
PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them


Abstract
:1. PARP Inhibitors for Cancer Treatment
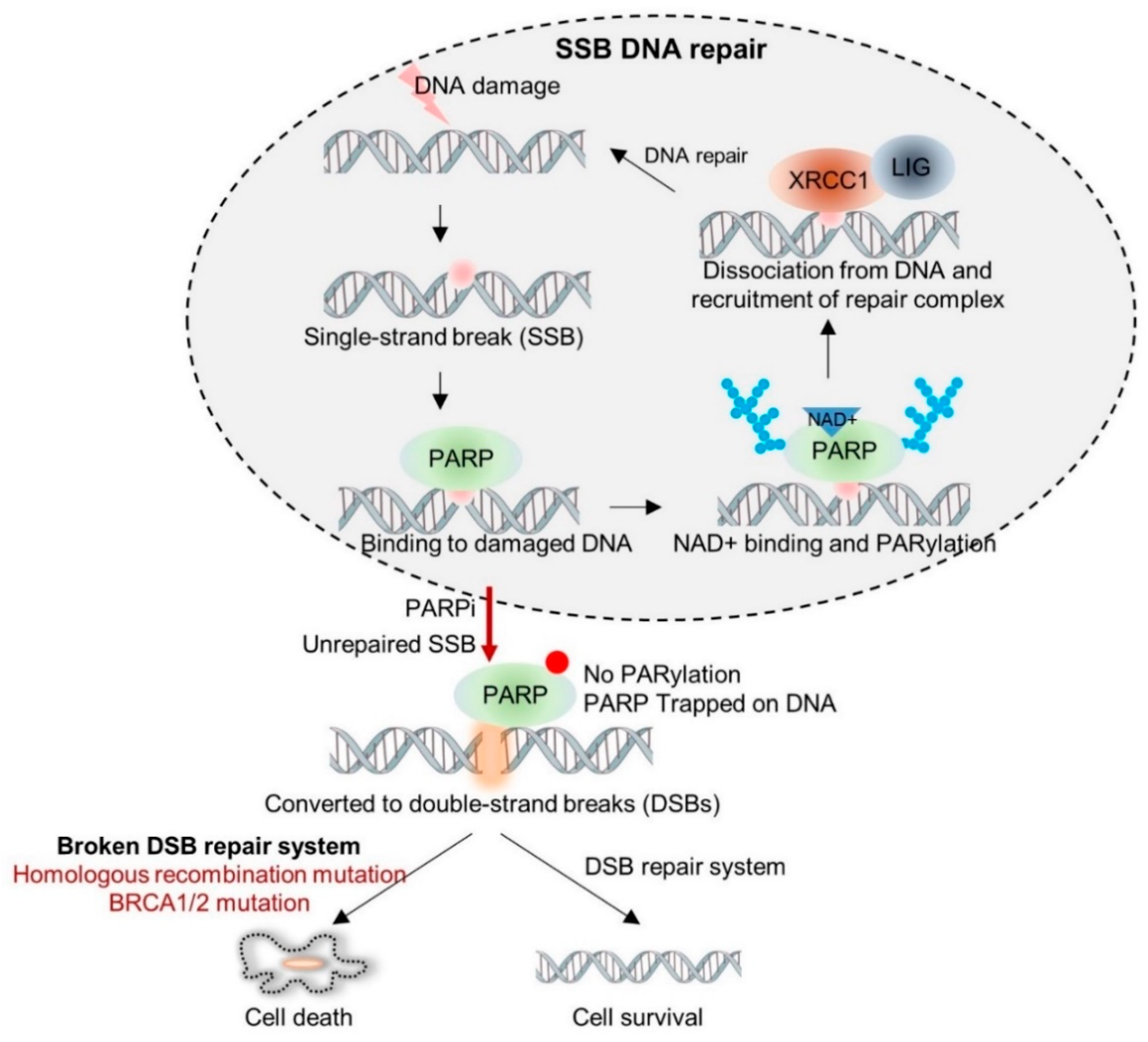
1.1. Synthetic Lethality of PARP Inhibitors
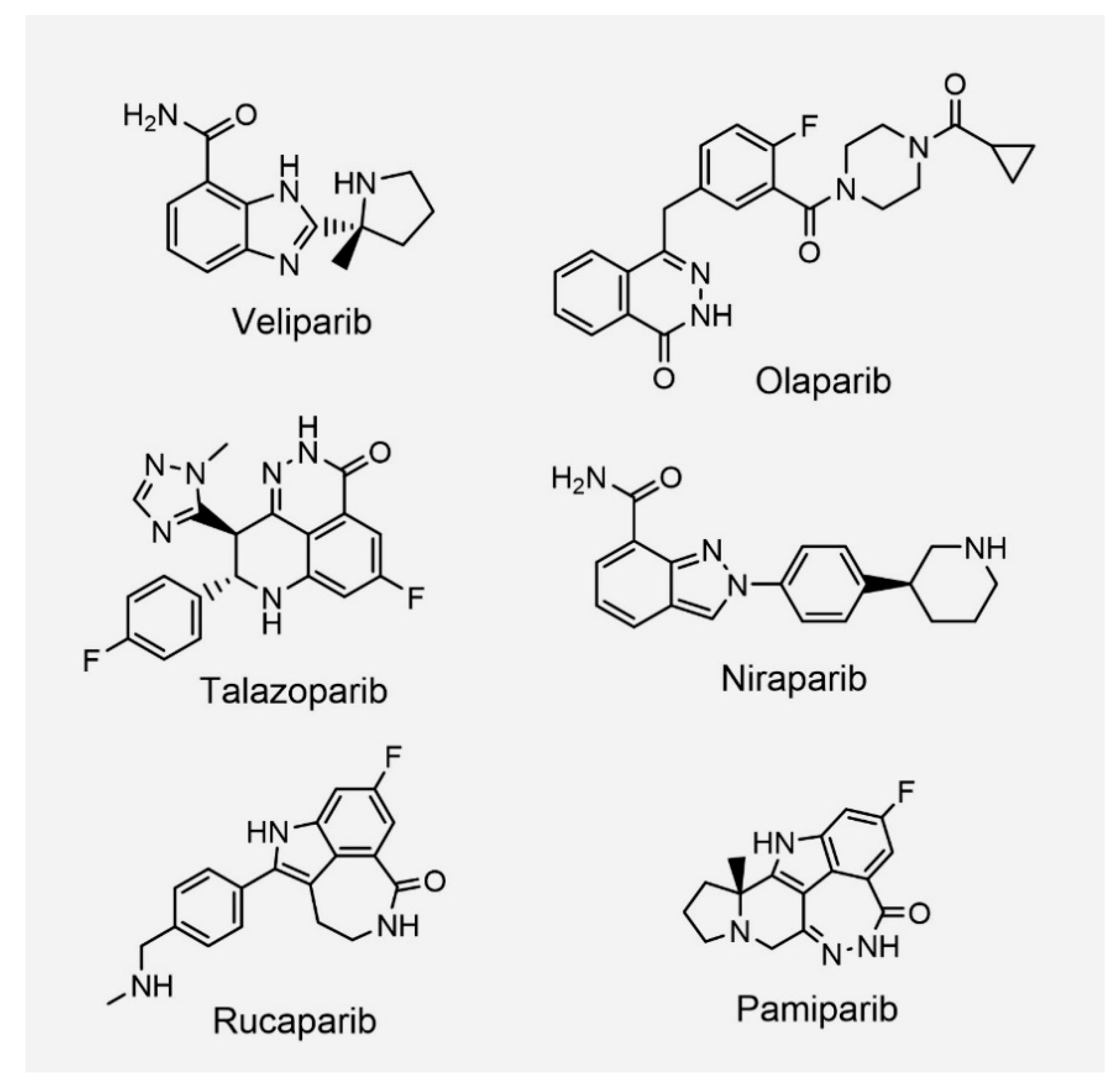
1.2. PARP Inhibitors from Bench to Bedside
2. PARP Inhibitors: Back to the Bench: PARPi Resistance
2.1. Reversion Mutations of HR Genes
2.2. Restoration of HR via Inactivation of Non-Homologous End Joining Proteins
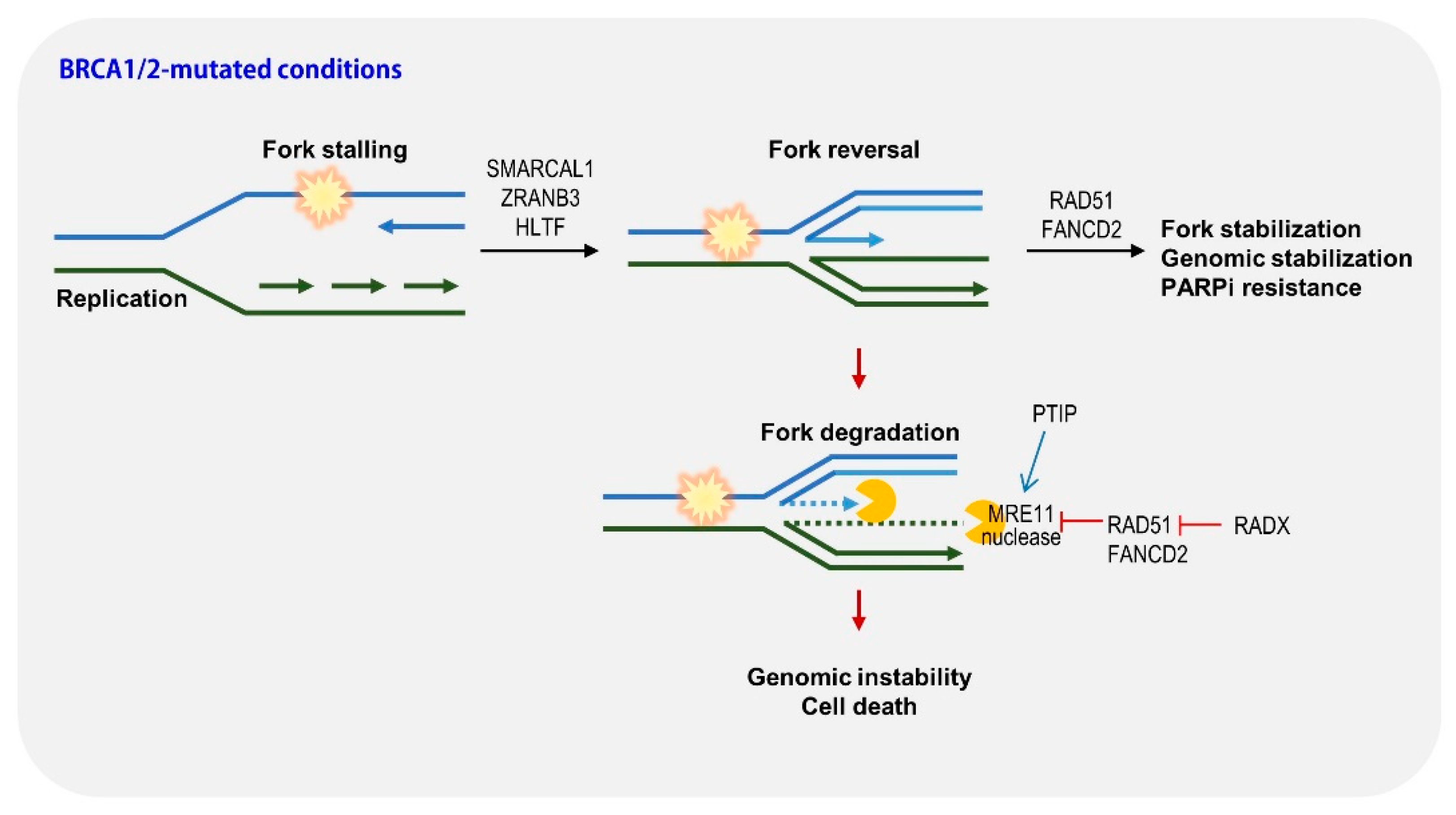
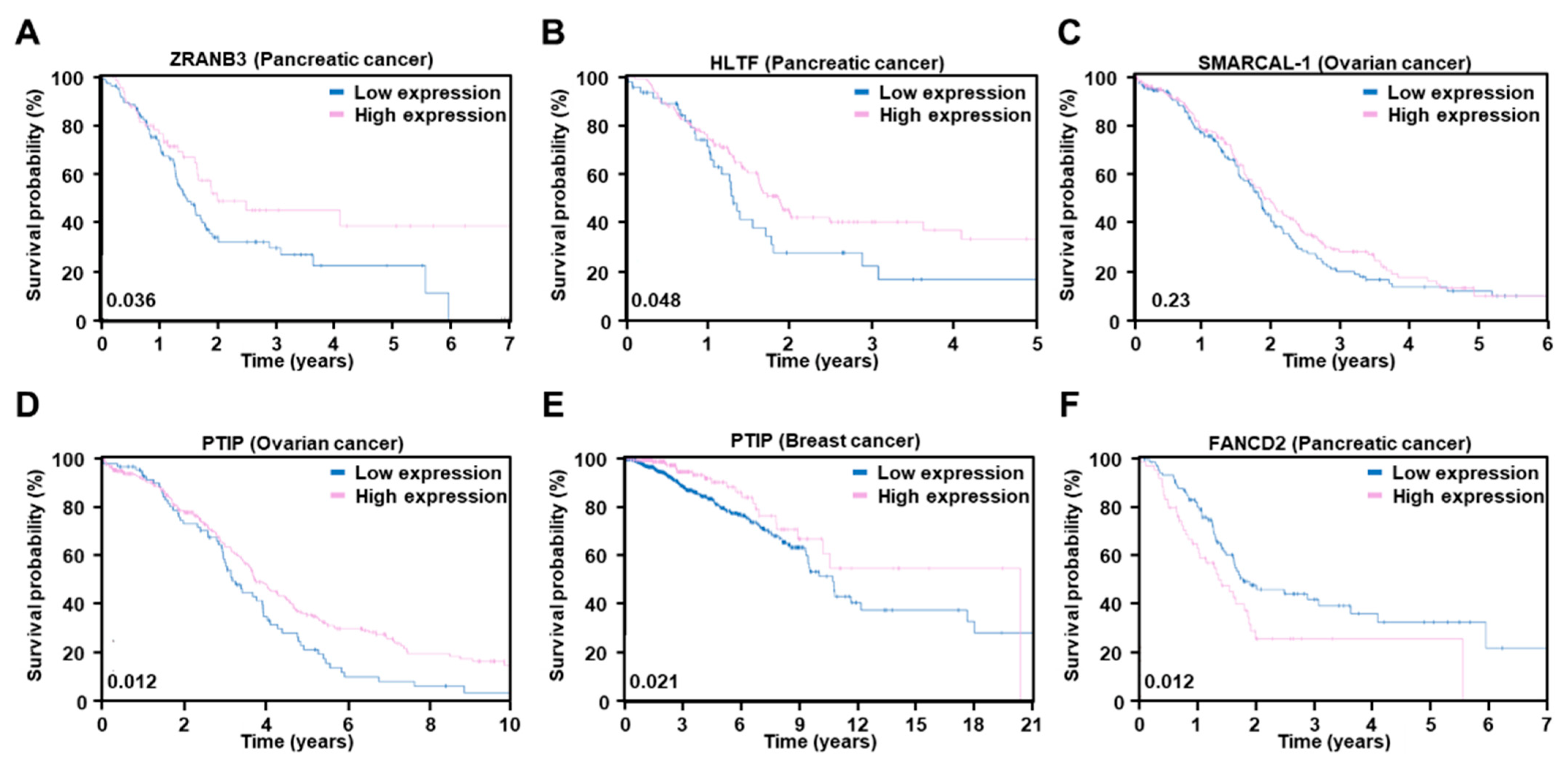
2.3. Restoration of Replication Fork Stability
2.4. PARP Mutations
3. Lessons from Bench: How Can We Overcome Resistance to PARPi or Expand the Use of PARPi?
3.1. PARPi Combination Approaches
3.2. Chemical Modifications of PARP Inhibitors
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. BioEssays News Rev. Mol. Cell. Dev. Biol. 2004, 26, 882–893. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Yap, T.A.; de Bono, J.S. Poly(ADP-ribose) polymerase inhibitors in cancer treatment: A clinical perspective. Eur. J. Cancer 2010, 46, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.M.; Belousova, E.A.; Kurgina, T.A.; Ukraintsev, A.A.; Vasil’eva, I.A.; Khodyreva, S.N.; Lavrik, O.I. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 2021, 11, 4849. [Google Scholar] [CrossRef]
- Izumi, T.; Mellon, I. Base Excision Repair and Nucleotide Excision Repair. In Genome Stability; Elsevier: Amsterdam, The Netherlands, 2021; pp. 293–322. [Google Scholar]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kustatscher, G.; Alabert, C.; Hodl, M.; Forne, I.; Volker-Albert, M.; Satpathy, S.; Beyer, T.E.; Mailand, N.; Choudhary, C.; et al. Proteome dynamics at broken replication forks reveal a distinct ATM-directed repair response suppressing DNA double-strand break ubiquitination. Mol. Cell 2021, 81, 1084–1099.e6. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly (ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res./DNA Repair 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Friebel, T.; Lynch, H.T.; Neuhausen, S.L.; van’t Veer, L.; Garber, J.E.; Evans, G.R.; Narod, S.A.; Isaacs, C.; Matloff, E.; et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: The PROSE Study Group. J. Clin. Oncol. 2004, 22, 1055–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebbeck, T.R.; Lynch, H.T.; Neuhausen, S.L.; Narod, S.A.; Van’t Veer, L.; Garber, J.E.; Evans, G.; Isaacs, C.; Daly, M.B.; Matloff, E. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N. Engl. J. Med. 2002, 346, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919849753. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Anscher, M.S.; Chang, E.; Gao, X.; Gong, Y.; Weinstock, C.; Bloomquist, E.; Adeniyi, O.; Charlab, R.; Zimmerman, S.; Serlemitsos-Day, M.; et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA-Mutated Metastatic Castrate-Resistant Prostate Cancer. Oncologist 2021, 26, 139–146. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs 2020, 38, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Arun, B.K.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bell-McGuinn, K.M.; Bach, B.A.; Kundu, M.G.; et al. Efficacy and safety of first-line veliparib and carboplatin-paclitaxel in patients with HER2- advanced germline BRCA+ breast cancer: Subgroup analysis of a randomised clinical trial. Eur. J. Cancer 2021, 154, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Novello, S.; Guclu, S.Z.; Bentsion, D.; Zvirbule, Z.; Szilasi, M.; Bernabe, R.; Syrigos, K.; Byers, L.A.; Clingan, P. Veliparib in Combination With Platinum-Based Chemotherapy for First-Line Treatment of Advanced Squamous Cell Lung Cancer: A Randomized, Multicenter Phase III Study. J. Clin. Oncol. 2021, 39, 3633–3644. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- Kruger, A.; Burkle, A.; Hauser, K.; Mangerich, A. Real-time monitoring of PARP1-dependent PARylation by ATR-FTIR spectroscopy. Nat. Commun. 2020, 11, 2174. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Ettl, J.; Quek, R.G.W.; Lee, K.H.; Rugo, H.S.; Hurvitz, S.; Goncalves, A.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: Patient-reported outcomes from the EMBRACA phase III trial. Ann. Oncol. 2018, 29, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Matulonis, U.A.; Malander, S.; Hudgens, S.; Sehouli, J.; del Campo, J.M.; Berton-Rigaud, D.; Banerjee, S.; Scambia, G.; Berek, J.S.; et al. Quality of life in patients with recurrent ovarian cancer treated with niraparib versus placebo (ENGOT-OV16/NOVA): Results from a double-blind, phase 3, randomised controlled trial. Lancet Oncol. 2018, 19, 1117–1125. [Google Scholar] [CrossRef]
- Del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M. Niraparib maintenance therapy in patients with recurrent ovarian cancer after a partial response to the last platinum-based chemotherapy in the ENGOT-OV16/NOVA trial. J. Clin. Oncol. 2019, 37, 2968. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Colombo, N.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; Leary, A.; et al. The effect of age on efficacy, safety and patient-centered outcomes with rucaparib: A post hoc exploratory analysis of ARIEL3, a phase 3, randomized, maintenance study in patients with recurrent ovarian carcinoma. Gynecol. Oncol. 2020, 159, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.R.; Scambia, G.; et al. Rucaparib for patients with platinum-sensitive, recurrent ovarian carcinoma (ARIEL3): Post-progression outcomes and updated safety results from a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 710–722. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Tjokrowidjaja, A.; Lee, C.K.; Friedlander, M.; Gebski, V.; Gladieff, L.; Ledermann, J.; Penson, R.; Oza, A.; Korach, J.; Huzarski, T.; et al. Concordance between CA-125 and RECIST progression in patients with germline BRCA-mutated platinum-sensitive relapsed ovarian cancer treated in the SOLO2 trial with olaparib as maintenance therapy after response to chemotherapy. Eur. J. Cancer 2020, 139, 59–67. [Google Scholar] [CrossRef]
- Friedlander, M.; Gebski, V.; Gibbs, E.; Davies, L.; Bloomfield, R.; Hilpert, F.; Wenzel, L.B.; Eek, D.; Rodrigues, M.; Clamp, A.; et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): A placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018, 19, 1126–1134. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, M.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Lisyanskaya, A.; Sonke, G.S.; Gourley, C.; Banerjee, S.; et al. Patient-centred outcomes and effect of disease progression on health status in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation receiving maintenance olaparib or placebo (SOLO1): A randomised, phase 3 trial. Lancet Oncol. 2021, 22, 632–642. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef]
- Nelson, A.C.; Holt, J.T. Impact of RING and BRCT domain mutations on BRCA1 protein stability, localization and recruitment to DNA damage. Radiat. Res. 2010, 174, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [Green Version]
- Pilie, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [Green Version]
- Ame, J.C.; Fouquerel, E.; Gauthier, L.R.; Biard, D.; Boussin, F.D.; Dantzer, F.; de Murcia, G.; Schreiber, V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J. Cell Sci. 2009, 122, 1990–2002. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- van Wietmarschen, N.; Nussenzweig, A. Mechanism for synthetic lethality in BRCA-deficient cancers: No longer lagging behind. Mol. Cell 2018, 71, 877–878. [Google Scholar] [CrossRef] [Green Version]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [Green Version]
- Dungrawala, H.; Bhat, K.P.; Le Meur, R.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Yekezare, M.; Gómez-González, B.; Diffley, J.F. Controlling DNA replication origins in response to DNA damage–inhibit globally, activate locally. J. Cell Sci. 2013, 126, 1297–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méchali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef]
- Haynes, B.; Murai, J.; Lee, J.M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Carrassa, L.; Damia, G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.t.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Barkauskaite, E.; Brassington, A.; Tan, E.S.; Warwicker, J.; Dunstan, M.S.; Banos, B.; Lafite, P.; Ahel, M.; Mitchison, T.J.; Ahel, I.; et al. Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nat. Commun. 2013, 4, 2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocaña, A.; Pandiella, A. Proteolysis targeting chimeras (PROTACs) in cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 189. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Yang, J.; Chen, Y.; Zhou, P.; Wang, Y.; Du, W.; Zhao, L.; Chen, Y. Discovery of SK-575 as a Highly Potent and Efficacious Proteolysis-Targeting Chimera Degrader of PARP1 for Treating Cancers. J. Med. Chem. 2020, 63, 11012–11033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chang, X.; Zhang, C.; Zeng, S.; Liang, M.; Ma, Z.; Wang, Z.; Huang, W.; Shen, Z. Identification of probe-quality degraders for Poly(ADP-ribose) polymerase-1 (PARP-1). J. Enzym. Inhib. Med. Chem. 2020, 35, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef]
- Kim, C.; Chen, C.; Yu, Y. Avoid the trap: Targeting PARP1 beyond human malignancy. Cell Chem. Biol. 2021, 28, 456–462. [Google Scholar] [CrossRef]
- Wang, S.; Han, L.; Han, J.; Li, P.; Ding, Q.; Zhang, Q.J.; Liu, Z.P.; Chen, C.; Yu, Y. Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nat. Chem. Biol. 2019, 15, 1223–1231. [Google Scholar] [CrossRef]
- Go, A.; Jang, J.W.; Lee, W.; Ha, J.D.; Kim, H.J.; Nam, H.J. Augmentation of the antitumor effects of PARP inhibitors in triple-negative breast cancer via degradation by hydrophobic tagging modulation. Eur. J. Med. Chem. 2020, 204, 112635. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs-A Strategy for Overcoming Anticancer Drug Resistance? Molecules 2021, 26, 2601. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, M.; Wang, Q.; Han, Z. Dual targeting, a new strategy for novel PARP inhibitor discovery. Drug Discov. Ther. 2021, 15, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmañà, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast CancerCombination of PI3K-and PARP-Inhibitors to Treat Breast Cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
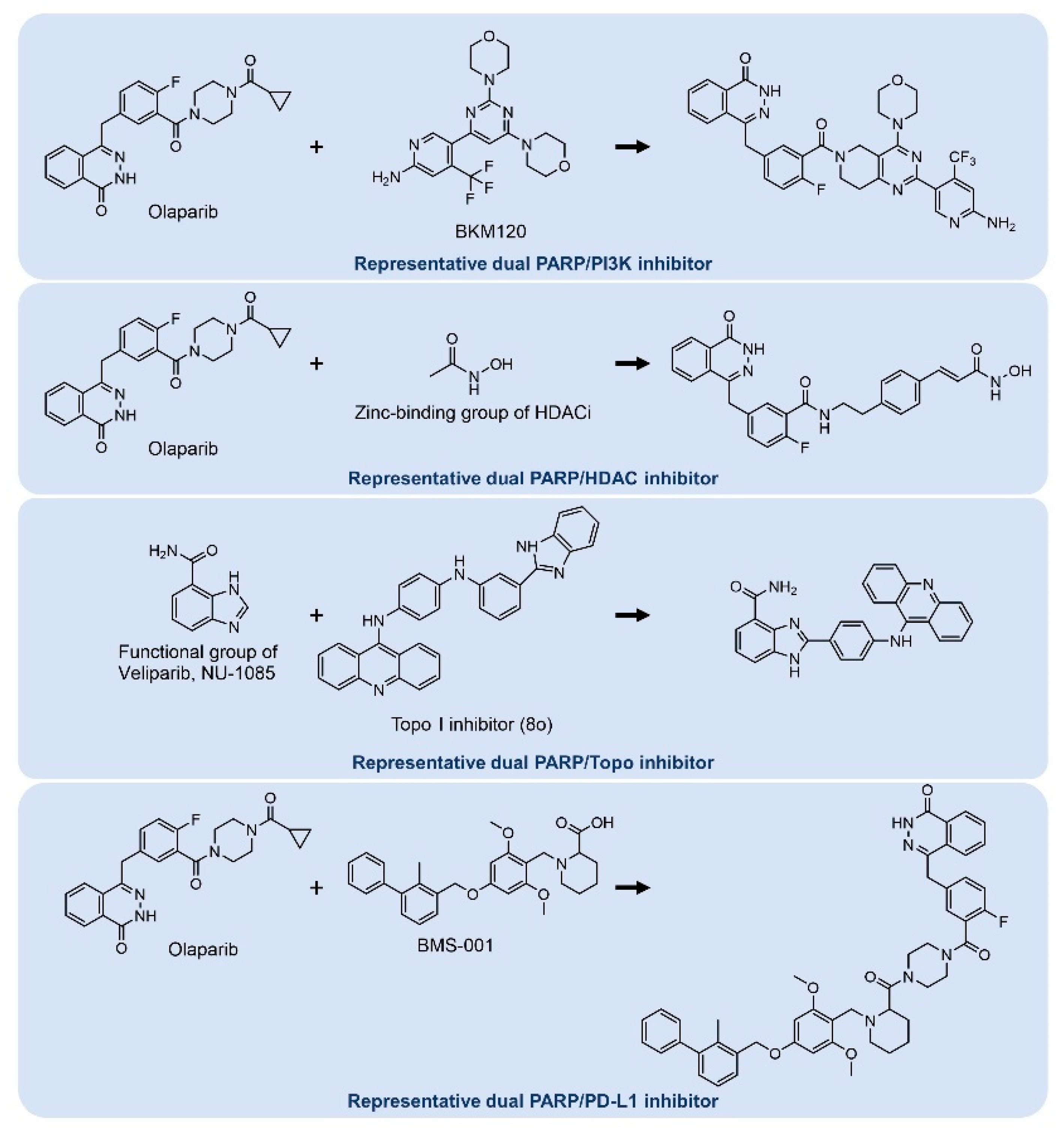
- Wang, J.; Li, H.; He, G.; Chu, Z.; Peng, K.; Ge, Y.; Zhu, Q.; Xu, Y. Discovery of novel dual poly (ADP-ribose) polymerase and phosphoinositide 3-kinase inhibitors as a promising strategy for cancer therapy. J. Med. Chem. 2019, 63, 122–139. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, A.; Im, S.A.; Kim, D.K.; Song, S.H.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Kim, T.Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 33. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Chen, S.; Sun, Q.; Wang, N.; Li, D.; Miao, S.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Olaparib hydroxamic acid derivatives as dual PARP and HDAC inhibitors for cancer therapy. Bioorganic Med. Chem. 2017, 25, 4100–4109. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Chen, S.; Chen, C.; Chen, J.; Chen, C.; Dai, Q.; Gao, C.; Jiang, Y. Design, synthesis and biological evaluation of 4-amidobenzimidazole acridine derivatives as dual PARP and Topo inhibitors for cancer therapy. Eur. J. Med. Chem. 2017, 138, 1135–1146. [Google Scholar] [CrossRef]
- Stewart, R.A.; Pilie, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofori, S.; Awuah, S.G. Small-Molecule Poly(ADP-ribose) Polymerase and PD-L1 Inhibitor Conjugates as Dual-Action Anticancer Agents. ACS Omega 2019, 4, 12584–12597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Ji, Y.; Lodhi, N.; Kotova, E.; Pinnola, A.D.; Golovine, K.; Makhov, P.; Pechenkina, K.; Kolenko, V.; Tulin, A.V. Non-NAD-Like poly(ADP-Ribose) Polymerase-1 Inhibitors effectively Eliminate Cancer in vivo. EBioMedicine 2016, 13, 90–98. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Identifier | Drug | Setting Conditions | With Chemo | Efficacy (Ref.) |
|---|---|---|---|---|
| Breast cancer | ||||
| NCT02163694 | Veliparib | Her2-negative advanced breast cancer; Germline BRCA1/2 mutated | Yes; Combination with carboplatin + paclitaxel | Increase in PFS compared with placebo in a germline BRCA1/2 mutation [25,26] |
| NCT02000622 | Olaparib | Her2-negative metastatic breast cancer patients; Germline BRCA 1/2 mutated | No | Benefit over standard chemotherapy in PFS [20,32] |
| NCT01945775 | Talazoparib | Advanced or metastatic breast cancer; Germline BRCA mutated | No | Benefit over standard chemotherapy in PFS [29,33] Approved by U.S. FDA in 2019 |
| NCT01905592 | Niraparib | Advanced or metastatic breast cancer | No | No significant differences between niparparib and standard chemotherapy in PFS and OS |
| Ovarian cancer | ||||
| NCT01847274 | Niraparib | Ovarian cancer; platinum-based chemotherapy sensitive | No | Increase in PFS compared with placebo regardless of the presence or absence of HRD [23,34,35] |
| NCT02655016 | Niraparib | Advanced ovarian cancer (Stage III or IV); Patients with clinical complete response or partial response following completion of platinum-based chemotherapy course. | No | Increase in PFS compared with placebo regardless of the presence or absence of HDR [36] Approved by U.S. FDA in 2020 for the maintenance treatment |
| NCT02470585 | Veliparib | Advanced ovarian cancer (Stage III or IV); Patients after surgery | Yes; Combination with first-line chemotherapy | Increase in PFS compared with placebo regardless of the presence or absence of HRD [27] |
| NCT01968213 | Rucaparib | Ovarian, fallopian, peritoneal cancer; Patients with clinical complete response or partial response following completion of platinum-based chemotherapy course. | No | Increase in PFS, CFI, TFST, TSST, and PSF2 compared with placebo in recurrent ovarian cancer regardless of the presence or absence of HDR [37,38,39] Approved by U.S. FDA in 2018 for the maintenance treatment |
| NCT01874353 | Olaparib | Relapsed high grade serous ovarian cancer; platinum-based chemotherapy sensitive; BRCA1/2 mutated | No | Increase in PFS and OS compared with placebo [40,41,42,43] Approved by U.S. FDA in 2017 for the maintenance treatment |
| NCT01844986 | Olaparib | Advanced Ovarian Cancer (Stage III, IV); BRCA1/2 mutated; Patients with clinical complete response or partial response following completion of platinum-based chemotherapy course. | No | Increase in PFS compared with placebo regardless of the presence or absence of HRD [44,45] |
| Lung cancer | ||||
| NCT02106546 | Veliparib | Advanced or metastatic squamous non-small cell lung cancer (NSCLC); | Yes; Combination with carboplatin + paclitaxel | Favorable OS in the 52-gene expression histology classifier (LP52)-positive population by veliparib; Favorable OS in the LP52-negative population by placebo [28] |
| Pancreatic cancer | ||||
| NCT02184195 | Olaparib | Metastatic pancreatic cancer; Germline BRCA1/2 mutated; No progression during first-line platinum-based chemotherapy | No | Increase in PFS compared to placebo with a germline BRCA1/2 mutation [21] Approved by U.S. FDA in 2019 for the maintenance treatment |
| PROTAC | ||||
|---|---|---|---|---|
| PARP Binder | E3 Ligase Binder | Tested Cell | Note | Ref. |
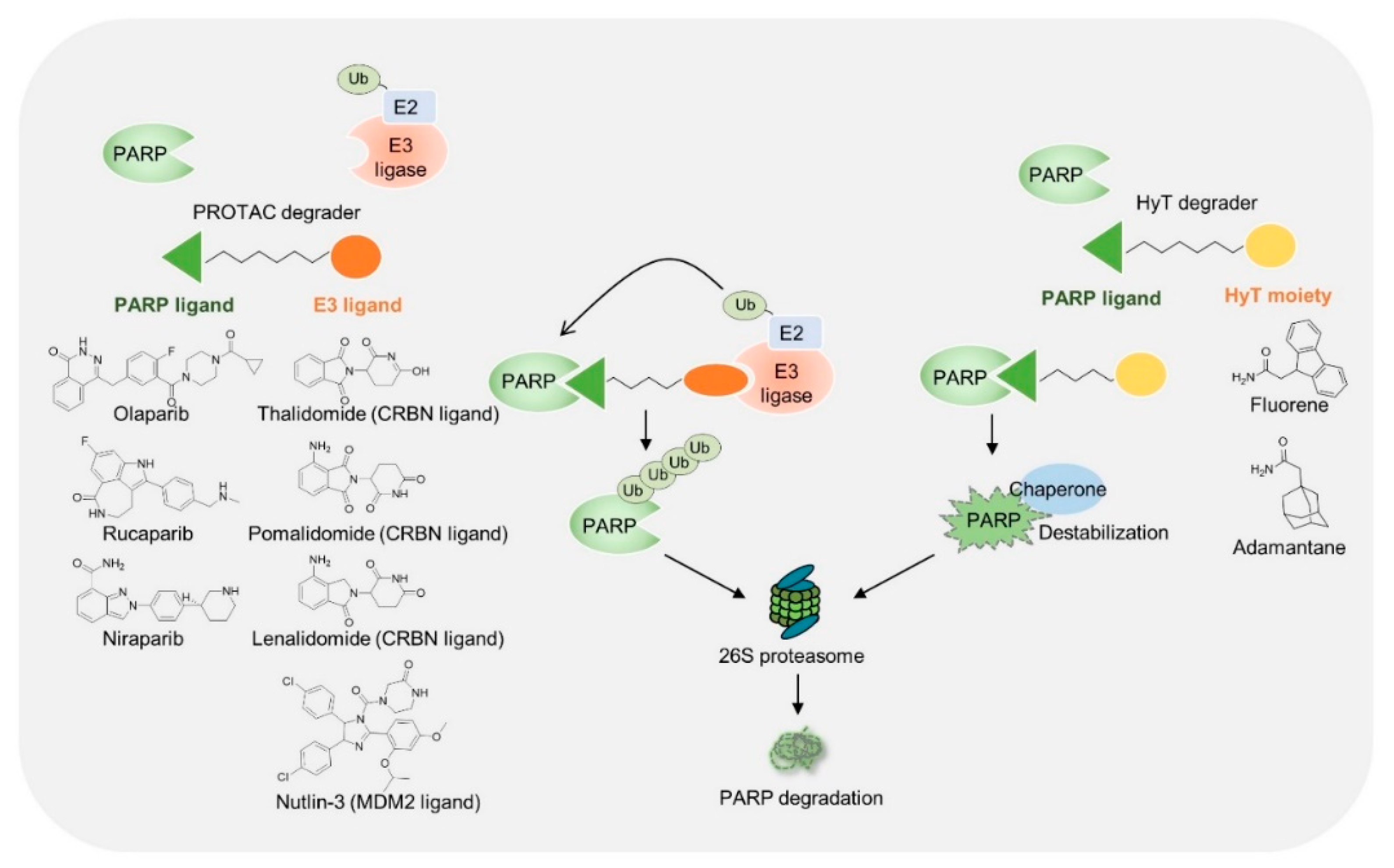
| Olaparib | CRBN ligand | MDA-MB-436 (BRCA1 mutated breast cancer cells), Capan-1 (BRCA2 mutated pancreatic cancer cells), SW620 (colon cancer cell) | Inhibition of tumor growth, Xenograft assay | [77] |
| Rucaparib | CRBN ligand | Primary rat neonatal cardiomyocytes, C2C12 (myoblast) | PARP1 non-trapping, No genotoxic induced cell death | [81] |
| Olaparib | CRBN ligand | SW620 | Increased apoptosis | [78] |
| Niraparib | MDM2 ligand | MDA-MB-231 (TNBC) | Induction of PARP1 cleavage, increased apoptosis | [79] |
| Hydrophobic Tagging | ||||
| PARP Binder | Hydrophobic Moiety | Tested Cell | Note | Ref. |
| Olaparib | Fluorene | MDA-MB-231, MDA-MB-468 (TNBC), HCC1937 (BRCA1 mutated breast cancer cells) | Increased apoptosis and ER stress | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. https://doi.org/10.3390/ijms23158412
Kim D, Nam HJ. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. International Journal of Molecular Sciences. 2022; 23(15):8412. https://doi.org/10.3390/ijms23158412
Chicago/Turabian StyleKim, Dongha, and Hye Jin Nam. 2022. "PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them" International Journal of Molecular Sciences 23, no. 15: 8412. https://doi.org/10.3390/ijms23158412
APA StyleKim, D., & Nam, H. J. (2022). PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. International Journal of Molecular Sciences, 23(15), 8412. https://doi.org/10.3390/ijms23158412