S.R.; Helbig, K.J.; Beard, M.R.; Hughes, J.; Pomerantz, R.T.; Thomas, P.Q. Large Deletions Induced by Cas9 Cleavage. Nature 2018, 560, E8–E9. [Google Scholar] [CrossRef] [Green Version]
- Callén, E.; Casado, J.A.; Tischkowitz, M.D.; Bueren, J.A.; Creus, A.; Marcos, R.; Dasí, A.; Estella, J.M.; Muñoz, A.; Ortega, J.J.; et al. A Common Founder Mutation in FANCA Underlies the World’s Highest Prevalence of Fanconi Anemia in Gypsy Families from Spain. Blood 2005, 105, 1946–1949. [Google Scholar] [CrossRef] [Green Version]
- Macé-Aimé, G.; Couvé, S.; Khassenov, B.; Rosselli, F.; Saparbaev, M.K. The Fanconi Anemia Pathway Promotes DNA Glycosylase-Dependent Excision of Interstrand DNA Crosslinks. Environ. Mol. Mutagen. 2010, 51, 508–519. [Google Scholar] [CrossRef]
- Kurata, M.; Wolf, N.K.; Lahr, W.S.; Weg, M.T.; Kluesner, M.G.; Lee, S.; Hui, K.; Shiraiwa, M.; Webber, B.R.; Moriarity, B.S. Highly Multiplexed Genome Engineering Using CRISPR/Cas9 gRNA Arrays. PLoS ONE 2018, 13, e0198714. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.-M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.-T.; Kim, H.S.; Kim, D.-E.; Lee, H.; Chung, E.; et al. Adenine Base Editing in Mouse Embryos and an Adult Mouse Model of Duchenne Muscular Dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef]
- Kluesner, M.G.; Lahr, W.S.; Lonetree, C.-L.; Smeester, B.A.; Qiu, X.; Slipek, N.J.; Claudio Vázquez, P.N.; Pitzen, S.P.; Pomeroy, E.J.; Vignes, M.J.; et al. CRISPR-Cas9 Cytidine and Adenosine Base Editing of Splice-Sites Mediates Highly-Efficient Disruption of Proteins in Primary and Immortalized Cells. Nat. Commun. 2021, 12, 2437. [Google Scholar] [CrossRef]
- Pinto, F.O.; Leblanc, T.; Chamousset, D.; Le Roux, G.; Brethon, B.; Cassinat, B.; Larghero, J.; de Villartay, J.-P.; Stoppa-Lyonnet, D.; Baruchel, A.; et al. Diagnosis of Fanconi Anemia in Patients with Bone Marrow Failure. Haematologica 2009, 94, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcourt, M.F.; Penketh, P.G.; Hodnick, W.F.; Johnson, D.A.; Sherman, D.H.; Rockwell, S.; Sartorelli, A.C. Mitomycin Resistance in Mammalian Cells Expressing the Bacterial Mitomycin C Resistance Protein MCRA. Proc. Natl. Acad. Sci. USA 1999, 96, 10489–10494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluesner, M.G.; Nedveck, D.A.; Lahr, W.S.; Garbe, J.R.; Abrahante, J.E.; Webber, B.R.; Moriarity, B.S. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J 2018, 1, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Kucharski, M.; Mrowiec, P.; Ocłoń, E. Current Standards and Pitfalls Associated with the Transfection of Primary Fibroblast Cells. Biotechnol. Prog. 2021, 37, e3152. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Nicoletti, E.; Rao, G.; Bueren, J.A.; Río, P.; Navarro, S.; Surrallés, J.; Choi, G.; Schwartz, J.D. Mosaicism in Fanconi Anemia: Concise Review and Evaluation of Published Cases with Focus on Clinical Course of Blood Count Normalization. Ann. Hematol. 2020, 99, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; van Twest, S.; Leis, A.; Bythell-Douglas, R.; Murphy, V.J.; Sharp, M.; Parker, M.W.; Crismani, W.; Deans, A.J. Monoubiquitination by the Human Fanconi Anemia Core Complex Clamps FANCI:FANCD2 on DNA in Filamentous Arrays. eLife 2020, 9, e54128. [Google Scholar] [CrossRef]
- Bagby, G.C., Jr. The Genetic Basis of Fanconi Anemia; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- ClinVar ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=fanconi+anemia (accessed on 26 April 2022).
- Arbab, M.; Shen, M.W.; Mok, B.; Wilson, C.; Matuszek, Ż.; Cassa, C.A.; Liu, D.R. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell 2020, 182, 463–480.e30. [Google Scholar] [CrossRef]
- Demirci, S.; Leonard, A.; Tisdale, J.F. Hematopoietic Stem Cells from Pluripotent Stem Cells: Clinical Potential, Challenges, and Future Perspectives. Stem Cells Transl. Med. 2020, 9, 1549–1557. [Google Scholar] [CrossRef]
- Wattanapanitch, M. Recent Updates on Induced Pluripotent Stem Cells in Hematological Disorders. Stem Cells Int. 2019, 2019, 5171032. [Google Scholar] [CrossRef]
- Hung, S.S.C.; Chrysostomou, V.; Li, F.; Lim, J.K.H.; Wang, J.-H.; Powell, J.E.; Tu, L.; Daniszewski, M.; Lo, C.; Wong, R.C.; et al. AAV-Mediated CRISPR/Cas Gene Editing of Retinal Cells In Vivo. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3470–3476. [Google Scholar] [CrossRef]
- Li, C.; Wang, H.; Georgakopoulou, A.; Gil, S.; Yannaki, E.; Lieber, A. In Vivo HSC Gene Therapy Using a Bi-Modular HDAd5/35++ Vector Cures Sickle Cell Disease in a Mouse Model. Mol. Ther. 2021, 29, 822–837. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Villiger, L.; Rothgangl, T.; Witzigmann, D.; Oka, R.; Lin, P.J.C.; Qi, W.; Janjuha, S.; Berk, C.; Ringnalda, F.; Beattie, M.B.; et al. In Vivo Cytidine Base Editing of Hepatocytes without Detectable off-Target Mutations in RNA and DNA. Nat. Biomed. Eng. 2021, 5, 179–189. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Mishra, A.; Gil, S.; Hawkins, R.D.; Yannaki, E.; Lieber, A. In Vivo HSPC Gene Therapy with Base Editors Allows for Efficient Reactivation of Fetal γ-Globin in β-YAC Mice. Blood Adv. 2021, 5, 1122–1135. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, W.; Nguyen, G.N.; Zhang, C.; Zeng, C.; Yan, J.; Du, S.; Hou, X.; Li, W.; Jiang, J.; et al. Functionalized Lipid-like Nanoparticles for in Vivo mRNA Delivery and Base Editing. Sci. Adv. 2020, 6, eabc2315. [Google Scholar] [CrossRef]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered Virus-like Particles for Efficient in Vivo Delivery of Therapeutic Proteins. Cell 2022, 185, 250–265.e16. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.-A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef]
- Richter, R.; Forssmann, W.; Henschler, R. Current Developments in Mobilization of Hematopoietic Stem and Progenitor Cells and Their Interaction with Niches in Bone Marrow. Transfus. Med. Hemother. 2017, 44, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-Seq Enables Genome-Wide Profiling of off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Martufi, M.; Good, R.B.; Rapiteanu, R.; Schmidt, T.; Patili, E.; Tvermosegaard, K.; New, M.; Nanthakumar, C.B.; Betts, J.; Blanchard, A.D.; et al. Single-Step, High-Efficiency CRISPR-Cas9 Genome Editing in Primary Human Disease-Derived Fibroblasts. CRISPR J. 2019, 2, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sipe, C.J.; Kluesner, M.G.; Bingea, S.P.; Lahr, W.S.; Andrew, A.A.; Wang, M.; DeFeo, A.P.; Hinkel, T.L.; Laoharawee, K.; Wagner, J.E.; et al. Correction of Fanconi Anemia Mutations Using Digital Genome Engineering. Int. J. Mol. Sci. 2022, 23, 8416. https://doi.org/10.3390/ijms23158416
Sipe CJ, Kluesner MG, Bingea SP, Lahr WS, Andrew AA, Wang M, DeFeo AP, Hinkel TL, Laoharawee K, Wagner JE, et al. Correction of Fanconi Anemia Mutations Using Digital Genome Engineering. International Journal of Molecular Sciences. 2022; 23(15):8416. https://doi.org/10.3390/ijms23158416
Chicago/Turabian StyleSipe, Christopher J., Mitchell G. Kluesner, Samuel P. Bingea, Walker S. Lahr, Aneesha A. Andrew, Minjing Wang, Anthony P. DeFeo, Timothy L. Hinkel, Kanut Laoharawee, John E. Wagner, and et al. 2022. "Correction of Fanconi Anemia Mutations Using Digital Genome Engineering" International Journal of Molecular Sciences 23, no. 15: 8416. https://doi.org/10.3390/ijms23158416
APA StyleSipe, C. J., Kluesner, M. G., Bingea, S. P., Lahr, W. S., Andrew, A. A., Wang, M., DeFeo, A. P., Hinkel, T. L., Laoharawee, K., Wagner, J. E., MacMillan, M. L., Vercellotti, G. M., Tolar, J., Osborn, M. J., McIvor, R. S., Webber, B. R., & Moriarity, B. S. (2022). Correction of Fanconi Anemia Mutations Using Digital Genome Engineering. International Journal of Molecular Sciences, 23(15), 8416. https://doi.org/10.3390/ijms23158416