
Therapeutic Vaccines for HPV-Associated Oropharyngeal and Cervical Cancer: The Next De-Intensification Strategy?




Abstract
:1. Introduction
2. Epidemiology and Virus Transmission
3. Structure and Oncogenesis of HPV
4. Molecular Pathogenesis of HPV-Related Cancer
5. Prophylactic Vaccines
6. Use of Prophylactic Vaccines to Prevent Recurrent Disease
7. Therapeutic Vaccines
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morand, G.B.; Diaconescu, A.; Ibrahim, I.; Lamarche, G.; Ruas, J.S.; Dalfen, J.; Hier, M.P.; Alaoui-Jamali, M.A.; Maschietto, M.; da Silva, S.D. Molecular prognostic indicators in HPV-positive oropharyngeal cancer: An updated review. Clin. Exp. Metastasis 2022, 39, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Pytynia, K.B.; Dahlstrom, K.R.; Sturgis, E.M. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol. 2014, 50, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L. Human Papillomavirus-Associated Head and NECK Cancer Is a Distinct Epidemiologic, Clinical, and Molecular Entity, Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2004; pp. 744–754. [Google Scholar]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.; Hoffmann, J.M.; Pouymayou, B.; Dappen, M.B.; Morand, G.B.; Guckenberger, M.; Gregoire, V.; Balermpas, P.; Unkelbach, J. Detailed patient-individual reporting of lymph node involvement in oropharyngeal squamous cell carcinoma with an online interface. Radiother. Oncol. 2022, 169, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Morand, G.B.; Sultanem, K.; Mascarella, M.A.; Hier, M.P.; Mlynarek, A.M. Historical Perspective: How the Discovery of HPV Virus Led to the Utilization of a Robot. Front. Oral. Health 2022, 3, 912861. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Arnovitz, E.; Frenkiel, S.; Hier, M.; Zeitouni, A.; Kost, K.; Mlynarek, A.; Black, M.; MacDonald, C.; Richardson, K.; et al. Psychosocial outcomes of human papillomavirus (HPV)- and non-HPV-related head and neck cancers: A longitudinal study. Psychooncology 2022, 31, 185–197. [Google Scholar] [CrossRef]
- Fischer, C.A.; Zlobec, I.; Green, E.; Probst, S.; Storck, C.; Lugli, A.; Tornillo, L.; Wolfensberger, M.; Terracciano, L.M. Is the improved prognosis of p16 positive oropharyngeal squamous cell carcinoma dependent of the treatment modality? Int. J. Cancer 2010, 126, 1256–1262. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Rabkin, C.S.; Biggar, R.J.; Melbye, M.; Curtis, R.E. Second Primary Cancers following Anal and Cervical Carcinoma: Evidence of Shared Etiologic Factors. Am. J. Epidemiol. 1992, 136, 54–58. [Google Scholar] [CrossRef]
- Hemminki, K.; Dong, C.; Frisch, M. Tonsillar and other upper aerodigestive tract cancers among cervical cancer patients and their husbands. Eur. J. Cancer Prev. 2000, 9, 433–437. [Google Scholar] [CrossRef]
- Gillison, M.L.; Castellsagué, X.; Chaturvedi, A.; Goodman, M.T.; Snijders, P.; Tommasino, M.; Arbyn, M.; Franceschi, S. Eurogin Roadmap: Comparative epidemiology of HPV infection and associated cancers of the head and neck and cervix. Int. J. Cancer 2014, 134, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Berman, T.A.; Schiller, J.T. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer 2017, 123, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Baboci, L.; Holzinger, D.; Boscolo-Rizzo, P.; Tirelli, G.; Spinato, R.; Lupato, V.; Fuson, R.; Schmitt, M.; Michel, A.; Halec, G.; et al. Low prevalence of HPV-driven head and neck squamous cell carcinoma in North-East Italy. Papillomavirus Res. 2016, 2, 133–140. [Google Scholar] [CrossRef] [PubMed]
- LeConte, B.A.; Szaniszlo, P.; Fennewald, S.M.; Lou, D.I.; Qiu, S.; Chen, N.-W.; Lee, J.H.; Resto, V.A. Differences in the viral genome between HPV-positive cervical and oropharyngeal cancer. PLoS ONE 2018, 13, e0203403. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Bagheri, A.; D’Souza, G. Epidemiology of oral human papillomavirus infection. Oral Oncol. 2014, 50, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.K.; Graubard, B.I.; Broutian, T.; Pickard, R.K.; Tong, Z.-y.; Xiao, W.; Kahle, L.; Gillison, M.L. NHANES 2009–2012 findings: Association of sexual behaviors with higher prevalence of oral oncogenic human papillomavirus infections in U.S. men. Cancer Res. 2015, 75, 2468–2477. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, G.; Agrawal, Y.; Halpern, J.; Bodison, S.; Gillison, M.L. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J. Infect. Dis. 2009, 199, 1263–1269. [Google Scholar] [CrossRef] [Green Version]
- Dunne, E.F.; Park, I.U. HPV and HPV-associated diseases. Infect. Dis. Clin. 2013, 27, 765–778. [Google Scholar] [CrossRef]
- Woodman, C.B.; Collins, S.; Winter, H.; Bailey, A.; Ellis, J.; Prior, P.; Yates, M.; Rollason, T.P.; Young, L.S. Natural history of cervical human papillomavirus infection in young women: A longitudinal cohort study. Lancet 2001, 357, 1831–1836. [Google Scholar] [CrossRef]
- Fakhry, C.; Rosenthal, B.T.; Clark, D.P.; Gillison, M.L. Associations between oral HPV16 infection and cytopathology: Evaluation of an oropharyngeal “pap-test equivalent” in high-risk populations. Cancer Prev. Res. 2011, 4, 1378–1384. [Google Scholar] [CrossRef] [Green Version]
- Kreimer, A.R.; Johansson, M.; Waterboer, T.; Kaaks, R.; Chang-Claude, J.; Drogen, D.; Tjønneland, A.; Overvad, K.; Quirós, J.R.; González, C.A. Evaluation of human papillomavirus antibodies and risk of subsequent head and neck cancer. J. Clin. Oncol. 2013, 31, 2708. [Google Scholar] [CrossRef]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [Green Version]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [Green Version]
- Schiller, J.T.; Lowy, D.R. Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat. Rev. Microbiol. 2012, 10, 681–692. [Google Scholar] [CrossRef]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M. Immunobiology of HPV and HPV vaccines. Gynecol. Oncol. 2008, 109 (Suppl. 2), S15–S21. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Fausch, S.C.; Da Silva, D.M.; Rudolf, M.P.; Kast, W.M. Human papillomavirus virus-like particles do not activate Langerhans cells: A possible immune escape mechanism used by human papillomaviruses. J. Immunol. 2002, 169, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.J.; Koutsky, L.A.; Wipf, G.C.; Christensen, N.D.; Lee, S.-K.; Kuypers, J.; Kiviat, N.; Galloway, D.A. The natural history of human papillomavirus type 16 capsid antibodies among a cohort of university women. J. Infect. Dis. 1996, 174, 927–936. [Google Scholar] [CrossRef]
- Herdman, M.T.; Pett, M.R.; Roberts, I.; Alazawi, W.O.; Teschendorff, A.E.; Zhang, X.-Y.; Stanley, M.A.; Coleman, N. Interferon-β treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, J.; Bertrand, M.; Brydon, L.; Gagné, H.; Hauck, B.; Mayrand, M.-H.; McFaul, S.; Power, P.; Schepansky, A.; Straszak-Suri, M.; et al. Colposcopic Management of Abnormal Cervical Cytology and Histology. J. Obstet. Gynaecol. Can. 2012, 34, 1188–1202. [Google Scholar] [CrossRef]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muench, P.; Probst, S.; Schuetz, J.; Leiprecht, N.; Busch, M.; Wesselborg, S.; Stubenrauch, F.; Iftner, T. Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res. 2010, 70, 6913–6924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crook, T.; Vousden, K. Properties of p53 mutations detected in primary and secondary cervical cancers suggest mechanisms of metastasis and involvement of environmental carcinogens. EMBO J. 1992, 11, 3935–3940. [Google Scholar] [CrossRef]
- Meyers, J.M.; Spangle, J.M.; Munger, K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 2013, 87, 4762–4767. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Fujii, T.; Kawachi, H.; Miki, Y.; Omura, K.; Morita, K.-I.; Kayamori, K.; Katsube, K.-I.; Yamaguchi, A. Reduction of NOTCH1 expression pertains to maturation abnormalities of keratinocytes in squamous neoplasms. Lab. Investig. 2012, 92, 688–702. [Google Scholar] [CrossRef] [Green Version]
- Banks, L.; Barnett, S.; Crook, T. HPV-16 E7 functions at the G1 to S phase transition in the cell cycle. Oncogene 1990, 5, 833–837. [Google Scholar]
- Sano, T.; Oyama, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. Expression Status of p16 Protein Is Associated with Human Papillomavirus Oncogenic Potential in Cervical and Genital Lesions. Am. J. Pathol. 1998, 153, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J.; Parton, A.; Hartley, K.; Banks, L.; Crook, T.; Stanley, M.; Crawford, L. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 1990, 178, 254–262. [Google Scholar] [CrossRef]
- Bohm, S.; Wilczynski, S.; Pfister, H.; Iftner, T. The predominant mRNA class in HPV16-infected genital neoplasias does not encode the e6 or the E7 protein. Int. J. Cancer 1993, 55, 791–798. [Google Scholar] [CrossRef]
- Williams, V.M.; Filippova, M.; Filippov, V.; Payne, K.J.; Duerksen-Hughes, P. Human papillomavirus type 16 E6* induces oxidative stress and DNA damage. J. Virol. 2014, 88, 6751–6761. [Google Scholar] [CrossRef] [Green Version]
- Rosenberger, S.; Arce, J.D.-C.; Langbein, L.; Steenbergen, R.D.M.; Rösl, F. Alternative splicing of human papillomavirus type-16 E6/E6* early mRNA is coupled to EGF signaling via Erk1/2 activation. Proc. Natl. Acad. Sci. USA 2010, 107, 7006–7011. [Google Scholar] [CrossRef] [Green Version]
- Villa, L.; Costa, R.; Petta, C.; Andrade, R.; Paavonen, J.; Iversen, O.; Olsson, S.; Høye, J.; Steinwall, M.; Riis-Johannessen, G. High sustained efficacy of a prophylactic quadrivalent human papillomavirus types 6/11/16/18 L1 virus-like particle vaccine through 5 years of follow-up. Br. J. Cancer 2006, 95, 1459–1466. [Google Scholar] [CrossRef]
- Smith, J.F.; Brownlow, M.; Brown, M.; Kowalski, R.; Esser, M.T.; Ruiz, W.; Barr, E.; Brown, D.R.; Bryan, J.T. Antibodies from women immunized with Gardasil® cross-neutralize HPV 45 pseudovirions. Hum. Vaccines 2007, 3, 109–115. [Google Scholar] [CrossRef]
- Bruni, L.; Saura-Lázaro, A.; Montoliu, A.; Brotons, M.; Alemany, L.; Diallo, M.S.; Afsar, O.Z.; LaMontagne, D.S.; Mosina, L.; Contreras, M.; et al. HPV vaccination introduction worldwide and WHO and UNICEF estimates of national HPV immunization coverage 2010–2019. Prev. Med. 2021, 144, 106399. [Google Scholar] [CrossRef] [PubMed]
- WHO. Human papillomavirus vaccines: WHO position paper, May 2017–Recommendations. Vaccine 2017, 35, 5753–5755. [Google Scholar]
- Meites, E.; Szilagyi, P.G.; Chesson, H.W.; Unger, E.R.; Romero, J.R.; Markowitz, L.E. Human papillomavirus vaccination for adults: Updated recommendations of the Advisory Committee on Immunization Practices. Am. J. Transplant. 2019, 19, 3202–3206. [Google Scholar] [CrossRef] [Green Version]
- Ghelardi, A.; Parazzini, F.; Martella, F.; Pieralli, A.; Bay, P.; Tonetti, A.; Svelato, A.; Bertacca, G.; Lombardi, S.; Joura, E.A. SPERANZA project: HPV vaccination after treatment for CIN2+. Gynecol. Oncol. 2018, 151, 229–234. [Google Scholar] [CrossRef]
- Lichter, K.; Krause, D.; Xu, J.; Tsai, S.H.L.; Hage, C.; Weston, E.; Eke, A.; Levinson, K. Adjuvant human papillomavirus vaccine to reduce recurrent cervical dysplasia in unvaccinated women: A systematic review and meta-analysis. Obstet. Gynecol. 2020, 135, 1070–1083. [Google Scholar] [CrossRef]
- Suk, R.; Mahale, P.; Sonawane, K.; Sikora, A.G.; Chhatwal, J.; Schmeler, K.M.; Sigel, K.; Cantor, S.B.; Chiao, E.Y.; Deshmukh, A.A. Trends in Risks for Second Primary Cancers Associated With Index Human Papillomavirus–Associated Cancers. JAMA Netw. Open 2018, 1, e181999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.H.; Jin, H.T.; Park, S.H.; Youn, J.I.; Sung, Y.-C. Optimal induction of HPV DNA vaccine-induced CD8+ T cell responses and therapeutic antitumor effect by antigen engineering and electroporation. Vaccine 2009, 27, 5906–5912. [Google Scholar] [CrossRef]
- Paolini, F.; Curzio, G.; Cordeiro, M.N.; Massa, S.; Mariani, L.; Pimpinelli, F.; de Freitas, A.C.; Franconi, R.; Venuti, A. HPV 16 E5 oncoprotein is expressed in early stage carcinogenesis and can be a target of immunotherapy. Hum. Vaccines Immunother. 2017, 13, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, C.; Cohen, R.B.; Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Knoblock, D.M.; Bauml, J.M.; Weinstein, G.S.; Lin, A.; Boyer, J.; et al. Immunotherapy Targeting HPV16/18 Generates Potent Immune Responses in HPV-Associated Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunwitz, C.; Salomon, N.; Vascotto, F.; Selmi, A.; Bukur, T.; Diken, M.; Kreiter, S.; Türeci, Ö.; Sahin, U. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. OncoImmunology 2019, 8, e1629259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16–related cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.W.; Hur, S.-Y.; Woo, J.W.; Kim, Y.-M.; Lim, M.C.; Park, S.Y.; Seo, S.S.; No, J.H.; Kim, B.-G.; Lee, J.-K. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: Interim results of a single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1653–1660. [Google Scholar] [CrossRef]
- Gerritsen, W.R.; Melief, C.J.; Welters, M.; Vergote, I.; Kroep, J.R.; Kenter, G.; Ottevanger, N.; Tjalma, W.A.; Denys, H.; van Poelgeest, M.I. Association of T cell responses after vaccination with HPV16 long peptides for late stage cervical cancer with prolonged survival. J. Clin. Oncol. 2017, 35, 5525. [Google Scholar] [CrossRef]
- Ferris, R.L. Immunology and Immunotherapy of Head and Neck Cancer. J. Clin. Oncol. 2015, 33, 3293–3304. [Google Scholar] [CrossRef]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An open-label randomised controlled phase 3 trial. Lancet 2019, 393, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Jordan, R.C.; Zhao, W.; Sturgis, E.M.; Burtness, B. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. [Google Scholar] [CrossRef]
- Wu, S.Y.; Yom, S.S. Current Standards for Organ Preservation in Locoregionally Advanced Non-nasopharyngeal Head and Neck Cancer and Evolving Strategies for Favorable-Risk and Platinum-Ineligible Populations. Curr. Treat. Options Oncol. 2019, 20, 89. [Google Scholar] [CrossRef]
- Tao, Y.; Aupérin, A.; Sun, X.; Sire, C.; Martin, L.; Coutte, A.; Lafond, C.; Miroir, J.; Liem, X.; Rolland, F.; et al. Avelumab-cetuximab-radiotherapy versus standards of care in locally advanced squamous-cell carcinoma of the head and neck: The safety phase of a randomised phase III trial GORTEC 2017-01 (REACH). Eur. J. Cancer 2020, 141, 21–29. [Google Scholar] [CrossRef]
- Yu, Y.; Zakeri, K.; Lee, N. Javelin Head Neck 100: Should we combine immunotherapy with radiation therapy? Oncotarget 2021, 12, 2223–2226. [Google Scholar] [CrossRef]
- Lee, N.Y.; Ferris, R.L.; Psyrri, A.; Haddad, R.I.; Tahara, M.; Bourhis, J.; Harrington, K.; Chang, P.M.; Lin, J.C.; Razaq, M.A.; et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2021, 22, 450–462. [Google Scholar] [CrossRef]
- Wei, J.; Montalvo-Ortiz, W.; Yu, L.; Krasco, A.; Ebstein, S.; Cortez, C.; Lowy, I.; Murphy, A.J.; Sleeman, M.A.; Skokos, D. Sequence of αPD-1 relative to local tumor irradiation determines the induction of abscopal antitumor immune responses. Sci. Immunol. 2021, 6, eabg0117. [Google Scholar] [CrossRef]
- Sadeghi, N.; Li, N.W.; Taheri, M.R.; Easley, S.; Siegel, R.S. Neoadjuvant chemotherapy and transoral surgery as a definitive treatment for oropharyngeal cancer: A feasible novel approach. Head Neck 2016, 38, 1837–1846. [Google Scholar] [CrossRef]
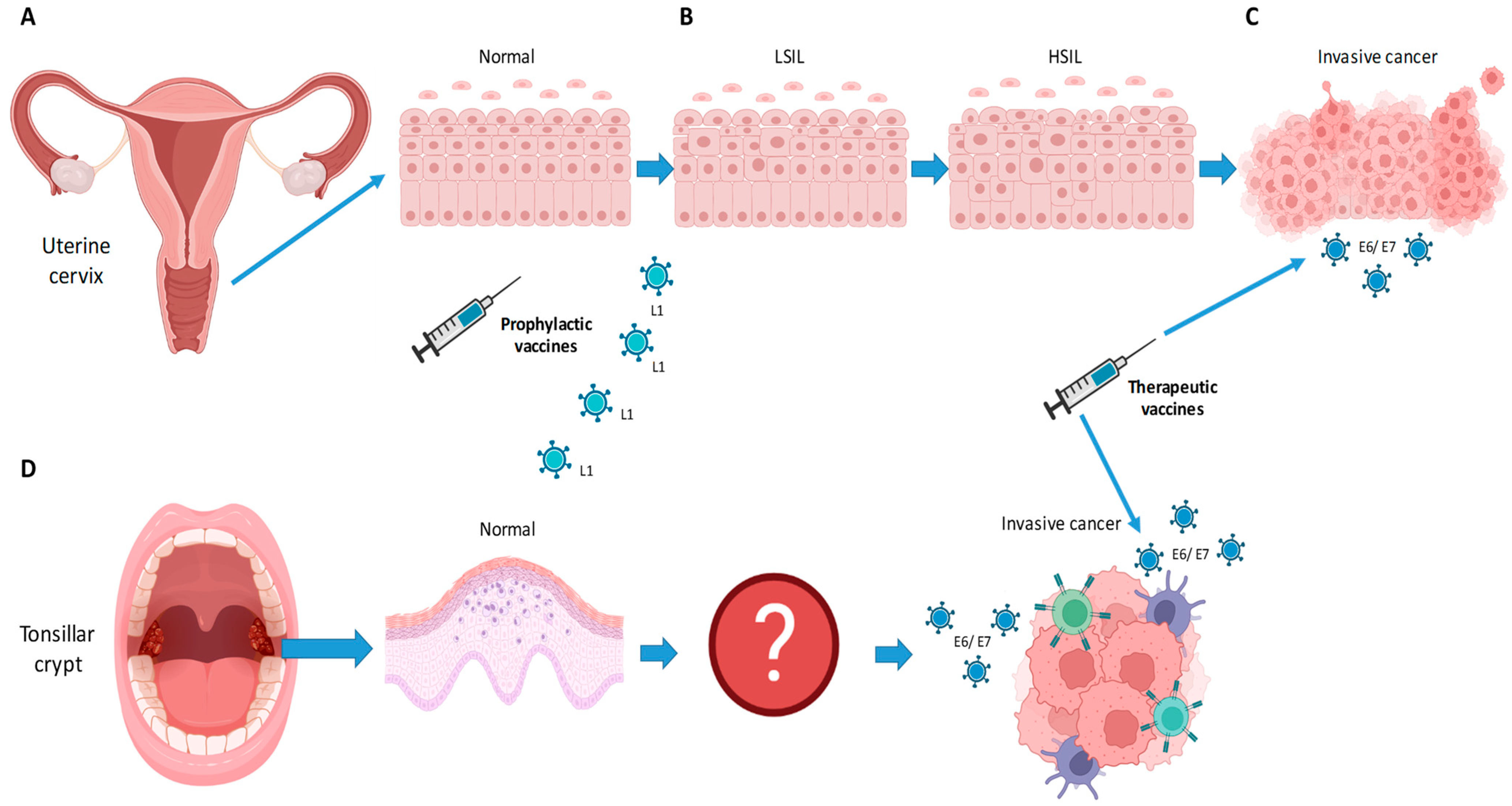


{kind=link}
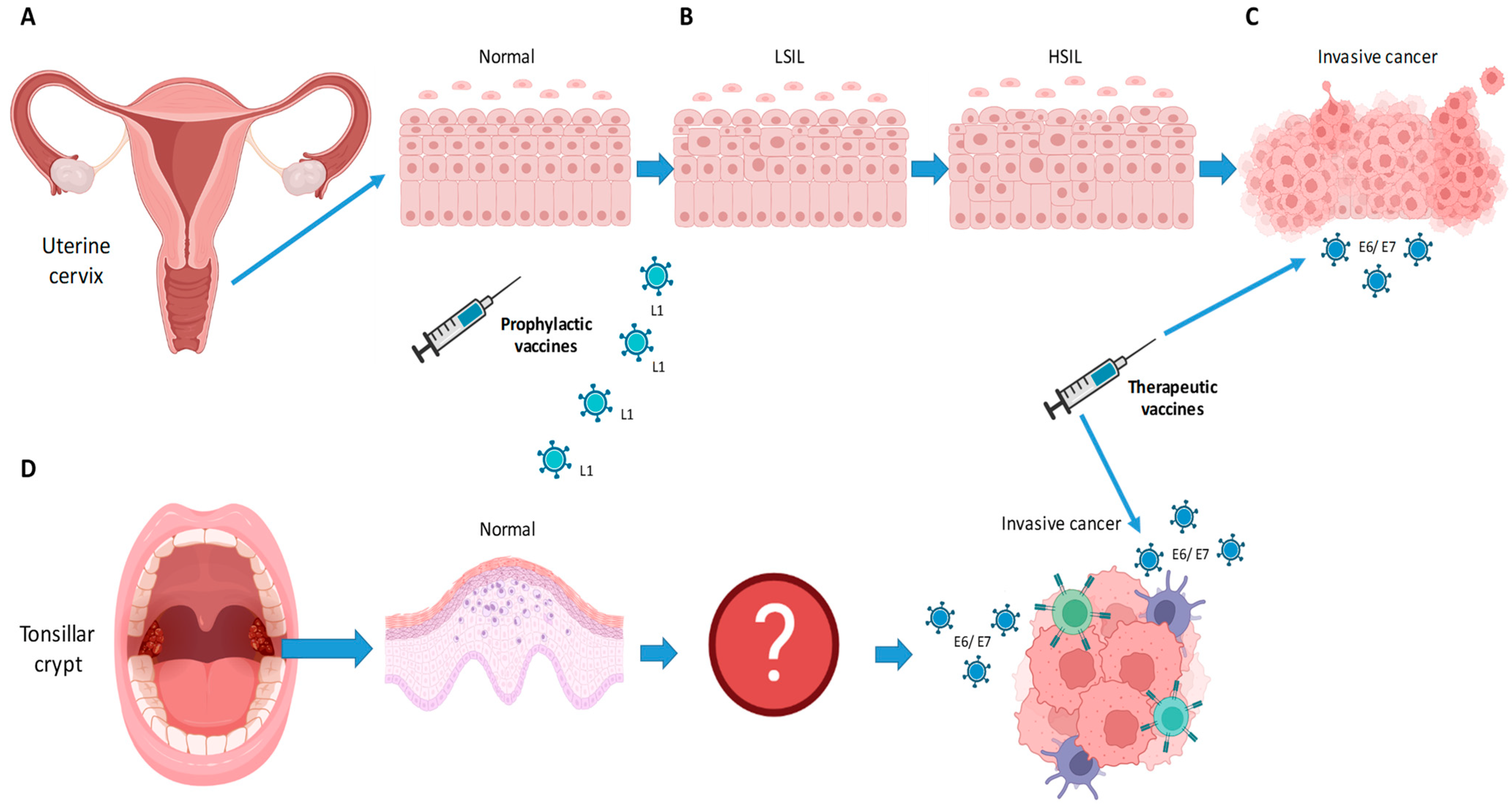
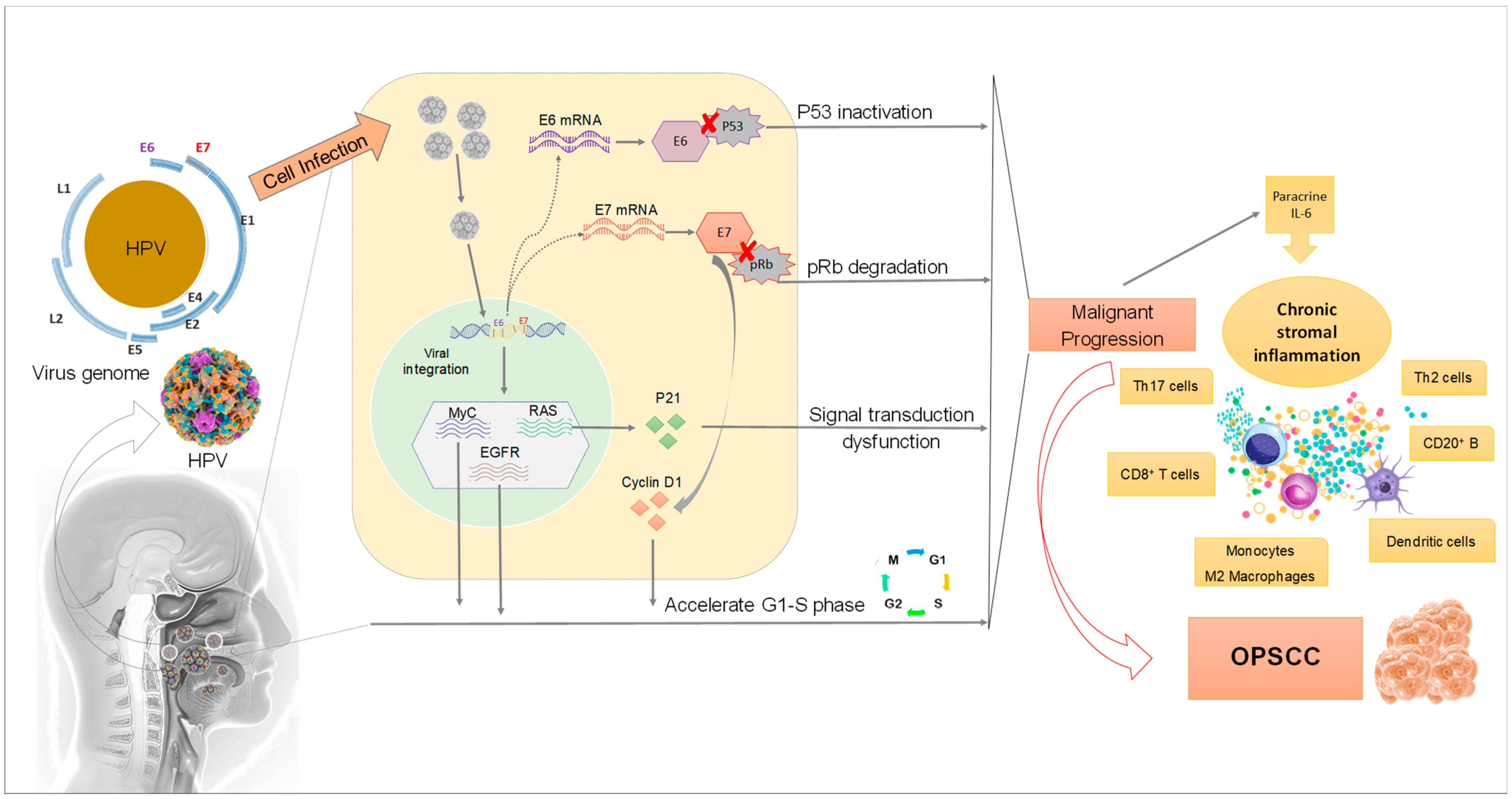{kind=link}
| Characteristics | Cervical Cancer | Oropharyngeal Cancer |
|---|---|---|
| Etiology | HPV > 99% | HPV, smoking, alcohol |
| Developing/developed country | 9:1 | 1:3 |
| Trends | Decreasing in developed countries Increasing in developing countries | Increasing |
| HPV16 | 61% of all cases | >90% of HPV-related cases |
| HPV18 | 10% of all cases | 2% of HPV-related cases |
| E6/E7 | Present | Present |
| Rb/p53 | Wt | Wt |
| Transmission | Sexual | Sexual (incl. French kissing) |
| Latency | Shortto CIN | >10 years (to SCC) |
| Natural history | CIN1, CIN2, CIN3, CIS, invasive SCC (median age 49 y) | Unknown (median age 54 y) |
| Screening | Yes, Pap smear | “Oro-pap“ smear does not work |
| Effect of “primary“ vaccination? | Yes (20 years) | Yes (20 years) |
| Effect of “secondary“ vaccination? | Yes (CIN 2+) | Unknown |
| Five-year survival rates | 68% | 95% |
| Vaccine Name | 9vHPV | 4vHPV | 2vHPV |
|---|---|---|---|
| Brand name | Gardasil 9, Merck | Gardasil, Merck | Cervarix, GlaxoSmithKline |
| Vaccine type | Nonavalent | Quadrivalent | Bivalent |
| Serotypes covered | HPV 6, 11, 16, 18, 31, 33, 45, 52, 58 | HPV 6, 11, 16, 18 | HPV 16, 18 |
| Manufacturing | Saccharomyces cerevisiae (Baker’s yeast)—expressing L1 | Saccharomyces cerevisiae (Baker’s yeast)—expressing L1 | Trichoplusia ni insect cell line infected with L1 encoding recombinant baculovirus |
| Adjuvant | 500 µg amorphous aluminum hydroxyphosphate sulfate | 225 µg amorphous aluminum hydroxyphosphate sulfate | 500 µg aluminum hydroxide 50 µg 3-O-desacyl-4′ monophosphoryl lipid A |
| Available on market (USA) | 2014–present | 2006–2016 | 2009–2016 |
| Target age | 11–12 years old, licensed for 9–26 years old (up to 45 years old) | ||
| Target group | Females since 2006, males since 2011 | ||
| Schedule | For persons < 15 years-old, two doses (0 and 6–12 months) For persons > 15 years-old, three doses (0, 1–2, and 6 months) | ||
| Administration | Intramuscularly (deltoid muscle), 0.5 mL vial, storage 2–8 °C | ||
| Side effects | Local: injection-site-related pain, swelling, and erythema Systemic: headache, dizziness, myalgia, arthralgia, and gastrointestinal symptoms No evidence for Guillain-Barré Syndrome, complex regional pain syndrome (CRPS) and/or postural orthostatic tachycardia syndrome (POTS) [48]. | ||
| Contraindications | Hypersensitivity to yeast | Hypersensitivity to yeast | Anaphylactic latex allergy |
| Therapeutic Setting | Trial Name | Country | Institution | PI | Population | Intervention | Comparison | Outcome |
|---|---|---|---|---|---|---|---|---|
| Definitive/Curative Setting | NCT05232851 | USA | Mayo | David M Routman | 24 patients with locally advanced HPV-positive oropharyngeal cancer | Neoadjuvant Pembrolizumab + Liposomal HPV-16 E6/E7 Multipeptide Vaccine PDS0101 | Neoadjuvant Pembrolizumab alone | PFS and OS |
| NCT02405221 | USA | Johns Hopkins | Stéphanie Gaillard | 14 patients with history of HPV positive cervical cancer | Adjuvant L2E6E7 vaccination (TA-CIN) | Single arm open label | Recurrence rate | |
| NCT04369937 | USA | Pittsburgh | Dan Zandberg | 50 patients with intermediate-risk HPV positive oropharyngeal cancer | Radiation + Cisplatin + Pembrolizumab + E6/E7 vaccination (ISA 101) | Single arm open label | PFS | |
| Recurrent/Metastatic Setting | NCT02426892 [58] | USA | MD Anderson | Massarelli | 34 patients with incurable HPV+ solid tumors | Nivolumab + HPV E6/7 vaccination (ISA 101) | Single arm open label | Overall response rate |
| NCT03444376 [59] | South Korea | Pohang | Soo-Young Hur | 60 patients with advanced HPV16 or HPV18-positive cervical cancer | Pembrolizumab + HPV E6/E7 vaccination (GX-188E) | Single arm open label | Overall response rate | |
| CerviISA NCT02128126 | Belgium/Germany/Netherlands | Multicentric | Winald Gerritsen | 93 patients with advanced HPV16-positive cervical cancer | Carboplatin and Paclitaxel with or without Bevacizumab + HPV E6/7 vaccination (ISA 101) | Single arm open label | Overall response rate | |
| NCT04405349 | Central Europe | Multicentric | Nykode Therapeutics | 50 patients with unresectable HPV-positive cervical cancer. | Atezolizumab + HPV E6/7 vaccination (VB10.16) | Single arm open label | Overall response rate | |
| NCT03260023 | USA/France/Spain | Multicenter | Transgene | 150 patients with HPV-positive unresectable malignancies | Avelumab + HPV E6/7 vaccination (TG4001) | Avelumab alone | Overall response rate | |
| NCT04180215 | USA | Multicenter | Hookipa Biotech | 200 patients with HPV16-positive cancer, unresectable | HPV E6/7 vaccination, i.v. and intratumoral | Single arm, open label | Overall response rate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morand, G.B.; Cardona, I.; Cruz, S.B.S.C.; Mlynarek, A.M.; Hier, M.P.; Alaoui-Jamali, M.A.; da Silva, S.D. Therapeutic Vaccines for HPV-Associated Oropharyngeal and Cervical Cancer: The Next De-Intensification Strategy? Int. J. Mol. Sci. 2022, 23, 8395. https://doi.org/10.3390/ijms23158395
Morand GB, Cardona I, Cruz SBSC, Mlynarek AM, Hier MP, Alaoui-Jamali MA, da Silva SD. Therapeutic Vaccines for HPV-Associated Oropharyngeal and Cervical Cancer: The Next De-Intensification Strategy? International Journal of Molecular Sciences. 2022; 23(15):8395. https://doi.org/10.3390/ijms23158395
Chicago/Turabian StyleMorand, Grégoire B., Isabel Cardona, Sara Brito Silva Costa Cruz, Alex M. Mlynarek, Michael P. Hier, Moulay A. Alaoui-Jamali, and Sabrina Daniela da Silva. 2022. "Therapeutic Vaccines for HPV-Associated Oropharyngeal and Cervical Cancer: The Next De-Intensification Strategy?" International Journal of Molecular Sciences 23, no. 15: 8395. https://doi.org/10.3390/ijms23158395
APA StyleMorand, G. B., Cardona, I., Cruz, S. B. S. C., Mlynarek, A. M., Hier, M. P., Alaoui-Jamali, M. A., & da Silva, S. D. (2022). Therapeutic Vaccines for HPV-Associated Oropharyngeal and Cervical Cancer: The Next De-Intensification Strategy? International Journal of Molecular Sciences, 23(15), 8395. https://doi.org/10.3390/ijms23158395