
The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V

 , , and
, , and 
Abstract
1. Structure, Physiology, and Function of the Vascular Endothelium
1.1. Overview
1.2. Morphological and Functional Heterogeneity of the Endothelium
1.3. Biomarkers of Endothelial Function
1.4. The Endothelium and Coagulation
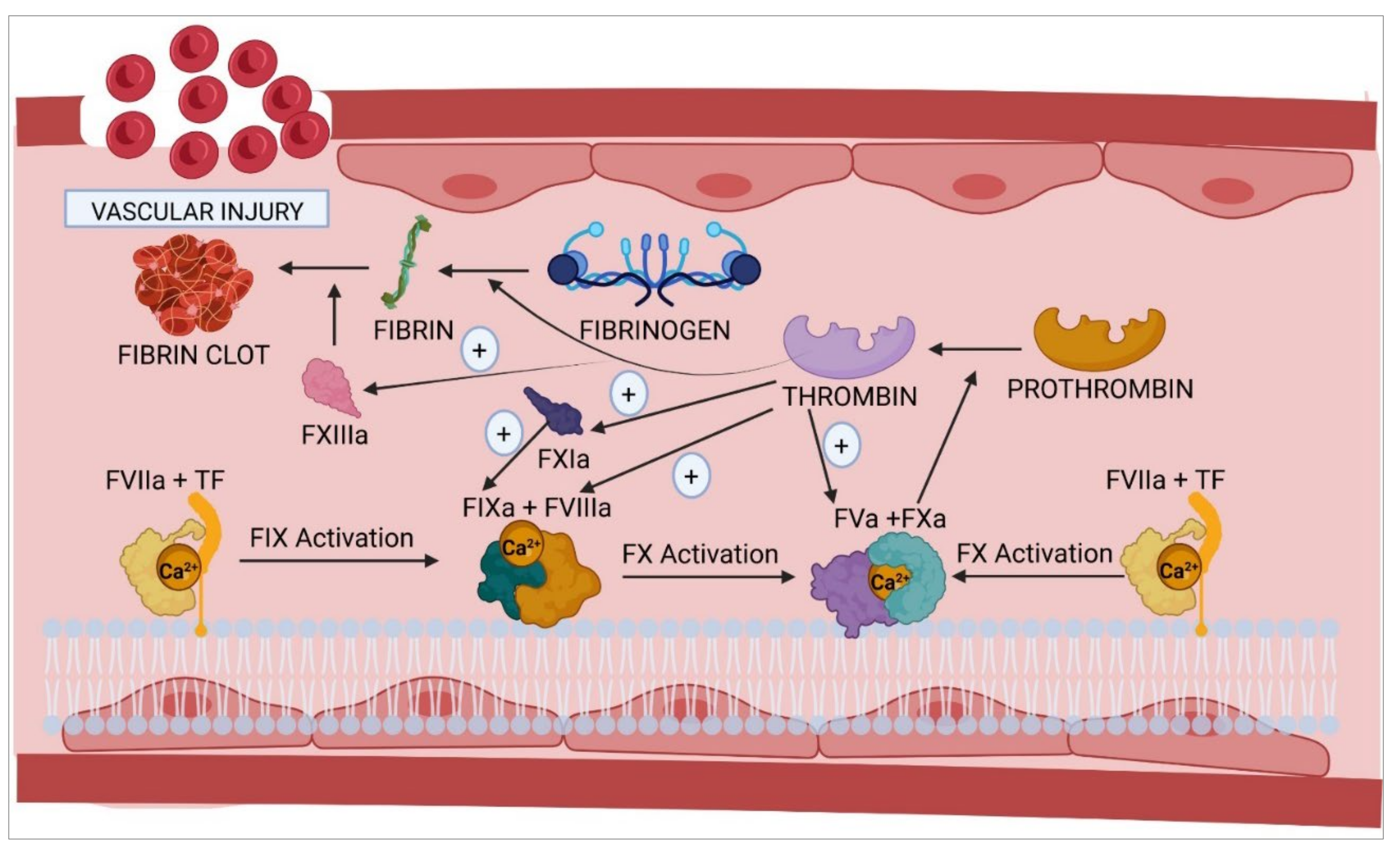
2. Hemostasis: An Overview
Hemostasis during the Neonatal Period
3. Homeostasis of Hemostasis
4. Molecular Structure and Clotting Function
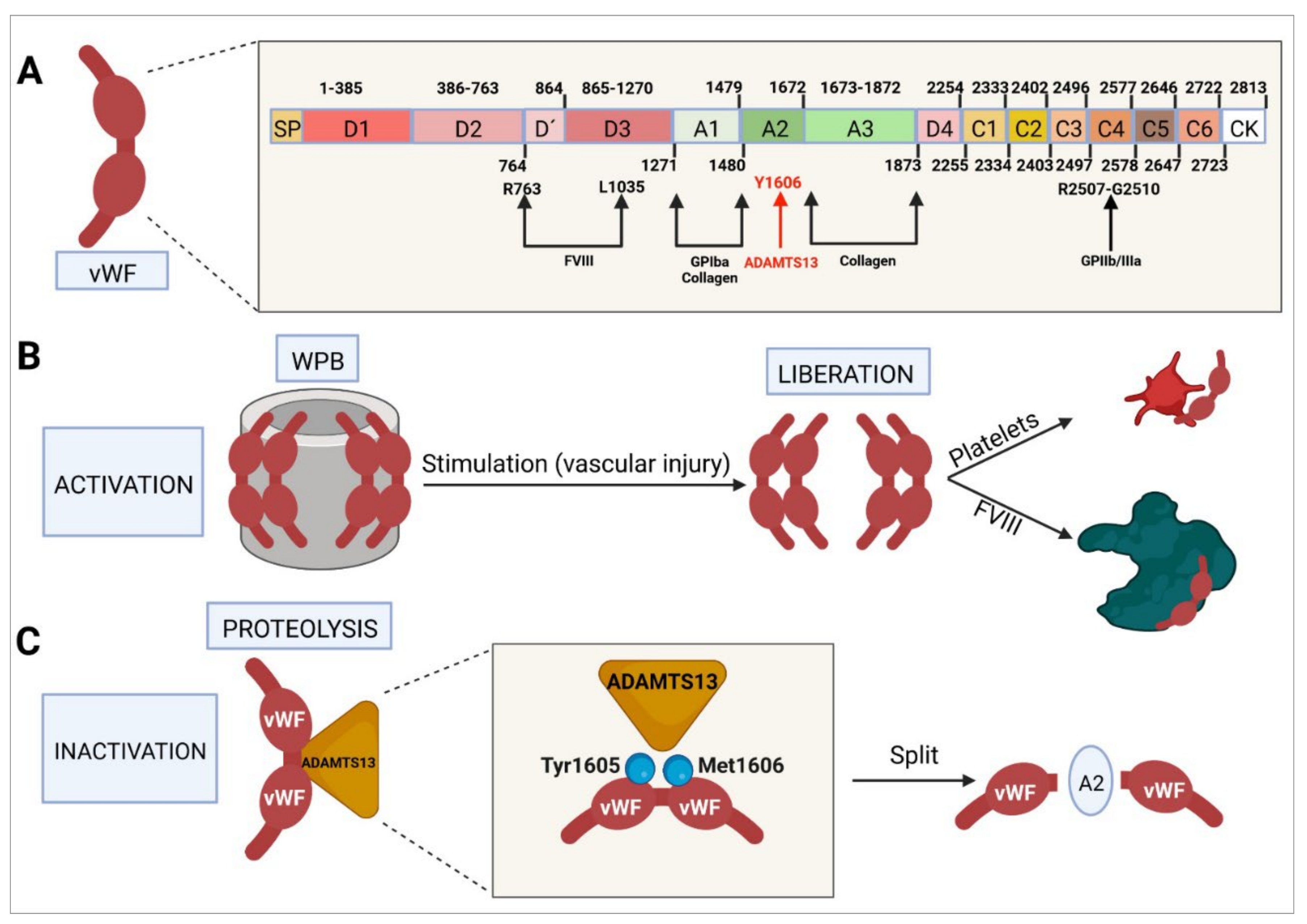
4.1. Molecular Structure of von Willebrand Factor
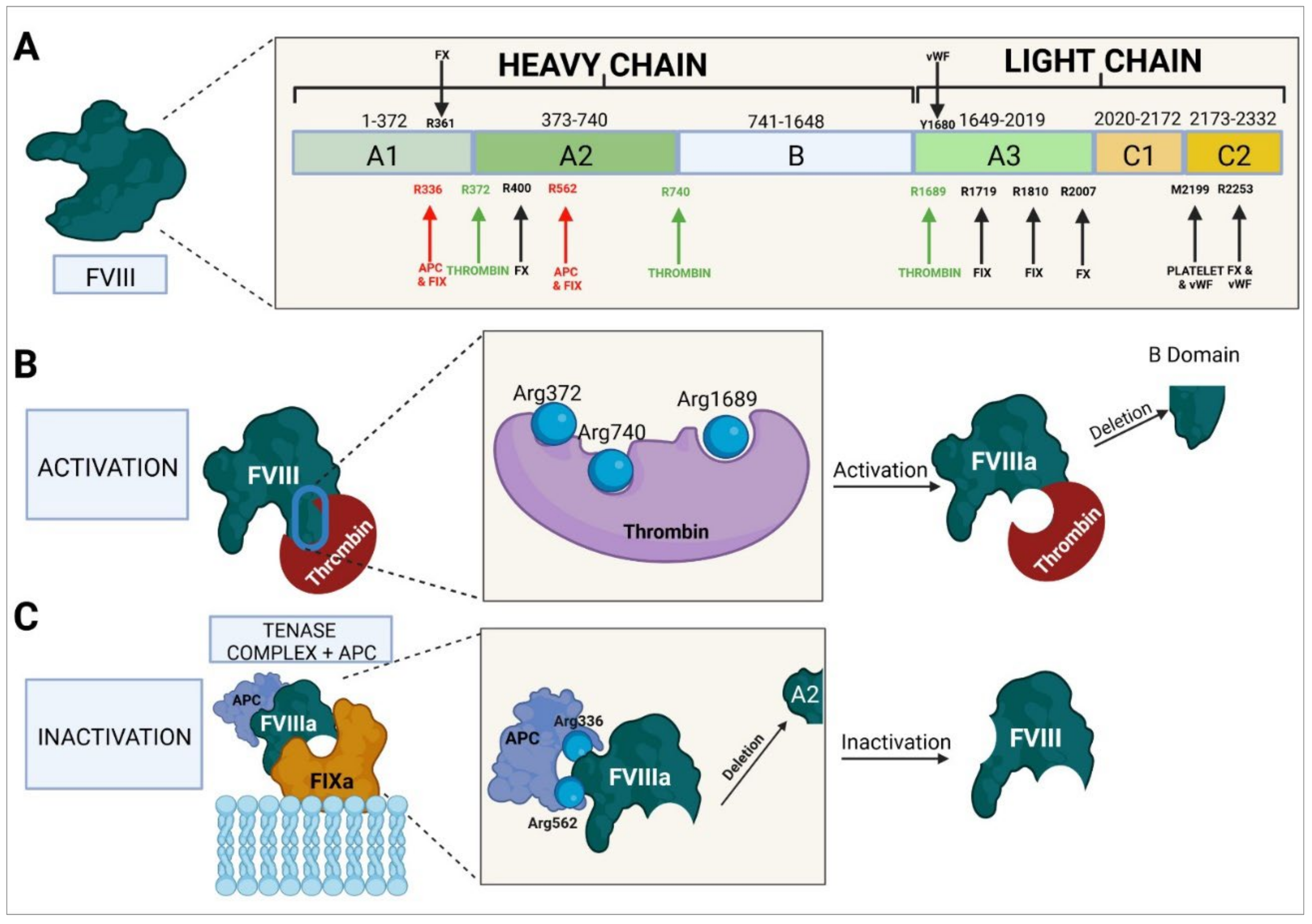
4.2. Molecular Structure of Factor VIII
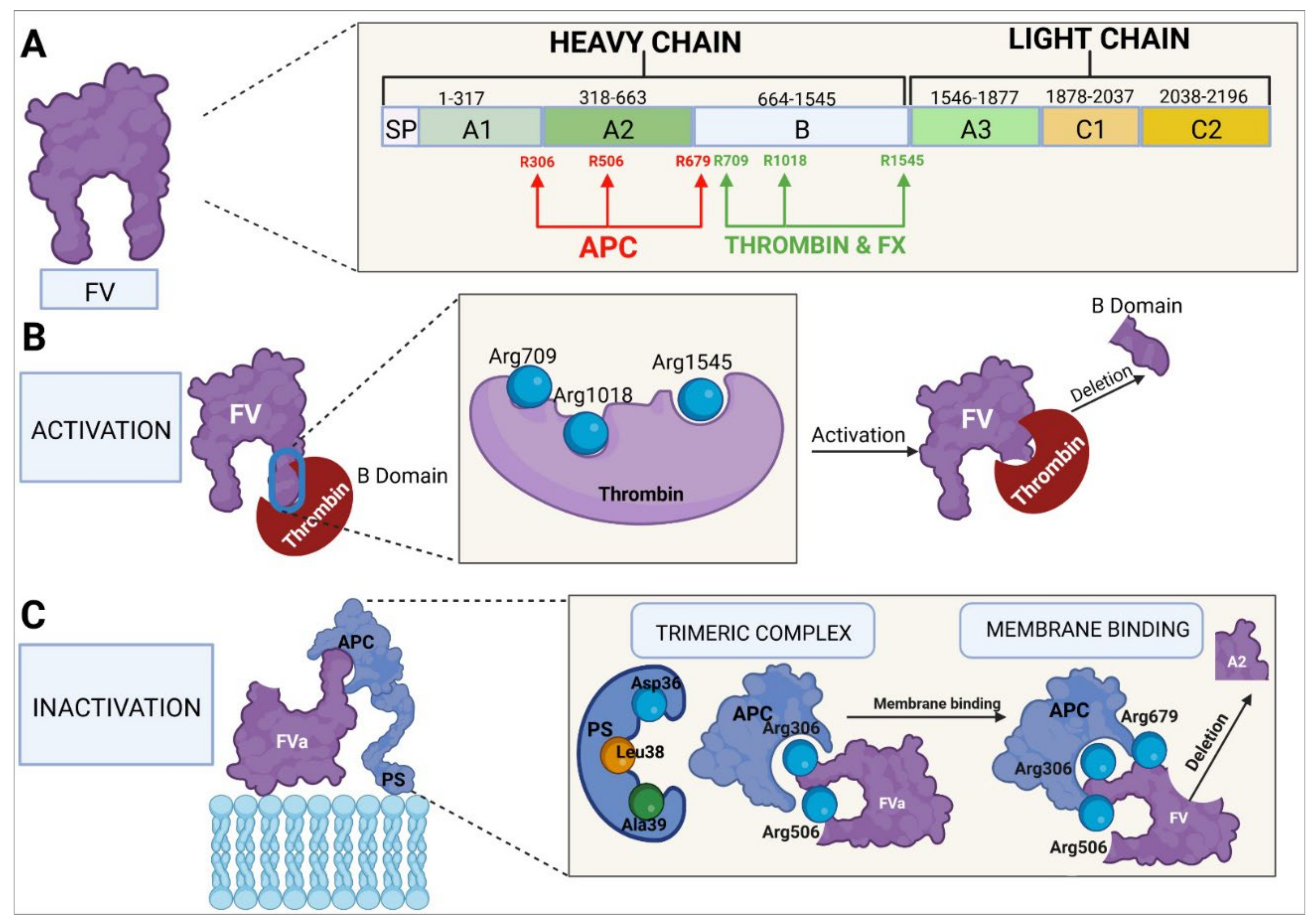
4.3. Molecular Structure of Factor V
5. Clotting Factor Biosynthesis
5.1. Von Willebrand Factor
5.2. Factor VIII
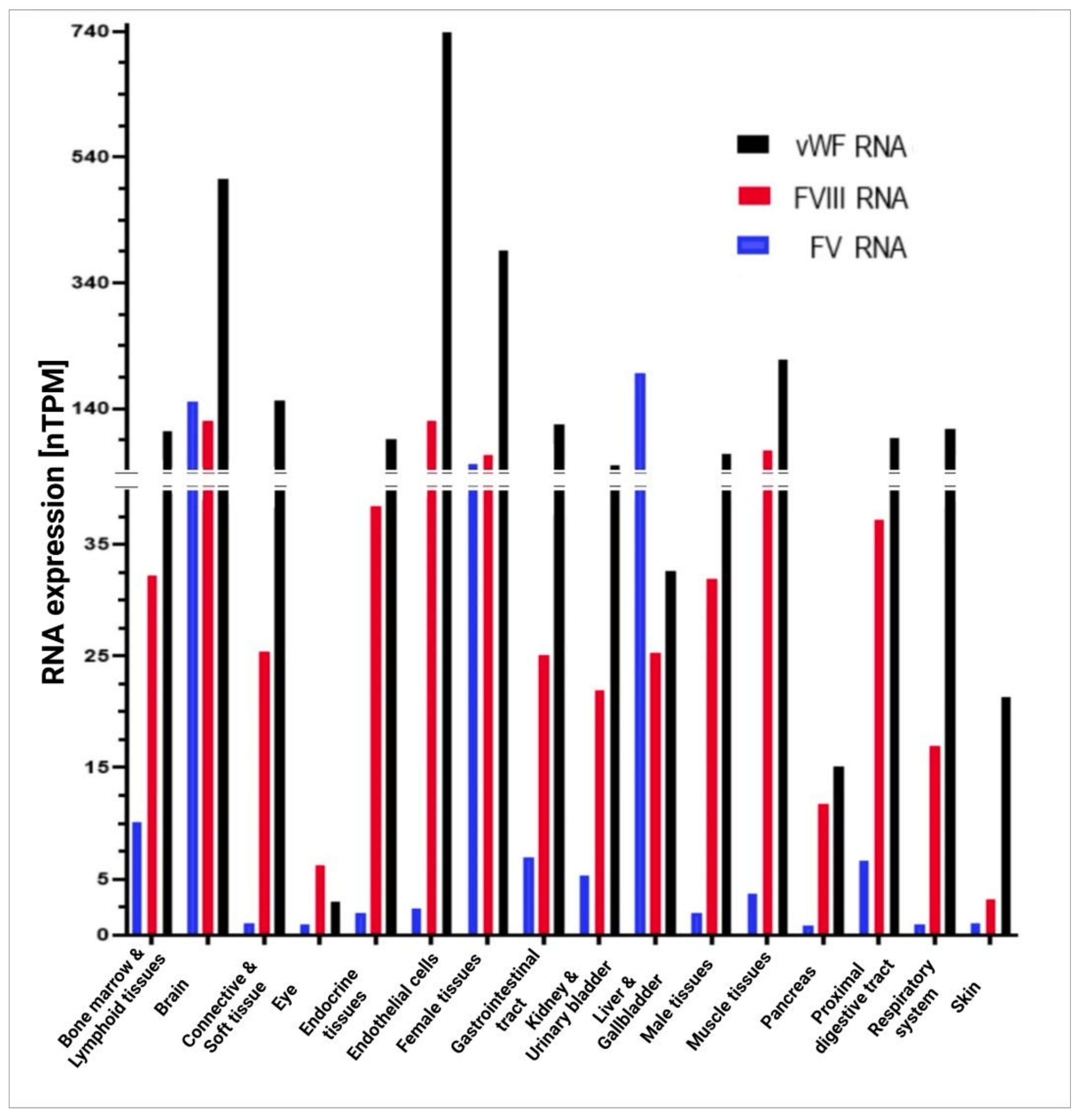
5.3. Factor V
6. Dysregulation of Homeostasis
6.1. Coagulopathies
6.1.1. Von Willebrand’s Disease
6.1.2. Hemophilia A
6.1.3. Factor V Deficiency
6.1.4. Combined Factor V and Factor VIII Deficiency
6.2. Thrombophilias
6.3. Vascular Pathologies Associated with Coagulation Factors
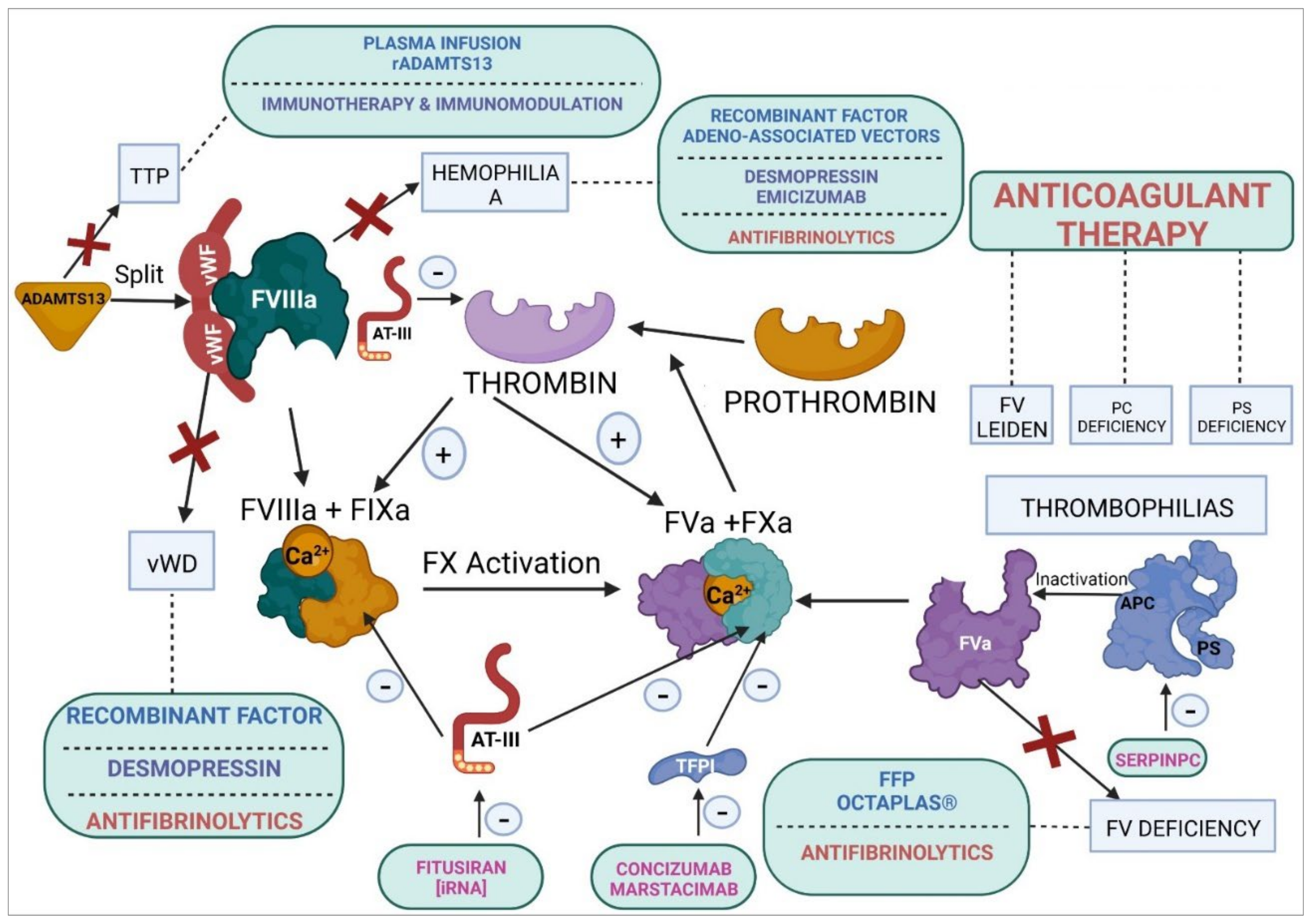
7. Homeostasis-Modifying Treatments in Coagulation
7.1. Alteration of the Functionality of Coagulation Factors
7.1.1. von Willebrand’s Disease
7.1.2. Hemophilia A
7.1.3. Factor V Deficiency
7.2. Rebalancing Therapies
7.3. Thrombophilias
7.3.1. Factor V Leiden Thrombophilia
7.3.2. PC Deficiency
7.3.3. PS Deficiency
7.3.4. Thrombotic Thrombocytopenic Purpura
7.3.5. Disseminated Intravascular Coagulation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chang, Y.; Wei, W. Endothelial dysfunction and inflammation: Immunity in rheumatoid arthritis. Mediat. Inflamm. 2016, 2016, 6813016. [Google Scholar] [CrossRef] [PubMed]
- Norooznezhad, A.H.; Mansouri, K. Endothelial cell dysfunction, coagulation, and angiogenesis in coronavirus disease 2019 (COVID-19). Microvasc. Res. 2021, 137, 104188. [Google Scholar] [CrossRef]
- Tang, Y.; Gan, X.; Curran, E.; Lamberti, G.; Krynska, B.; Kiani, M.F.; Wang, B. Targeted delivery of vascular endothelial growth factor improves stem cell therapy in a rat myocardial infarction model. Nanomedicine 2014, 10, 1711–1718. [Google Scholar] [CrossRef]
- Tang, Y.; Soroush, F.; Tong, Z.; Kiani, M.F.; Wang, B. Targeted multidrug delivery system to overcome chemoresistance in breast cancer. Int. J. Nanomed. 2017, 12, 671–681. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Brenner, J.S.; Greineder, C.; Shuvaev, V.; Muzykantov, V. Endothelial nanomedicine for the treatment of pulmonary disease. Expert Opin. Drug Deliv. 2015, 12, 239–261. [Google Scholar] [CrossRef]
- Glassman, P.M.; Myerson, J.W.; Ferguson, L.T.; Kiseleva, R.Y.; Shuvaev, V.V.; Brenner, J.S.; Muzykantov, V.R. Targeting drug delivery in the vascular system: Focus on endothelium. Adv. Drug Deliv. Rev. 2020, 157, 96–117. [Google Scholar] [CrossRef]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.; Vane, J. Human Arterial and Venous Tissues Generate Prostacyclin (Prostaglandin X), a Potent Inhibitor of Platelet Aggregation. Lancet 1977, 309, 18–20. [Google Scholar] [CrossRef]
- Weksler, B.B.; Marcus, A.J.; Jaffe, E.A. Synthesis of prostaglandin I2 (prostacyclin) by cultured human and bovine endothelial cells. Proc. Natl. Acad. Sci. USA 1977, 74, 3922–3926. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.; Sessa, W.C. eNOS derived nitric oxide regulates endothelial barrier function via VE cadherin and Rho GTPases. J. Cell Sci. 2013, 126, 5541–5552. [Google Scholar] [CrossRef]
- Yang, Q.; Wijerathne, H.; Langston, J.C.; Kiani, M.F.; Kilpatrick, L.E. Emerging Approaches to Understanding Microvascular Endothelial Heterogeneity: A Roadmap for Developing Anti-Inflammatory Therapeutics. Int. J. Mol. Sci. 2021, 22, 7770. [Google Scholar] [CrossRef]
- Oliver, G.; Srinivasan, R.S. Endothelial cell plasticity: How to become and remain a lymphatic endothelial cell. Development 2010, 137, 363–372. [Google Scholar] [CrossRef]
- Qiu, J.; Hirschi, K.K. Endothelial cell development and its application to regenerative medicine. Circ. Res. 2019, 125, 489–501. [Google Scholar] [CrossRef]
- Sriram, G.; Tan, J.Y.; Islam, I.; Rufaihah, A.J.; Cao, T. Efficient differentiation of human embryonic stem cells to arterial and venous endothelial cells under feeder-and serum-free conditions. Stem Cell Res. Ther. 2015, 6, 261. [Google Scholar] [CrossRef]
- Guo, Z.; Mo, Z. Regulation of endothelial cell differentiation in embryonic vascular development and its therapeutic potential in cardiovascular diseases. Life Sci. 2021, 276, 119406. [Google Scholar] [CrossRef]
- Chi, J.-T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; Van De Rijn, M.; Botstein, D. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef]
- Young, E.W.; Simmons, C.A. Macro-and microscale fluid flow systems for endothelial cell biology. Lab Chip 2010, 10, 143–160. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, eaaw4361. [Google Scholar] [CrossRef]
- Cavaillon, J.M.; Singer, M.; Skirecki, T. Sepsis therapies: Learning from 30 years of failure of translational research to propose new leads. EMBO Mol. Med. 2020, 12, e10128. [Google Scholar] [CrossRef]
- Brubaker, D.K.; Lauffenburger, D.A. Translating preclinical models to humans. Science 2020, 367, 742–743. [Google Scholar] [CrossRef]
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019, 30, 123–142. [Google Scholar] [CrossRef]
- Kalla, D.; Kind, A.; Schnieke, A. Genetically engineered pigs to study cancer. Int. J. Mol. Sci. 2020, 21, 488. [Google Scholar] [CrossRef] [PubMed]
- Perlman, H.; Budinger, G.R.; Ward, P.A. Humanizing the mouse: In defense of murine models of critical illness. Am. J. Respir. Crit. Care Med. 2013, 187, 898–900. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Gonçalves, L.; Seiça, R. Methods to evaluate vascular function: A crucial approach towards predictive, preventive, and personalised medicine. EPMA J. 2022, 13, 209–235. [Google Scholar] [CrossRef]
- Ruparelia, N.; Choudhury, R. Inflammation and atherosclerosis: What is on the horizon? Heart 2020, 106, 80–85. [Google Scholar] [CrossRef]
- Badimon, L.; Romero, J.C.; Cubedo, J.; Borrell-Pages, M. Circulating biomarkers. Thromb. Res. 2012, 130, S12–S15. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Hidalgo, A.; Peired, A.J.; Wild, M.; Vestweber, D.; Frenette, P.S. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 2007, 26, 477–489. [Google Scholar] [CrossRef]
- Verma, S.; Anderson, T.J. Fundamentals of endothelial function for the clinical cardiologist. Circulation 2002, 105, 546–549. [Google Scholar] [CrossRef]
- Yeh, E.T.; Willerson, J.T. Coming of age of C-reactive protein: Using inflammation markers in cardiology. Circulation 2003, 107, 370–371. [Google Scholar] [CrossRef]
- Wenzel, F.; Baertl, A.; Zimmermann, N.; Hohlfeld, T.; Giers, G.; Oldenburg, J.; Assert, R. Different behaviour of soluble CD40L concentrations can be reflected by variations of preanalytical conditions. Clin. Hemorheol. Microcirc. 2008, 39, 417–422. [Google Scholar] [CrossRef]
- Szmitko, P.E.; Wang, C.H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New markers of inflammation and endothelial cell activation: Part I. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [CrossRef]
- Dowsett, L.; Higgins, E.; Alanazi, S.; Alshuwayer, N.A.; Leiper, F.C.; Leiper, J. ADMA: A key player in the relationship between vascular dysfunction and inflammation in atherosclerosis. J. Clin. Med. 2020, 9, 3026. [Google Scholar] [CrossRef]
- Kielstein, J.T.; Fliser, D. The past, presence and future of ADMA in nephrology. Nephrol. Ther. 2007, 3, 47–54. [Google Scholar] [CrossRef]
- Boger, R.H.; Sullivan, L.M.; Schwedhelm, E.; Wang, T.J.; Maas, R.; Benjamin, E.J.; Schulze, F.; Xanthakis, V.; Benndorf, R.A.; Vasan, R.S. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 2009, 119, 1592–1600. [Google Scholar] [CrossRef]
- Caglar, K.; Yilmaz, M.I.; Sonmez, A.; Cakir, E.; Kaya, A.; Acikel, C.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Bilgi, C.; et al. ADMA, proteinuria, and insulin resistance in non-diabetic stage I chronic kidney disease. Kidney Int. 2006, 70, 781–787. [Google Scholar] [CrossRef]
- Kobayashi, S.; Oka, M.; Maesato, K.; Ikee, R.; Mano, T.; Hidekazu, M.; Ohtake, T. Coronary artery calcification, ADMA, and insulin resistance in CKD patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1289–1295. [Google Scholar] [CrossRef]
- Leong, T.; Zylberstein, D.; Graham, I.; Lissner, L.; Ward, D.; Fogarty, J.; Bengtsson, C.; Bjorkelund, C.; Thelle, D.; Swedish-Irish-Norwegian, C. Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenburg. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 961–967. [Google Scholar] [CrossRef]
- Stuhlinger, M.C.; Oka, R.K.; Graf, E.E.; Schmolzer, I.; Upson, B.M.; Kapoor, O.; Szuba, A.; Malinow, M.R.; Wascher, T.C.; Pachinger, O.; et al. Endothelial dysfunction induced by hyperhomocyst(e)inemia: Role of asymmetric dimethylarginine. Circulation 2003, 108, 933–938. [Google Scholar] [CrossRef]
- Yoo, J.H.; Lee, S.C. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 2001, 158, 425–430. [Google Scholar] [CrossRef]
- Sena, C.M.; Bento, C.F.; Pereira, P.; Seica, R. Diabetes mellitus: New challenges and innovative therapies. EPMA J. 2010, 1, 138–163. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and endotelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic. Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Bar, A.; Targosz-Korecka, M.; Suraj, J.; Proniewski, B.; Jasztal, A.; Marczyk, B.; Sternak, M.; Przybyło, M.; Kurpińska, A.; Walczak, M.; et al. Degradation of glycocalyx and multiple manifestations of endothelial dysfunction coincide in the early phase of endothelial dysfunction before atherosclerotic plaque development in apolipoprotein E/low-density lipoprotein receptor-deficient mice. J. Am. Heart Assoc. 2019, 8, e011171. [Google Scholar] [CrossRef] [PubMed]
- Pennathur, S.; Heinecke, J.W. Oxidative stress and endothelial dysfunction in vascular disease. Curr. Diab. Rep. 2007, 7, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. Oxidative biomarkers in the diagnosis and prognosis of cardiovascular disease. Am. J. Cardiol. 2006, 98, 9P–17P. [Google Scholar] [CrossRef] [PubMed]
- Ferratini, M.; Ripamonti, V.; Masson, S.; Grati, P.; Racca, V.; Cuccovillo, I.; Raimondi, E.; Capomolla, S.; Macchi, C.; Coruzzi, P.; et al. Pentraxin-3 predicts functional recovery and 1-year major adverse cardiovascular events after rehabilitation of cardiac surgery patients. J. Cardiopulm. Rehabil. Prev. 2012, 32, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Baumann, K.; Przybilla, D.; Schmitz, T.; Flender, A.; Paul, K.; Alhusseiny, A.; Nickenig, G.; Werner, N. Endothelial microparticles reduce ICAM-1 expression in a microRNA-222-dependent mechanism. J. Cell Mol. Med. 2015, 19, 2202–2214. [Google Scholar] [CrossRef]
- Berckmans, R.J.; Nieuwland, R.; Boing, A.N.; Romijn, F.P.; Hack, C.E.; Sturk, A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb. Haemost. 2001, 85, 639–649. [Google Scholar] [CrossRef]
- Wen, B.; Combes, V.; Bonhoure, A.; Weksler, B.B.; Couraud, P.O.; Grau, G.E. Endotoxin-induced monocytic microparticles have contrasting effects on endothelial inflammatory responses. PLoS ONE 2014, 9, e91597. [Google Scholar] [CrossRef]
- Williams, M.S.; Rogers, H.L.; Wang, N.Y.; Ziegelstein, R.C. Do platelet derived microparticles play a role in depression, inflammation, and acute coronary syndrome? Psychosomatics 2014, 55, 252–260. [Google Scholar] [CrossRef]
- Toth, B.; Liebhardt, S.; Steinig, K.; Ditsch, N.; Rank, A.; Bauerfeind, I.; Spannagl, M.; Friese, K.; Reininger, A.J. Platelet-derived microparticles and coagulation activation in breast cancer patients. Thromb. Haemost. 2008, 100, 663–669. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, D.; Darbousset, R.; Farge, D.; Debourdeau, P.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin. Oncol. 2014, 41, 346–358. [Google Scholar] [CrossRef]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef]
- Nawroth, P.; Kisiel, W.; Stern, D. The role of endothelium in the homeostatic balance of haemostasis. Clin. Haematol. 1985, 14, 531–546. [Google Scholar] [CrossRef]
- Wang, C.L.; Miyata, T.; Weksler, B.; Rubin, A.L.; Stenzel, K.H. Collagen-induced platelet aggregation and release. I Effects of side-chain modifications and role of arginyl residues. Biochim. Biophys. Acta 1978, 544, 555–567. [Google Scholar] [CrossRef]
- Hoffman, M.; Monroe III, D.M. A Cell-based Model of Hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [CrossRef]
- Hoffman, M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev. 2003, 17, S1–S5. [Google Scholar] [CrossRef]
- Chee, Y.L. Coagulation. J. R. Coll. Physicians Edinb. 2014, 44, 42–45. [Google Scholar] [CrossRef]
- Winter, W.E.; Flax, S.D.; Harris, N.S. Coagulation Testing in the Core Laboratory. Lab. Med. 2017, 48, 295–313. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Hrachovinová, I. Diagnostic strategies in disorders of hemostasis. Vnitr. Lek. 2018, 64, 537–544. [Google Scholar] [CrossRef]
- Stassen, J.; Arnout, J.; Deckmyn, H. The Hemostatic System. Curr. Med. Chem. 2004, 11, 2245–2260. [Google Scholar] [CrossRef]
- Broos, K.; Feys, H.B.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at work in primary hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism action of platelets and crucial blood coagulation pathways in hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Lasne, D.; Jude, B.; Susen, S. From normal to pathological hemostasis. Can. J. Anesth. Can. Anesth. 2006, 53, S2–S11. [Google Scholar] [CrossRef][Green Version]
- Furie, B.; Furie, B.C. In vivo thrombus formation. J. Thromb. Haemost. 2007, 5, 12–17. [Google Scholar] [CrossRef]
- Monagle, P.; Massicotte, P. Developmental haemostasis: Secondary haemostasis. Semin. Fetal Neonatal Med. 2011, 16, 294–300. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Intrinsic Pathway of Coagulation and Thrombosis: Insights from Animal Models. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 331–338. [Google Scholar] [CrossRef]
- Hoffman, M.; Monroe, D.M. Coagulation 2006: A Modern View of Hemostasis. Hematol. Oncol. Clin. N. Am. 2007, 21, 1–11. [Google Scholar] [CrossRef]
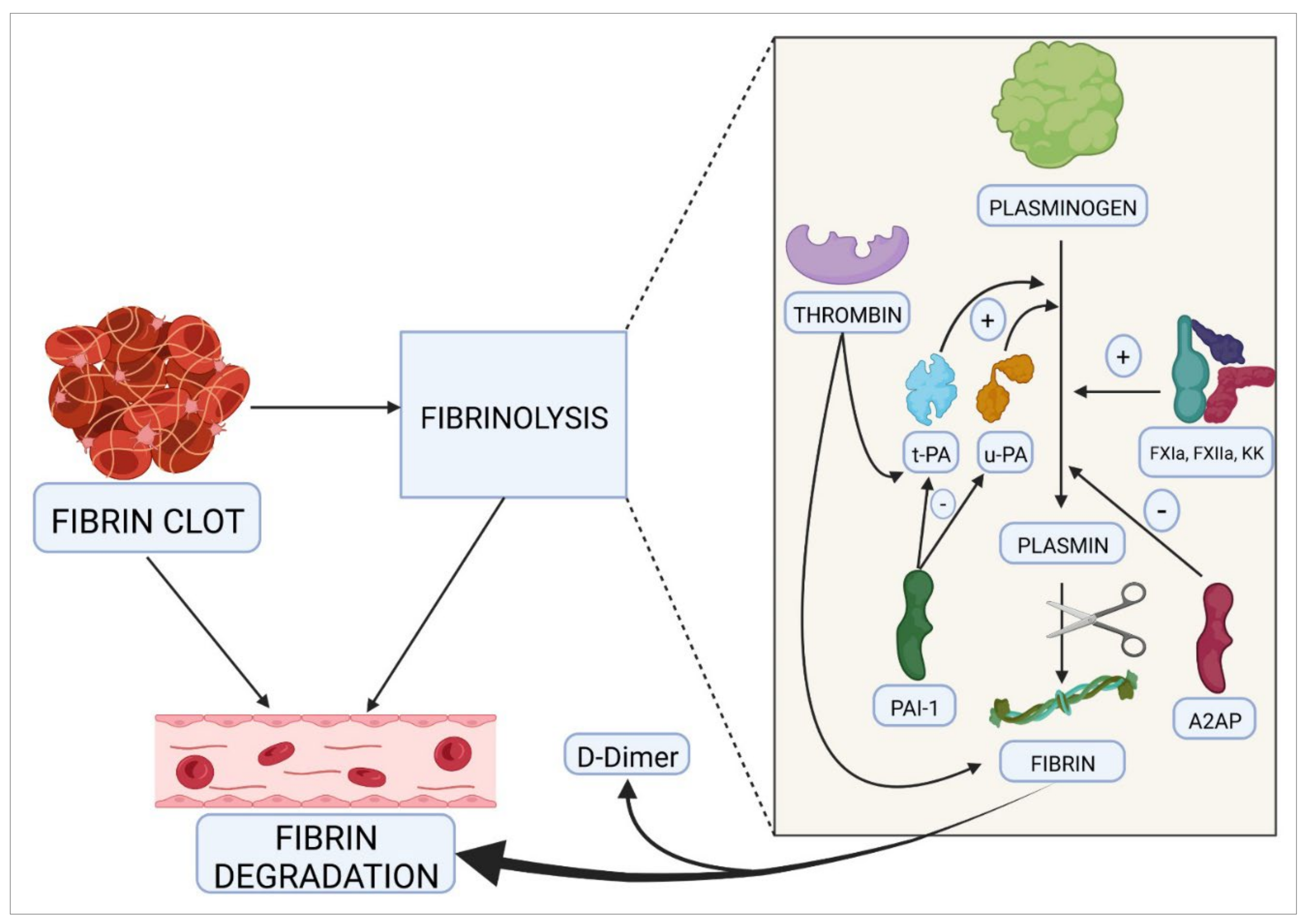
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Rey-Formoso, V.; Barreto-Mota, R.; Soares, H. Developmental hemostasis in the neonatal period. World J. Pediatr. 2022, 18, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ignjatovic, V.; Mertyn, E.; Monagle, P. The coagulation system in children: Developmental and pathophysiological considerations. Semin. Thromb. Hemost. 2011, 37, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Monagle, P.; Ignjatovic, V.; Savoia, H. Hemostasis in neonates and children: Pitfalls and dilemmas. Blood Rev. 2010, 24, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Guzzetta, N.A.; Miller, B.E. Principles of hemostasis in children: Models and maturation. Paediatr. Anaesth. 2011, 21, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jaffray, J.; Young, G. Developmental hemostasis: Clinical implications from the fetus to the adolescent. Pediatr. Clin. N. Am. 2013, 60, 1407–1417. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Osmond, M.; Bapir, M.; Riddell, A.; Smith, C.; Lee, C.A.; Kadir, R.A. The effect of labour on the coagulation system in the term neonate. Haemophilia 2013, 19, 533–538. [Google Scholar] [CrossRef]
- Strauss, T.; Sidlik-Muskatel, R.; Kenet, G. Developmental hemostasis: Primary hemostasis and evaluation of platelet function in neonates. Semin. Fetal Neonatal Med. 2011, 16, 301–304. [Google Scholar] [CrossRef]
- Campbell, S.E.; Bolton-Maggs, P.H. Congenital and acquired bleeding disorders in infancy. Early Hum. Dev. 2015, 91, 637–642. [Google Scholar] [CrossRef]
- Neary, E.; McCallion, N.; Kevane, B.; Cotter, M.; Egan, K.; Regan, I.; Kirkham, C.; Mooney, C.; Coulter-Smith, S.; Ní Áinle, F. Coagulation indices in very preterm infants from cord blood and postnatal samples. J. Thromb. Haemost. 2015, 13, 2021–2030. [Google Scholar] [CrossRef]
- Nowak-Gottl, U.; Limperger, V.; Bauer, A.; Kowalski, D.; Kenet, G. Bleeding issues in neonates and infants-update 2015. Thromb. Res. 2015, 135, S41–S43. [Google Scholar] [CrossRef]
- Bhat, R.; Monagle, P. The preterm infant with thrombosis. Arch. Dis. Child Fetal Neonatal Ed. 2012, 97, F423–F428. [Google Scholar] [CrossRef]
- Israels, S.J.; Rand, M.L.; Michelson, A.D. Neonatal platelet function. Semin. Thromb. Hemost. 2003, 29, 363–372. [Google Scholar] [CrossRef]
- Lippi, G.; Franchini, M.; Montagnana, M.; Guidi, G.C. Coagulation testing in pediatric patients: The young are not just miniature adults. Semin. Thromb. Hemost. 2007, 33, 816–820. [Google Scholar] [CrossRef]
- Lippi, G.; Salvagno, G.L.; Rugolotto, S.; Chiaffoni, G.P.; Padovani, E.M.; Franchini, M.; Guidi, G.C. Routine coagulation tests in newborn and young infants. J. Thromb. Thrombolysis 2007, 24, 153–155. [Google Scholar] [CrossRef]
- Monagle, P.; Barnes, C.; Ignjatovic, V.; Furmedge, J.; Newall, F.; Chan, A.; De Rosa, L.; Hamilton, S.; Ragg, P.; Robinson, S.; et al. Developmental haemostasis. Impact for clinical haemostasis laboratories. Thromb. Haemost. 2006, 95, 362–372. [Google Scholar] [CrossRef]
- Chalmers, E.A. Neonatal coagulation problems. Arch. Dis. Child Fetal Neonatal Ed. 2004, 89, F475–F478. [Google Scholar] [CrossRef]
- Andrew, M.; Paes, B.; Milner, R.; Johnston, M.; Mitchell, L.; Tollefsen, D.M.; Powers, P. Development of the human coagulation system in the full-term infant. Blood 1987, 70, 165–172. [Google Scholar] [CrossRef]
- Sokou, R.; Konstantinidi, A.; Stefanaki, C.; Tsantes, A.G.; Parastatidou, S.; Lampropoulou, K.; Katsaras, G.; Tavoulari, E.; Iacovidou, N.; Kyriakou, E.; et al. Thromboelastometry: Studying hemostatic profile in small for gestational age neonates-a pilot observational study. Eur. J. Pediatr. 2019, 178, 551–557. [Google Scholar] [CrossRef]
- Mitsiakos, G.; Karametou, M.; Gkampeta, A.; Karali, C.; Papathanasiou, A.E.; Papacharalambous, E.; Babacheva, E.; Papadakis, E.; Yupsani, A.; Chatziioannidis, I.; et al. Effectiveness and safety of 4-factor prothrombin complex concentrate (4PCC) in neonates with intractable bleeding or severe coagulation disturbances: A retrospective study of 37 cases. J. Pediatr. Hematol. Oncol. 2019, 41, e135–e140. [Google Scholar] [CrossRef]
- Bernhard, H.; Rosenkranz, A.; Petritsch, M.; Kofeler, H.; Rehak, T.; Novak, M.; Muntean, W. Phospholipid content, expression and support of thrombin generation of neonatal platelets. Acta Paediatr. 2009, 98, 251–255. [Google Scholar] [CrossRef]
- Diaz-Miron, J.; Miller, J.; Vogel, A.M. Neonatal hematology. Semin. Pediatr. Surg. 2013, 22, 199–204. [Google Scholar] [CrossRef]
- Haley, K.M.; Recht, M.; McCarty, O.J. Neonatal platelets: Mediators of primary hemostasis in the developing hemostatic system. Pediatr. Res. 2014, 76, 230–237. [Google Scholar] [CrossRef]
- Faraoni, D.; O’Leary, J.D. Understanding developmental hemostasis through the use of viscoelastic tests of whole blood coagulation. Minerva Anestesiol. 2017, 83, 347–349. [Google Scholar] [CrossRef]
- Jaffray, J.; Young, G.; Ko, R.H. The bleeding newborn: A review of presentation, diagnosis, and management. Semin. Fetal Neonatal Med. 2016, 21, 44–49. [Google Scholar] [CrossRef]
- Mitsiakos, G.; Papaioannou, G.; Papadakis, E.; Chatziioannidis, E.; Giougi, E.; Karagianni, P.; Evdoridou, J.; Malindretos, P.; Athanasiou, M.; Athanassiadou, F.; et al. Haemostatic profile of full-term, healthy, small for gestational age neonates. Thromb. Res. 2009, 124, 288–291. [Google Scholar] [CrossRef]
- Konstantinidi, A.; Sokou, R.; Parastatidou, S.; Lampropoulou, K.; Katsaras, G.; Boutsikou, T.; Gounaris, A.K.; Tsantes, A.E.; Iacovidou, N. Clinical application of thromboelastography/thromboelastometry (TEG/TEM) in the neonatal population: A narrative review. Semin. Thromb. Hemost. 2019, 45, 449–457. [Google Scholar] [CrossRef]
- Mitsiakos, G.; Giougi, E.; Chatziioannidis, I.; Karagianni, P.; Papadakis, E.; Tsakalidis, C.; Papaioannou, G.; Malindretos, P.; Nikolaidis, N. Haemostatic profile of healthy premature small for gestational age neonates. Thromb. Res. 2010, 126, 103–106. [Google Scholar] [CrossRef]
- Cannon, W.B. Organization for physiological homeostasis. Physiol. Rev. 1929, 9, 399–431. [Google Scholar] [CrossRef]
- Cannon, W.B. The Wisdom of the Body; The Norton Library: New York, NY, USA, 1932. [Google Scholar]
- De Luca, L.A., Jr. A critique on the theory of homeostasis. Physiol. Behav. 2022, 247, 113712. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue factor: An essential mediator of hemostasis and trigger of thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef]
- Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Investig. 2012, 122, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Anticoagulant SERPINs: Endogenous Regulators of Hemostasis and Thrombosis. Front. Cardiovasc. Med. 2022, 9, 878199. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E. Tissue factor pathway inhibitor: Multiple anticoagulant activities for a single protein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.H.; Zlokovic, B.V.; Mosnier, L.O. Protein C anticoagulant and cytoprotective pathways. Int. J. Hematol. 2012, 95, 333–345. [Google Scholar] [CrossRef]
- Díaz-Hung, M.L.; Hetz, C. Proteostasis and resilience: On the interphase between individual’s and intracellular stress. Trends Endocrinol. Metab. 2022, 33, 305–317. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef]
- Rai, M.; Curley, M.; Coleman, Z.; Demontis, F. Contribution of proteases to the hallmarks of aging and to age-related neurodegeneration. Aging Cell 2022, 21, e13603. [Google Scholar] [CrossRef]
- Rai, M.; Coleman, Z.; Curley, M.; Nityanandam, A.; Platt, A.; Robles-Murguia, M.; Jiao, J.; Finkelstein, D.; Wang, Y.D.; Xu, B.; et al. Proteasome stress in skeletal muscle mounts along-range protective response that delays retinal and brain aging. Cell Metab. 2021, 33, 1137–1154. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Chuang, C.L.; Demontis, F. Systemic manifestation and contribution of peripheral tissues to Huntington’s disease pathogenesis. Ageing Res. Rev. 2021, 69, 101358. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Nicolaes, G.A.F.; Dahlbäck, B. Factor V and Thrombotic Disease: Description of a Janus-Faced Protein. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 530–538. [Google Scholar] [CrossRef]
- Segers, K.; Dahlbäck, B.; Nicolaes, G. Coagulation factor V and thrombophilia: Background and mechanisms. Thromb. Haemost. 2007, 98, 530–542. [Google Scholar] [CrossRef]
- Cramer, T.J.; Gale, A.J. The anticoagulant function of coagulation factor V. Thromb. Haemost. 2012, 107, 15–21. [Google Scholar] [CrossRef]
- Santamaria, S.; Reglińska-Matveyev, N.; Gierula, M.; Camire, R.M.; Crawley, J.T.B.; Lane, D.A.; Ahnström, J. Factor V has an anticoagulant cofactor activity that targets the early phase of coagulation. J. Biol. Chem. 2017, 292, 9335–9344. [Google Scholar] [CrossRef]
- Sankar, A.D.; Weyand, A.C.; Pipe, S.W. The evolution of recombinant factor replacement for hemophilia. Transfus. Apher. Sci. 2019, 58, 596–600. [Google Scholar] [CrossRef]
- Bleuez, C.; Koch, W.F.; Urbach, C.; Hollfelder, F.; Jermutus, L. Exploiting protease activation for therapy. Drug Discov. Today. 2022, 27, 1743–1754. [Google Scholar] [CrossRef]
- Hu, S.; Ma, L.; Dong, B.; Shan, Q.; Zhou, J.; Zhang, G.; Wang, M.; Cen, S.; Zhu, M.; Wang, J.; et al. A kind of HIV-1 protease inhibitors containing phenols with antiviral activity against DRV-resistant variants. Bioorg. Med. Chem. 2022, 64, 116760. [Google Scholar] [CrossRef]
- Liu, H.; Iketani, S.; Zask, A.; Khanizeman, N.; Bednarova, E.; Forouhar, F.; Fowler, B.; Hong, S.J.; Mohri, H.; Nair, M.S.; et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022, 13, 1891. [Google Scholar] [CrossRef]
- Antonopoulou, I.; Sapountzaki, E.; Rova, U.; Christakopoulos, P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput. Struct. Biotechnol. J. 2022, 20, 1306–1344. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An enigmatic zymogen. Blood 2021, 137, 2881–2889. [Google Scholar] [CrossRef]
- Huntington, J.A. Slow thrombin is zymogen-like. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 159–164. [Google Scholar] [CrossRef][Green Version]
- Al-Amer, O.M. The role of thrombin in haemostasis. Blood Coagul. Fibrinolysis 2022, 33, 145–148. [Google Scholar] [CrossRef]
- Naß, J.; Terglane, J.; Gerke, V. Weibel Palade Bodies: Unique Secretory Organelles of Endothelial Cells that Control Blood Vessel Homeostasis. Front. Cell Dev. Biol. 2021, 9, 813995. [Google Scholar] [CrossRef]
- Yasar, S.J.; Abdullah, O.; Fay, W.; Balla, S. Von Willebrand Factor Revisited. J. Interven. Cardiol. 2018, 31, 360–367. [Google Scholar] [CrossRef]
- Randi, A.M.; Laffan, M.A. Von Willebrand Factor and Angiogenesis: Basic and Applied Issues. J. Thromb. Haemost. 2017, 15, 13–20. [Google Scholar] [CrossRef]
- Leebeek, F.W.G.; Eikenboom, J.C.J. Von Willebrand’s Disease. N. Engl. J. Med. 2016, 375, 2067–2080. [Google Scholar] [CrossRef]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. Von Willebrand Factor Biosynthesis, Secretion, and Clearance: Connecting the Far Ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef]
- Zhou, Y.-F.; Eng, E.T.; Zhu, J.; Lu, C.; Walz, T.; Springer, T.A. Sequence and Structure Relationships within von Willebrand Factor. Blood 2012, 120, 449–458. [Google Scholar] [CrossRef]
- Wohner, N.; Kovács, A.; Machovich, R.; Kolev, K. Modulation of the von Willebrand Factor-Dependent Platelet Adhesion through Alternative Proteolytic Pathways. Thromb. Res. 2012, 129, e41–e46. [Google Scholar] [CrossRef] [PubMed]
- Mourik, M.; Eikenboom, J. Lifecycle of Weibel-Palade bodies. Hamostaseologie 2017, 37, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Brandherm, I.; Disse, J.; Zeuschner, D.; Gerke, V. cAMP-induced secretion of endothelial von Willebrand factor is regulated by a phosphorylation/dephosphorylation switch in annexin A2. Blood 2013, 122, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Holthenrich, A.; Gerke, V. Regulation of Von-Willebrand Factor Secretion from Endothelial Cells by the Annexin A2-S100A10 Complex. Int. J. Mol. Sci. 2018, 19, 1752. [Google Scholar] [CrossRef]
- Li, Z.; Lin, J.; Sulchek, T.; Cruz, M.A.; Wu, J.; Dong, J.; Zhu, C. Domain-Specific Mechanical Modulation of VWF–ADAMTS13 Interaction. Mol. Biol. Cell. 2019, 30, 1920–1929. [Google Scholar] [CrossRef]
- Ngo, J.C.K.; Huang, M.; Roth, D.A.; Furie, B.C.; Furie, B. Crystal Structure of Human Factor VIII: Implications for the Formation of the Factor IXa-Factor VIIIa complex. Structure 2008, 16, 597–606. [Google Scholar] [CrossRef]
- Jenkins, P.V.; Dill, J.L.; Zhou, Q.; Fay, P.J. Contribution of Factor VIIIa A2 and A3-C1-C2 Subunits to the Affinity for Factor IXa in Factor Xase. Biochemistry 2004, 43, 5094–5101. [Google Scholar] [CrossRef]
- Mazurkiewicz-Pisarek, A.; Płucienniczak, G.; Ciach, T.; Płucienniczak, A. The factor VIII protein and its function. Acta Biochim. Pol. 2016, 63, 11–16. [Google Scholar] [CrossRef]
- Takeyama, M.; Nogami, K.; Sasai, K.; Furukawa, S.; Shima, M. Contribution of Factor VIII A2 Domain Residues 400-409 to a Factor X-Interactive Site in the Factor Xase Complex. Thromb. Haemost. 2018, 118, 830–841. [Google Scholar] [CrossRef]
- Orfeo, T.; Elsman, R.; Gissel, M.; Mann, K.G.; Butenas, S. Activation, activity and inactivation of factor VIII in factor VIII products. Haemophilia 2016, 22, 462–473. [Google Scholar] [CrossRef]
- Terraube, V.; O’Donnell, J.S.; Jenkins, P.V. Factor VIII and von Willebrand Factor Interaction: Biological, Clinical and Therapeutic Importance. Haemophilia 2010, 16, 3–13. [Google Scholar] [CrossRef]
- Shen, L.; Dahlbäck, B. Factor V and protein S as synergistic cofactors to activated protein C in degradation of factor VIIIa. J. Biol. Chem. 1994, 269, 18735–18738. [Google Scholar] [CrossRef]
- Plantier, J.L.; Rolli, V.; Ducasse, C.; Dargaud, Y.; Enjolras, N.; Boukerche, H.; Négrier, C. Activated factor X cleaves factor VIII at arginine 562, limiting its cofactor efficiency. J. Thromb. Haemost. 2010, 8, 286–293. [Google Scholar] [CrossRef]
- Dahlbäck, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef]
- Peyvandi, F.; Duga, S.; Akhavan, S.; Mannucci, P.M. Rare coagulation deficiencies: Rare coagulation deficiencies. Haemophilia 2002, 8, 308–321. [Google Scholar] [CrossRef]
- Ruben, E.A.; Rau, M.J.; Fitzpatrick, J.; Di Cera, E. Cryo-EM structures of human coagulation factors V and Va. Blood 2021, 137, 3137–3144. [Google Scholar] [CrossRef]
- Stoilova-Mcphie, S.; Parmenter, C.D.J.; Segers, K.; Villoutreix, B.O.; Nicolaes, G.A.F. Defining the structure of membrane-bound human blood coagulation factor Va: Defining the structure of human blood coagulation factor Va. J. Thromb. Haemost. 2007, 6, 76–82. [Google Scholar] [CrossRef]
- Asselta, R.; Peyvandi, F. Factor V Deficiency. Semin. Thromb. Hemost. 2009, 35, 382–389. [Google Scholar] [CrossRef]
- Adams, T.E.; Hockin, M.F.; Mann, K.G.; Everse, S.J. The crystal structure of activated protein C-inactivated bovine factor Va: Implications for cofactor function. Proc. Natl. Acad. Sci. USA 2004, 101, 8918–8923. [Google Scholar] [CrossRef]
- Dahlbäck, B. Pro- and anticoagulant properties of factor V in pathogenesis of thrombosis and bleeding disorders.pdf. Int. J. Lab. Hemtol. 2016, 38 (Suppl. 1), 4–11. [Google Scholar] [CrossRef]
- Bukys, M.A.; Blum, M.A.; Kim, P.Y.; Brufatto, N.; Nesheim, M.E.; Kalafatis, M. Incorporation of Factor Va into Prothrombinase Is Required for Coordinated Cleavage of Prothrombin by Factor Xa. J. Biol. Chem. 2005, 280, 27393–27401. [Google Scholar] [CrossRef] [PubMed]
- Autin, L.; Steen, M.; Dahlbäck, B.; Villoutreix, B.O. Proposed structural models of the prothrombinase (FXa-FVa) complex. Proteins Struct. Funct. Bioinform. 2006, 63, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Hockin, M.F.; Cawthern, K.M.; Kalafatis, M.; Mann, K.G. A Model Describing the Inactivation of Factor Va by APC: Bond Cleavage, Fragment Dissociation, and Product Inhibition. Biochemistry 1999, 38, 6918–6934. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.H.; Fernández, J.A.; Gale, A.J.; Mosnier, L.O. Activated protein C. J. Thromb. Haemost. 2007, 5, 73–80. [Google Scholar] [CrossRef]
- Oto, J.; Fernández-Pardo, Á.; Miralles, M.; Plana, E.; España, F.; Navarro, S.; Medina, P. Activated protein C assays: A review. Clin. Chim. Acta 2020, 502, 227–232. [Google Scholar] [CrossRef]
- Nogami, K.; Shinozawa, K.; Ogiwara, K.; Matsumoto, T.; Amano, K.; Fukutake, K.; Midori, S. Novel FV mutation (W1920R, FVNara) associated with serious deep vein thrombosis and more potent APC resistance relative to FVLeiden. Blood 2014, 123, 2420–2428. [Google Scholar] [CrossRef]
- Van Doorn, P.; Rosing, J.; Duckers, C.; Hackeng, T.; Simioni, P.; Castoldi, E. Factor V Has Anticoagulant Activity in Plasma in the Presence of TFPIα: Difference between FV1 and FV2. Thromb. Haemost. 2018, 118, 1194–1202. [Google Scholar] [CrossRef]
- Forastiero, R. Inhibidores fisiológicos. Hematología 2017, 21, 43–47. Available online: https://www.studocu.com/es-ar/document/universidad-catolica-de-cuyo/fisiologia/inhibidores-fisiologicos/28318222 (accessed on 15 July 2022).
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef]
- Kashif, M.; Isermann, B. Role of the coagulation system in development. Thromb. Res. 2013, 131 (Suppl. 1), S14–S17. [Google Scholar] [CrossRef]
- Tesfamariam, B. Distinct characteristics of neonatal platelet reactivity. Pharmacol. Res. 2017, 123, 1–9. [Google Scholar] [CrossRef]
- Schlagenhauf, A.; Schweintzger, S.; Birner-Gruenberger, R.; Leschnik, B.; Muntean, W. Newborn platelets: Lower levels of protease-activated receptors cause hypoaggregability to thrombin. Platelets 2010, 21, 641–647. [Google Scholar] [CrossRef]
- Del Vecchio, A.; Sola, M.C. Performing and interpreting the bleeding time in the neonatal intensive care unit. Clin. Perinatol. 2000, 27, 643–654. [Google Scholar] [CrossRef]
- Nacoti, M.; Corbella, D.; Fazzi, F.; Rapido, F.; Bonanomi, E. Coagulopathy and transfusion therapy in pediatric liver transplantation. World J. Gastroenterol. 2016, 22, 2005–2023. [Google Scholar] [CrossRef]
- Israels, S.J.; Cheang, T.; McMillan-Ward, E.M.; Cheang, M. Evaluation of primary hemostasis in neonates with a new in vitro platelet function analyzer. J. Pediatr. 2001, 138, 116–119. [Google Scholar] [CrossRef]
- Coffin, J.D.; Harrison, J.; Schwartz, S.; Heimark, R. Angioblast Differentiation and Morphogenesis of the Vascular Endothelium in the Mouse Embryo. Dev. Biol. 1991, 148, 51–62. [Google Scholar] [CrossRef]
- Thomas, K.; Sutor, A.; Altinkaya, N.; Grohmann, A.; Zehenter, A.; Leititis, J. Von Willebrand Factor-Collagen Binding Activity Is Increased in Newborns and Infants. Acta Paediatr. 1995, 84, 697–700. [Google Scholar] [CrossRef]
- Roschitz, B.; Sudi, K.; Köstenberger, M.; Muntean, W. Shorter PFA-100® Closure Times in Neonates than in Adults: Role of Red Cells, White Cells, Platelets and von Willebrand Factor. Acta Paediatr. 2007, 90, 664–670. [Google Scholar] [CrossRef]
- Schmugge, M.; Dunn, M.S.; Amankwah, K.S.; Blanchette, V.S.; Freedman, J.; Rand, M.L. The Activity of the von Willebrand Factor Cleaving Protease ADAMTS-13 in Newborn Infants: VWF Cleaving Protease ADAMTS-13 in Neonates. J. Thromb. Haemost. 2004, 2, 228–233. [Google Scholar] [CrossRef]
- Punzalan, R.C.; Gottschall, J.L. Use and future investigations of recombinant and plasma-derived coagulation and anticoagulant products in the neonate. Transfus. Med. Rev. 2016, 30, 189–196. [Google Scholar] [CrossRef]
- Acharya, S.S. Rare bleeding disorders in children: Identification and primary care management. Pediatrics 2013, 132, 882–892. [Google Scholar] [CrossRef]
- Bi, L.; Lawler, A.M.; Antonarakis, S.E.; High, K.A.; Gearhart, J.D.; Kazazian, H.H. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 1995, 10, 119–121. [Google Scholar] [CrossRef]
- Bhat, V.; Olmer, M.; Joshi, S.; Durden, D.L.; Cramer, T.J.; Barnes, R.F.; Ball, S.T.; Hughes, T.H.; Silva, M.; Luck, J.V.; et al. Vascular remodeling underlies rebleeding in hemophilic arthropathy. Am. J. Hematol. 2015, 90, 1027–1035. [Google Scholar] [CrossRef]
- Cadé, M.; Muñoz-Garcia, J.; Babuty, A.; Paré, L.; Cochonneau, D.; Fekir, K.; Chatelais, M.; Heymann, M.F.; Lokajczyk, A.; Boisson-Vidal, C.; et al. FVIII regulates the molecular profile of endothelial cells: Functional impact on the blood barrier and macrophage behavior. Cell Mol. Life Sci. 2022, 79, 145. [Google Scholar] [CrossRef]
- Serrano, L.J.; Cañete, A.; Garcia-Leal, T.; Tomás-Gallardo, L.; Flores, A.I.; de la Torre, P.; Liras, A.; Sánchez, M.J. Searching for a Cell-Based Therapeutic Tool for Haemophilia A within the Embryonic/Foetal Liver and the Aorta-Gonads-Mesonephros Region. Thromb. Haemost. 2018, 118, 1370–1381. [Google Scholar] [CrossRef]
- Hayakawa, M.; Sakata, A.; Hayakawa, H.; Matsumoto, H.; Hiramoto, T.; Kashiwakura, Y.; Baatartsogt, N.; Fukushima, N.; Sakata, Y.; Suzuki-Inoue, K.; et al. Characterization and visualization of murine coagulation factor VIII-producing cells in vivo. Sci. Rep. 2021, 11, 14824. [Google Scholar] [CrossRef]
- Bontempo, F.A.; Lewis, J.H.; Gorenc, T.J.; Spero, J.A.; Ragni, M.V.; Scott, J.P.; Starzl, T.E. Liver transplantation in hemophilia A. Blood 1987, 69, 1721–1724. [Google Scholar] [CrossRef]
- Follenzi, A.; Benten, D.; Novikoff, P.; Faulkner, L.; Raut, S.; Gupta, S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J. Clin. Investig. 2008, 118, 935–945. [Google Scholar] [CrossRef][Green Version]
- Shahani, T.; Lavend’homme, R.; Luttun, A.; Saint-Remy, J.M.; Peerlinck, K.; Jacquemin, M. Activation of human endothelial cells from specific vascular beds induces the release of a FVIII storage pool. Blood 2010, 115, 4902–4909. [Google Scholar] [CrossRef]
- Shahani, T.; Covens, K.; Lavend’homme, R.; Jazouli, N.; Sokal, E.; Peerlinck, K.; Jacquemin, M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014, 12, 36–42. [Google Scholar] [CrossRef]
- Everett, L.A.; Cleuren, A.C.; Khoriaty, R.N.; Ginsburg, D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood 2014, 123, 3697–3705. [Google Scholar] [CrossRef] [PubMed]
- Fahs, S.A.; Hille, M.T.; Shi, Q.; Weiler, H.; Montgomery, R.R. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood 2014, 123, 3706–3713. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Dinh, T.T.; Rajaraman, A.; Lee, M.; Scholz, A.; Czupalla, C.J.; Kiefel, H.; Zhu, L.; Xia, L.; Morser, J.; et al. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood 2016, 128, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Zanolini, D.; Merlin, S.; Feola, M.; Ranaldo, G.; Amoruso, A.; Gaidano, G.; Zaffaroni, M.; Ferrero, A.; Brunelleschi, S.; Valente, G.; et al. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica 2015, 100, 881–892. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Wasley, L.C.; Davies, M.V.; Wise, R.J.; Israel, D.I.; Dorner, A.J. Effect of von Willebrand factor coexpression on the synthesis and secretion of factor VIII in Chinese hamster ovary cells. Mol. Cell Biol. 1989, 9, 1233–1242. [Google Scholar] [CrossRef]
- Turner, N.A.; Moake, J.L. Factor VIII Is Synthesized in Human Endothelial Cells, Packaged in Weibel-Palade Bodies and Secreted Bound to ULVWF Strings. PLoS ONE 2015, 10, e0140740. [Google Scholar] [CrossRef]
- Banno, K.; Yoder, M.C. Endothelial Stem and Progenitor Cells for Regenerative Medicine. Curr. Stem Cell Rep. 2019, 5, 101–108. [Google Scholar] [CrossRef]
- Lin, Y.; Chang, L.; Solovey, A.; Healey, J.F.; Lollar, P.; Hebbel, R.P. Use of blood outgrowth endothelial cells for gene therapy for hemophilia A. Blood 2002, 99, 457–462. [Google Scholar] [CrossRef]
- Matsui, H. Endothelial progenitor cell-based therapy for hemophilia A. Int. J. Hematol. 2012, 95, 119–124. [Google Scholar] [CrossRef]
- Gao, K.; Kumar, P.; Cortez-Toledo, E.; Hao, D.; Reynaga, L.; Rose, M.; Wang, C.; Farmer, D.; Nolta, J.; Zhou, J.; et al. Potential long-term treatment of hemophilia A by neonatal co-transplantation of cord blood-derived endothelial colony-forming cells and placental mesenchymal stromal cells. Stem Cell Res. Ther. 2019, 10, 34. [Google Scholar] [CrossRef]
- Campioni, D.; Zauli, G.; Gambetti, S.; Campo, G.; Cuneo, A.; Ferrari, R.; Secchiero, P. In vitro characterization of circulating endothelial progenitor cells isolated from patients with acute coronary syndrome. PLoS ONE 2013, 8, e56377. [Google Scholar] [CrossRef]
- Stem, C.; Rodman, C.; Ramamurthy, R.M.; George, S.; Meares, D.; Farland, A.; Atala, A.; Doering, C.B.; Spencer, H.T.; Porada, C.D.; et al. Investigating Optimal Autologous Cellular Platforms for Prenatal or Perinatal Factor VIII Delivery to Treat Hemophilia A. Front. Cell Dev. Biol. 2021, 9, 678117. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Naito, H.; Suehiro, J.I.; Lin, Y.; Kawaji, H.; Iba, T.; Kouno, T.; Ishikawa-Kato, S.; Furuno, M.; Takara, K.; et al. CD157 Marks Tissue-Resident Endothelial Stem Cells with Homeostatic and Regenerative Properties. Cell Stem Cell 2018, 22, 384–397.e6. [Google Scholar] [CrossRef]
- Bleyer, W.A.; Hakami, N.; Shepard, T.H. The development of hemostasis in the human fetus and newborn infant. J. Pediatr. 1971, 79, 838–853. [Google Scholar] [CrossRef]
- Dube, B.; Dube, R.K.; Bhargava, V.; Kolindewala, J.K.; Kota, V.L.; Das, B.K. Hemostatic parameters in newborn--I. Effect of gestation and rate of intrauterine growth. Thromb. Haemost. 1986, 55, 47–50. [Google Scholar] [CrossRef]
- Terwiel, J.; Veltkamp, J.J.; Bertina, R.M.; Muller, H.P. Coagulation Factors in the Human Fetus of about 20 Weeks of Gestational Age. Br. J. Haematol. 1980, 45, 641–650. [Google Scholar] [CrossRef]
- Terwiel, J.P.; Veltkamp, J.J.; Bertina, R.M.; van der Linden, I.K.; van Tilburg, N.H. Coagulation Factors in the Premature Infant Born after About 32 Weeks of Gestation. Neonatology 1985, 47, 9–18. [Google Scholar] [CrossRef]
- Poralla, C.; Traut, C.; Hertfelder, H.-J.; Oldenburg, J.; Bartmann, P.; Heep, A. The coagulation system of extremely preterm infants: Influence of perinatal risk factors on coagulation. J. Perinatol. 2012, 32, 869–873. [Google Scholar] [CrossRef]
- Hassan, H.J.; Leonardi, A.; Chelucci, C.; Mattia, G.; Macioce, G.; Guerriero, R.; Russo, G.; Mannucci, P.M.; Peschle, C. Blood coagulation factors in human embryonic-fetal development: Preferential expression of the FVII/tissue factor pathway. Blood 1990, 76, 1158–1164. [Google Scholar] [CrossRef]
- Andrew, M.; Paes, B.; Milner, R.; Johnston, M.; Mitchell, L.; Tollefsen, D.M.; Castle, V.; Powers, P. Development of the human coagulation system in the healthy premature infant. Blood 1988, 72, 1651–1657. [Google Scholar] [CrossRef]
- Reverdiau-Moalic, P.; Delahousse, B.; Body, G.; Bardos, P.; Leroy, J. Evolution of Blood Coagulation Activators and Inhibitors in the Healthy Human Fetus. Blood 1996, 88, 900–903. [Google Scholar] [CrossRef]
- Heikinheimo, R. Coagulation Studies with Fetal Blood. Neonatology 1964, 7, 319–327. [Google Scholar] [CrossRef]
- His, B.L.; Faulk, W.P.; Yeh, C.J.; McIntyre, J.A. Immunohistology of clotting factor V in human extraembryonic membranes. Placenta 1987, 8, 529–535. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Shapiro, S.S.; De Marco, L. Monoclonal immunoglobulin M lambda coagulation inhibitor with phospholipid specificity. Mechanism of a lupus anticoagulant. J. Clin. Investig. 1980, 66, 397–405. [Google Scholar] [CrossRef]
- Cui, J.; O’Shea, K.; Purkayastha, A.; Saunders, T.; Ginsburg, D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature 1996, 384, 66–68. [Google Scholar] [CrossRef]
- Yang, T.; Cui, J.; Taylor, J.; Yang, A.; Gruber, S.; Ginsburg, D. Rescue of Fatal Neonatal Hemorrhage in Factor V Deficient Mice by Low Level Transgene Expression. Thromb. Haemost. 2000, 83, 70–77. [Google Scholar] [CrossRef]
- Maroney, S.A.; Westrick, R.J.; Cleuren, A.C.; Martinez, N.D.; Siebert, A.E.; Zogg, M.; Ginsburg, D.; Weiler, H.; Mast, A.E. Tissue factor pathway inhibitor is required for cerebrovascular development in mice. Blood 2021, 137, 258–268. [Google Scholar] [CrossRef]
- Weyand, A.C.; Grzegorski, S.J.; Rost, M.S.; Lavik, K.I.; Ferguson, A.C.; Menegatti, M.; Richter, C.E.; Asselta, R.; Duga, S.; Peyvandi, F.; et al. Analysis of factor V in zebrafish demonstrates minimal levels needed for early hemostasis. Blood Adv. 2019, 3, 1670–1680. [Google Scholar] [CrossRef]
- Connolly, A.J.; Ishihara, H.; Kahn, M.L.; Farese, R.V., Jr.; Coughlin, S.R. Role of the thrombin receptor in development and evidence for a second receptor. Nature 1996, 381, 516–519. [Google Scholar] [CrossRef]
- Xue, J.; Wu, Q.; Westfield, L.A.; Tuley, E.A.; Lu, D.; Zhang, Q.; Shim, K.; Zheng, X.; Sadler, J.E. Incomplete embryonic lethality and fatal neonatal hemorrhage caused by prothrombin deficiency in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7603–7607. [Google Scholar] [CrossRef]
- Griffin, C.T. A Role for Thrombin Receptor Signaling in Endothelial Cells during Embryonic Development. Science 2001, 293, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.; Horsfall, W.; Conway, E.; Schuh, A. Early Embryonic Expression of Murine Coagulation System Components. Thromb. Haemost. 2000, 84, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.P.M.; Gonçalves, D.A.; de Sousa, R.X.; Mouro, M.G.; Higa, E.M.S.; Sperandio, L.P.; Vitoriano, C.M.; Rosa, E.B.S.; Santos, F.O.D.; de Queiroz, G.N.; et al. Disruption of Redox Homeostasis by Alterations in Nitric Oxide Synthase Activity and Tetrahydrobiopterin along with Melanoma Progression. Int. J. Mol. Sci. 2022, 23, 5979. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Han, L.; Li, J.; Chen, C. Acquired coagulation dysfunction resulting from vitamin K-dependent coagulation factor deficiency associated with rheumatoid arthritis: A case report. World J. Clin. Cases 2022, 10, 236–241. [Google Scholar] [CrossRef]
- Van Cott, E.M.; Khor, B.; Zehnder, J.L. Factor V Leiden. Am. J. Hematol. 2016, 91, 46–49. [Google Scholar] [CrossRef]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef]
- Tambuyzer, E.; Vandendriessche, B.; Austin, C.P.; Brooks, P.J.; Larsson, K.; Miller Needleman, K.I.; Valentine, J.; Davies, K.; Groft, S.C.; Preti, R.; et al. Therapies for rare diseases: Therapeutic modalities, progress and challenges ahead. Nat. Rev. Drug Discov. 2020, 19, 93–111. [Google Scholar] [CrossRef]
- Pogue, R.E.; Cavalcanti, D.P.; Shanker, S.; Andrade, R.V.; Aguiar, L.R.; de Carvalho, J.L.; Costa, F.F. Rare genetic diseases: Update on diagnosis, treatment and online resources. Drug Discov. Today 2018, 23, 187–195. [Google Scholar] [CrossRef]
- Rick, M.E.; Walsh, C.E.; Key, N.S. Congenital Bleeding Disorders. Hematol. Am. Soc. Hematol. Educ. Programm. 2003, 2003, 559–574. [Google Scholar] [CrossRef][Green Version]
- Brown, D.L. Congenital bleeding disorders. Curr. Probl. Pediatr. Adolesc. Health Care 2005, 35, 38–62. [Google Scholar] [CrossRef]
- Bhat, R.; Cabey, W. Evaluation and Management of Congenital Bleeding Disorders. Hematol. Oncol. Clin. N. Am. 2017, 31, 1105–1122. [Google Scholar] [CrossRef]
- Batsuli, G.; Kouides, P. Rare Coagulation Factor Deficiencies (Factors VII, X, V, and II). Hematol. Oncol. Clin. N. Am. 2021, 35, 1181–1196. [Google Scholar] [CrossRef]
- Nummi, V.; Lassila, R.; Joutsi-Korhonen, L.; Armstrong, E.; Szanto, T. Comprehensive Re-Evaluation of Historical von Willebrand Disease Diagnosis in Association with Whole Blood Platelet Aggregation and Function. Int. J. Lab. Hem. 2018, 40, 304–311. [Google Scholar] [CrossRef]
- Millar, C. Why and How Do We Classify von Willebrand Disease? Haemophilia 2015, 21, 407–410. [Google Scholar] [CrossRef]
- Flood, V.H.; Garcia, J.; Haberichter, S.L. The Role of Genetics in the Pathogenesis and Diagnosis of Type 1 Von Willebrand Disease. Curr. Opin. Hematol. 2019, 26, 331–335. [Google Scholar] [CrossRef]
- Woods, A.I.; Paiva, J.; Primrose, D.M.; Blanco, A.N.; Sánchez-Luceros, A. Type 2A and 2M von Willebrand Disease: Differences in Phenotypic Parameters According to the Affected Domain by Disease-Causing Variants and Assessment of Pathophysiological Mechanisms. Semin. Thromb. Hemost. 2021, 47, 862–874. [Google Scholar] [CrossRef]
- Kranzhöfer, D.; Pavlova, A.; Schneider, H.; Franck, P.; Glonnegger, H.; Büchsel, M.; Yoshimi-Nöllke, A.; Oldenburg, J.; Zieger, B. Type 2B von Willebrand Disease: Early Manifestation as Neonatal Thrombocytopenia. Hamostaseologie 2021, 41, 469–474. [Google Scholar] [CrossRef]
- Seidizadeh, O.; Peyvandi, F.; Mannucci, P.M. Von Willebrand Disease Type 2N: An Update. J. Thromb. Haemost. 2021, 19, 909–916. [Google Scholar] [CrossRef]
- Leebeek, F.W.G.; Atiq, F. How I Manage Severe von Willebrand Disease. Br. J. Haematol. 2019, 187, 418–430. [Google Scholar] [CrossRef]
- The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff (HGMD). Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 29 June 2022).
- James, P.D.; Notley, C.; Hegadorn, C.; Leggo, J.; Tuttle, A.; Tinlin, S.; Brown, C.; Andrews, C.; Labelle, A.; Chirinian, Y.; et al. The Mutational Spectrum of Type 1 von Willebrand Disease: Results from a Canadian Cohort Study. Blood 2007, 109, 145–154. [Google Scholar] [CrossRef]
- De Jong, A.; Eikenboom, J. Von Willebrand Disease Mutation Spectrum and Associated Mutation Mechanisms. Thromb. Res. 2017, 159, 65–75. [Google Scholar] [CrossRef]
- Chornenki, N.L.J.; Shanjer, M.; James, P.D. Vascular Abnormalities in Patients with von Willebrand Disease: A Scoping Review. J. Thromb. Haemost. 2021, 19, 2151–2160. [Google Scholar] [CrossRef]
- Sanders, Y.V.; Fijnvandraat, K.; Boender, J.; Mauser-Bunschoten, E.P.; van der Bom, J.G.; de Meris, J.; Smiers, F.J.; Granzen, B.; Brons, P.; Tamminga, R.Y.J.; et al. Bleeding Spectrum in Children with Moderate or Severe von Willebrand Disease: Relevance of Pediatric-Specific Bleeding: Bleeding Phenotype of Pediatric VWD Patients. Am. J. Hematol. 2015, 90, 1142–1148. [Google Scholar] [CrossRef]
- Kadir, R.A.; Edlund, M.; Von Mackensen, S. The Impact of Menstrual Disorders on Quality of Life in Women with Inherited Bleeding Disorders: Quatily of life in menorrhagia. Haemophilia 2010, 16, 832–839. [Google Scholar] [CrossRef]
- De Wee, E.M.; Mauser-Bunschoten, E.P.; Van Der Bom, J.G.; Degenaar-Dujardin, M.E.L.; Eikenboom, H.C.J.; Fijnvandraat, K.; De Goede-Bolder, A.; Laros-Van Gorkom, B.A.P.; Meijer, K.; Raat, H.; et al. Health-Related Quality of Life among Adult Patients with Moderate and Severe von Willebrand Disease: Quality of Life with von Willebrand Disease. J. Thromb. Haemost. 2010, 8, 1492–1499. [Google Scholar] [CrossRef]
- James, P.D.; Connell, N.T.; Ameer, B.; Di Paola, J.; Eikenboom, J.; Giraud, N.; Haberichter, S.; Jacobs-Pratt, V.; Konkle, B.; McLintock, C.; et al. ASH ISTH NHF WFH 2021 Guidelines on the Diagnosis of von Willebrand Disease. Blood Adv. 2021, 5, 280–300. [Google Scholar] [CrossRef]
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and Clinical Similarities and Differences. Haematologica 2019, 104, 1702–1709. [Google Scholar] [CrossRef]
- Tomeo, F.; Mariz, S.; Brunetta, A.L.; Stoyanova-Beninska, V.; Penttila, K.; Magrelli, A. Haemophilia, State of the Art and New Therapeutic Opportunities, a Regulatory Perspective. Br. J. Clin. Pharmacol. 2021, 87, 4183–4196. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.; Pasi, K.J. Haemophilias A and B. Lancet 2003, 361, 1801–1809. [Google Scholar] [CrossRef]
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Primers 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wang, H.L.; Chang, L.J. Molecular therapeutics of hemophilia A and B. Expert Rev. Hematol. 2022, 15, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Marlar, R.A.; Strandberg, K.; Shima, M.; Adcock, D.M. Clinical utility and impact of the use of the chromogenic vs. one-stage factor activity assays in haemophilia A and B. Eur. J. Haematol. 2020, 104, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sheen, C.R.; McDonald, M.A.; George, P.M.; Smith, M.P.; Morris, C.M. Genotyping the factor VIII intron 22 inversion locus using fluorescent in situ hybridization. Blood Cells Mol. Dis. 2011, 46, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Swystun, L.L.; James, P.D. Genetic diagnosis in hemophilia and von Willebrand disease. Blood Rev. 2017, 31, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, S.; Shiravand, Y.; Shams, M.; Safa, M.; Gholami, M.S.; Heydari, F.; Ahmadi, A.; Rashidpanah, J.; Dorgalaleh, A. A Comprehensive Overview of Coagulation Factor V and Congenital Factor V Deficiency. Semin. Thromb. Hemost. 2019, 45, 523–543. [Google Scholar] [CrossRef]
- Tabibian, S.; Dorgalaleh, A.; Camire, R.M. Congenital Factor V Deficiency. In Congenital Bleeding Disorders; Dorgalaleh, A., Ed.; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Cripe, L.D.; Moore, K.D.; Kane, W.H. Structure of the gene for human coagulation factor V. Biochemistry 1992, 31, 3777–3785. [Google Scholar] [CrossRef]
- Bos, M.H.A.; Camire, R.M. Blood coagulation factors V and VIII: Molecular Mechanisms of Procofactor Activation. J. Coagul. Disord. 2010, 2, 19–27. [Google Scholar]
- Girolami, A.; Cosi, E.; Ferrari, S.; Lombardi, A.M.; Girolami, B. New clotting disorders that cast new light on blood coagulation and may play a role in clinical practice. J. Thromb. Thrombolysis 2017, 44, 71–75. [Google Scholar] [CrossRef]
- Naderi, M.; Tabibian, S.; Alizadeh, S.; Hosseini, S.; Zaker, F.; Bamedi, T.; Shamsizadeh, M.; Dorgalaleh, A. Congenital Factor V Deficiency: Comparison of the Severity of Clinical Presentations among Patients with Rare Bleeding Disorders. Acta Haematol. 2015, 133, 148–154. [Google Scholar] [CrossRef]
- Jain, S.; Acharya, S.S. Management of rare coagulation disorders in 2018. Transfus. Apher. Sci. 2018, 57, 705–712. [Google Scholar] [CrossRef]
- Zhang, B. Recent developments in the understanding of the combined deficiency of FV and FVIII. Br. J. Haematol. 2009, 145, 15–23. [Google Scholar] [CrossRef]
- Zheng, C.; Zhang, B. Combined Deficiency of Coagulation Factors V and VIII: An Update. Semin. Thromb. Hemost. 2013, 39, 613–620. [Google Scholar] [CrossRef]
- Spreafico, M.; Peyvandi, F. Combined Factor V and Factor VIII Deficiency. Semin. Thromb. Hemost. 2009, 35, 390–399. [Google Scholar] [CrossRef]
- Zhang, B.; Zheng, C.; Zhu, M.; Tao, J.; Vasievich, M.P.; Baines, A.; Kim, J.; Schekman, R.; Kaufman, R.J.; Ginsburg, D. Mice Deficient in LMAN1 Exhibit FV and FVIII Deficiencies and Liver Accumulation of A1-Antitrypsin. Blood 2011, 118, 3384–3391. [Google Scholar] [CrossRef]
- Majid, Z.; Tahir, F.; Ahmed, J.; Bin Arif, T.; Haq, A. Protein C Deficiency as a Risk Factor for Stroke in Young Adults: A Review. Cureus 2020, 12, e7472. [Google Scholar] [CrossRef]
- Gupta, A.; Tun, A.M.; Gupta, K.; Tuma, F. Protein S Deficiency; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Akwaa, F.; Cataland, S.R.; Antun, A.G. How I Treat Immune Mediated Thrombotic Thrombocytopenic Purpura After Hospital Discharge. Blood 2022. online ahead of print. [Google Scholar] [CrossRef]
- Jenkins, P.V.; Rawley, O.; Smith, O.P.; O’Donnell, J.S. Elevated factor VIII levels and risk of venous thrombosis. Br. J. Haematol. 2012, 157, 653–663. [Google Scholar] [CrossRef]
- Applegate, J.S.; Gronefeld, D. Factor V Leiden. Radiol. Technol. 2019, 90, 259–273. [Google Scholar]
- Castaman, G.; Lunghi, B.; Missiaglia, E.; Bernardi, F.; Rodeghiero, F. Phenotypic homozygous activated protein C resistance associated with compound heterozygosity for Arg506Gln (factor V Leiden) and His1299Arg substitutions in factor V. Br. J. Haematol. 1997, 99, 257–261. [Google Scholar] [CrossRef]
- Eppenberger, D.; Nilius, H.; Anagnostelis, B.; Huber, C.A.; Nagler, M. Current Knowledge on Factor V Leiden Mutation as a Risk Factor for Recurrent Venous Thromboembolism: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 883986. [Google Scholar] [CrossRef]
- Dizon-Townson, D.; Miller, C.; Sibai, B.; Spong, C.Y.; Thom, E.; Wendel, G., Jr.; Wenstrom, K.; Samuels, P.; Cotroneo, M.A.; Moawad, A.; et al. The Relationship of the Factor V Leiden Mutation and Pregnancy Outcomes for Mother and Fetus. Obstet. Gynecol. 2005, 106, 517–524. [Google Scholar] [CrossRef]
- Moser, M.; Patterson, C. Thrombin and vascular development: A sticky subject. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 922–930. [Google Scholar] [CrossRef]
- Mackman, N.; Davis, G.E. Blood coagulation and blood vessel development: Is tissue factor the missing link? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2364–2366. [Google Scholar] [CrossRef]
- Randi, A.M. Angiogenesis and the ADAMTS13-VWF Balance. Blood 2017, 130, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.D.; Ferraro, F.; Paschalaki, K.E.; Dryden, N.H.; McKinnon, T.A.J.; Sutton, R.E.; Payne, E.M.; Haskard, D.O.; Hughes, A.D.; Cutler, D.F.; et al. Endothelial von Willebrand Factor Regulates Angiogenesis. Blood 2011, 117, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Galanis, E. Targeting Angiogenesis: Progress with Anti-VEGF Treatment with Large Molecules. Nat. Rev. Clin. Oncol. 2009, 6, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Cooke, E.J.; Wyseure, T.; Zhou, J.Y.; Gopal, S.; Nasamran, C.A.; Fisch, K.M.; Manon-Jensen, T.; Karsdal, M.A.; Mosnier, L.O.; von Drygalski, A. Mechanisms of vascular permeability and remodeling associated with hemarthrosis in factor VIII-deficient mice. J. Thromb. Haemost. 2019, 17, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.S.; Kaplan, R.N.; Macdonald, D.; Fabiyi, O.T.; DiMichele, D.; Lyden, D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood 2011, 117, 2484–2493. [Google Scholar] [CrossRef]
- Samuelson Bannow, B.; Recht, M.; Négrier, C.; Hermans, C.; Berntorp, E.; Eichler, H.; Mancuso, M.E.; Klamroth, R.; O’Hara, J.; Santagostino, E.; et al. Factor VIII: Long-established role in haemophilia A and emerging evidence beyond haemostasis. Blood Rev. 2019, 35, 43–50. [Google Scholar] [CrossRef]
- López-Sagaseta, J.; Montes, R.; Puy, C.; Díez, N.; Fukudome, K.; Hermida, J. Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. J. Thromb. Haemost. 2007, 5, 1817–1824. [Google Scholar] [CrossRef]
- Magisetty, J.; Pendurthi, U.R.; Esmon, C.T.; Rao, L.V.M. EPCR deficiency or function-blocking antibody protects against joint bleeding-induced pathology in hemophilia mice. Blood 2020, 135, 2211–2223. [Google Scholar] [CrossRef]
- Sen, P.; Gopalakrishnan, R.; Kothari, H.; Keshava, S.; Clark, C.A.; Esmon, C.T.; Pendurthi, U.R.; Rao, L.V. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood 2011, 117, 3199–3208. [Google Scholar] [CrossRef]
- Haralabopoulos, G.C.; Grant, D.S.; Kleinman, H.K.; Maragoudakis, M.E. Thrombin promotes endothelial cell alignment in Matrigel in vitro and angiogenesis in vivo. Am. J. Physiol. 1997, 273, C239–C245. [Google Scholar] [CrossRef]
- Yang, Y.; Xiao, L.; Chen, N.; Li, Y.; Deng, X.; Wang, L.; Sun, H.; Wu, J. Platelet-derived factor V promotes angiogenesis in a mouse hind limb ischemia model. J. Vasc. Surg. 2017, 65, 1180–1188. [Google Scholar] [CrossRef]
- Trigg, D.E.; Stergiotou, I.; Peitsidis, P.; Kadir, R.A. A Systematic Review: The Use of Desmopressin for Treatment and Prophylaxis of Bleeding Disorders in Pregnancy: The use of desmopressin in pregnancy. Haemophilia 2012, 18, 25–33. [Google Scholar] [CrossRef]
- Connell, N.T.; Flood, V.H.; Brignardello-Petersen, R.; Abdul-Kadir, R.; Arapshian, A.; Couper, S.; Grow, J.M.; Kouides, P.; Laffan, M.; Lavin, M.; et al. ASH ISTH NHF WFH 2021 Guidelines on the Management of von Willebrand Disease. Blood Adv. 2021, 5, 301–325. [Google Scholar] [CrossRef]
- Castaman, G. How I Treat von Willebrand Disease. Thromb. Res. 2020, 196, 618–625. [Google Scholar] [CrossRef]
- Sreeraman, S.; McKinlay, S.; Li, A.; Crowther, M.; Motalo, O. Efficacy of Parenteral Formulations of Desmopressin in the Treatment of Bleeding Disorders: A Systematic Review. Thromb. Res. 2022, 213, 16–26. [Google Scholar] [CrossRef]
- Zubairov, D.M.; Andrushko, I.A.; Zubairova, L.D.; Svintenok, G.Y. Study of the Mechanism of Hemostatic Effect of Desmopressin. Bull. Exp. Biol. Med. 2007, 144, 200–202. [Google Scholar] [CrossRef]
- White, B.; Lawler, P.; Riddell, A.; Nitu-Whalley, I.C.; Hermans, C.; Lee, C.A.; Brown, S.A. Response to Desmopressin of Factors XI, X and V in Patients with Factor VIII Deficiency and von Willebrand Disease: Effect of DDAVP on FXI, FX, FV in Haemophilia A and VWD. Br. J. Haematol. 2004, 126, 100–104. [Google Scholar] [CrossRef]
- Carcao, M.; Gouider, E.; Wu, R. Low Dose Prophylaxis and Antifibrinolytics: Options to Consider with Proven Benefits for Persons with Haemophilia. Haemophilia 2022, 28, 26–34. [Google Scholar] [CrossRef]
- Graf, L.; Yan, S.; Shen, M.C.; Balasa, V. A Systematic Review Evaluating the Efficacy and Factor Consumption of Long-Acting Recombinant Factor VIII Products for the Prophylactic Treatment of Hemophilia A. J. Med. Econ. 2020, 23, 1493–1498. [Google Scholar] [CrossRef]
- Ozelo, M.C.; Yamaguti-Hayakawa, G.G. Impact of novel hemophilia therapies around the world. Res. Pract. Thromb. Haemost. 2022, 6, e12695. [Google Scholar] [CrossRef]
- Lewandowska, M.; Nasr, S.; Shapiro, A.D. Therapeutic and technological advancements in haemophilia care: Quantum leaps forward. Haemophilia 2022, 28, 77–92. [Google Scholar] [CrossRef]
- Castro, H.E.; Briceño, M.F.; Casas, C.P.; Rueda, J.D. The History and Evolution of the Clinical Effectiveness of Haemophilia Type A Treatment: A Systematic Review. Indian J. Hematol. Blood Transfus. 2014, 30, 1–11. [Google Scholar] [CrossRef]
- Miesbach, W.; Eladly, F. Current and Future Options of Haemophilia A Treatments. Expert Opin. Biol. Ther. 2021, 21, 1395–1402. [Google Scholar] [CrossRef]
- Miesbach, W.; Schwäble, J.; Müller, M.M.; Seifried, E. Treatment Options in Hemophilia. Dtsch. Arztebl. Int. 2019, 116, 791–798. [Google Scholar] [CrossRef]
- Tiede, A. Half-life extended factor VIII for the treatment of hemophilia A. J. Thromb. Haemost. 2015, 13, S176–S179. [Google Scholar] [CrossRef]
- Kruse-Jarres, R.; Kempton, C.L.; Baudo, F.; Collins, P.W.; Knoebl, P.; Leissinger, C.A.; Tiede, A.; Kessler, C.M. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am. J. Hematol. 2017, 92, 695–705. [Google Scholar] [CrossRef]
- Bryant, P.; Boukouvala, A.; McDaniel, J.; Nance, D. Hemophilia A in Females: Considerations for Clinical Management. Acta Haematol. 2020, 143, 289–294. [Google Scholar] [CrossRef]
- Mahlangu, J.; Iorio, A.; Kenet, G. Emicizumab state-of-the-art update. Haemophilia 2022, 28, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, M.U.; Negrier, C.; Paz-Priel, I.; Chang, T.; Chebon, S.; Lehle, M.; Mahlangu, J.; Young, G.; Kruse-Jarres, R.; Mancuso, M.E.; et al. Long-term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1–4 studies. Blood 2021, 137, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Merchán, E.C.; De Pablo-Moreno, J.A.; Liras, A. Gene Therapy in Hemophilia: Recent Advances. Int. J. Mol. Sci. 2021, 22, 7647. [Google Scholar] [CrossRef] [PubMed]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Bolt, M.W.; Brady, J.T.; Whiteley, L.O.; Khan, K.N. Development challenges associated with rAAV-based gene therapies. J. Toxicol. Sci. 2021, 46, 57–68. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Single-Arm Study to Evaluate the Efficacy and Safety of Valoctocogene Roxaparvovec in Hemophilia a Patients at a Dose of 4E13 vg/kg (BMN270-302). ClinicalTrials.gov Identifier: NCT03392974. Available online: https://clinicaltrials.gov/ct2/show/NCT03392974?term=NCT03392974&draw=2&rank=1 (accessed on 29 June 2022).
- U.S. National Library of Medicine. Gene Therapy Study in Severe Haemophilia a Patients (270-201). ClinicalTrials.gov Identifier: NCT02576795. Available online: https://clinicaltrials.gov/ct2/show/NCT02576795?term=NCT02576795&draw=2&rank=1 (accessed on 29 June 2022).
- U.S. National Library of Medicine. Study to Evaluate the Efficacy and Safety of PF-07055480/Giroctocogene Fitelparvovec Gene Therapy in Moderately Severe to Severe Hemophilia A Adults (AFFINE). ClinicalTrials.gov Identifier: NCT04370054. Available online: https://clinicaltrials.gov/ct2/show/NCT04370054 (accessed on 29 June 2022).
- U.S. National Library of Medicine. Gene Therapy for Haemophilia A. (GO-8). ClinicalTrials.gov Identifier: NCT03001830. Available online: https://clinicaltrials.gov/ct2/show/NCT03001830 (accessed on 29 June 2022).
- U.S. National Library of Medicine. ASC618 Gene Therapy in Hemophilia A Patients. ClinicalTrials.gov Identifier: NCT04676048. Available online: https://clinicaltrials.gov/ct2/show/NCT04676048 (accessed on 29 June 2022).
- Menegatti, M.; Peyvandi, F. Treatment of rare factor deficiencies other than hemophilia. Blood 2019, 133, 415–424. [Google Scholar] [CrossRef]
- Heger, A.; Svae, T.E.; Neisser-Svae, A.; Jordan, S.; Behizad, M.; Römisch, J. Biochemical quality of the pharmaceutically licensed plasma OctaplasLG® after implementation of a novel prion protein (PrP Sc) removal technology and reduction of the solvent/detergent (S/D) process time. Vox Sang. 2009, 97, 219–225. [Google Scholar] [CrossRef]
- Peyvandi, F.; Menegatti, M. Treatment of rare factor deficiencies in 2016. Hematology 2016, 2016, 663–669. [Google Scholar] [CrossRef]
- Von Drygalski, A.; Bhat, V.; Gale, A.J.; Burnier, L.; Cramer, T.J.; Griffin, J.H.; Mosnier, L.O. An engineered factor Va prevents bleeding induced by anticoagulant wt activated protein C. PLoS ONE 2014, 9, e104304. [Google Scholar] [CrossRef]
- Gale, A.J.; Bhat, V.; Pellequer, J.L.; Griffin, J.H.; Mosnier, L.O.; Von Drygalski, A. Safety, Stability and Pharmacokinetic Properties of (super)Factor Va, a Novel Engineered Coagulation Factor V for Treatment of Severe Bleeding. Pharm. Res. 2016, 33, 1517–1526. [Google Scholar] [CrossRef]
- Joseph, B.C.; Miyazawa, B.Y.; Esmon, C.T.; Cohen, M.J.; von Drygalski, A.; Mosnier, L.O. An engineered activated factor V for the prevention and treatment of acute traumatic coagulopathy and bleeding in mice. Blood Adv. 2022, 6, 959–969. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. A Trial Evaluating Efficacy and Safety of Prophylactic Administration of Concizumab in Patients with Severe Haemophilia A Without Inhibitors (explorer™5). ClinicalTrials.gov Identifier: NCT03196297. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03196297?term=Concizumab&draw=2&rank=5 (accessed on 29 June 2022).
- U.S. National Library of Medicine. A Study of Marstacimab to Compare Prefilled Pen (PFP) Device to Prefilled Syringe (PFS) Device. ClinicalTrials.gov Identifier: NCT04832139. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04832139?term=Marstacimab&draw=2&rank=1 (accessed on 29 June 2022).
- Rezaie, A.R.; Giri, H. Anticoagulant and signaling functions of antithrombin. J. Thromb. Haemost. 2020, 18, 3142–3153. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Long-term Safety and Efficacy Study of Fitusiran in Patients with Hemophilia A or B, With or Without Inhibitory Antibodies to Factor VIII or IX (ATLAS-OLE). ClinicalTrials.gov Identifier: NCT03754790. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03754790?term=Fitusiran&draw=2&rank=3 (accessed on 29 June 2022).
- Polderdijk, S.G.I.; Huntington, J.A. Identification of serpins specific for activated protein C using a lysate-based screening assay. Sci. Rep. 2018, 8, 8793. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. The Safety and Tolerability of SerpinPC in Healthy Men and in Men With Severe Blood Disorders (Haemophilia A and B). ClinicalTrials.gov Identifier: NCT04073498. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04073498?term=SerpinPC&draw=2&rank=1 (accessed on 29 June 2022).
- Valanejad, S.M.; Davis, K.A. Direct Oral Anticoagulants in Select Patients with Hypercoagulable Disorders. Ann. Pharmacother. 2021, 55, 891–901. [Google Scholar] [CrossRef]
- Morrow, M.; Lynch-Smith, D. Factor V Leiden: Development of VTE in Surgery and Trauma Patients: A Systematic Review. Dimens. Crit. Care Nurs. 2022, 41, 190–199. [Google Scholar] [CrossRef]
- Khiralla, S.; Meadows, C.A. The Effect of Switching from Warfarin to Novel Oral Anticoagulants on Patients’ Satisfaction and the Travel Burden in a Rural Setting. Cureus 2022, 14, e24608. [Google Scholar] [CrossRef]
- Sukumar, S.; Lämmle, B.; Cataland, S.R. Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. J. Clin. Med. 2021, 10, 536. [Google Scholar] [CrossRef]
- Pascreau, T.; Zia-Chahabi, S.; Zuber, B.; Tcherakian, C.; Farfour, E.; Vasse, M. ADAMTS 13 deficiency is associated with abnormal distribution of von Willebrand factor multimers in patients with COVID-19. Thromb. Res. 2021, 204, 138–140. [Google Scholar] [CrossRef]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef]
- EU Clinical Trials Register. Clinical Trials for Recombinant ADAMTS. EudraCT Number: 2020-003348-10. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=recombinant+ADAMTS (accessed on 29 June 2022).
- Palandri, F.; Di Pietro, C.; Ricci, F.; Tazzari, P.L.; Randi, V.; Bartoletti, D.; Cavo, M.; Vianelli, N.; Auteri, G. Immune thrombotic thrombocytopenic purpura: Personalized therapy using ADAMTS-13 activity and autoantibodies. Res. Pract. Thromb. Haemost. 2021, 5, e12606. [Google Scholar] [CrossRef]
- Ikezoe, T. Advances in the diagnosis and treatment of disseminated intravascular coagulation in haematological malignancies. Int. J. Hematol. 2021, 113, 34–44. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Pathological Condition |
|---|---|
| Desmopressin Recombinant factors Tranexamic acid Aminocaproic acid | von Willebrand’s disease |
| Recombinant factors (half-life coagulation factors) Emicizumab Tranexamic acid Aminocaproic acid Desmopressin Valoctocogene roxaparvovec/giroctocogene fitelparovec (adeno-associated vectors) a | Hemophilia A |
| SUPERFVa b Octaplas® Fresh frozen plasma Tranexamic acid Aminocaproic acid Estrogen Progesterone | FV deficiency |
| Concizumab (anti-TFPI) a Marstacimab (anti-TFPI) a Fitusiran (anti-AT-III) a | Hemophilia A or B Other clotting disorders |
| SerpinPC (APC inhibitor) a | Hemophilia A and FV deficiency Other clotting disorders |
| DOACs Warfarin | Factor V Leiden thrombophilia |
| Heparin Warfarin Aspirin Clopidogrel | Protein C deficiency |
| Heparin Vitamin K antagonists Warfarin | Protein S deficiency |
| Fresh frozen plasma Prednisone Rituximab rADAMTS13 a Caplacizumab (anti-vWF) | Thrombotic thrombocytopenic purpura |
| Trans retinoic acid Tranexamic acid Recombinant human thrombomodulin c | Disseminated intravascular coagulation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Pablo-Moreno, J.A.; Serrano, L.J.; Revuelta, L.; Sánchez, M.J.; Liras, A. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. Int. J. Mol. Sci. 2022, 23, 8283. https://doi.org/10.3390/ijms23158283
De Pablo-Moreno JA, Serrano LJ, Revuelta L, Sánchez MJ, Liras A. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. International Journal of Molecular Sciences. 2022; 23(15):8283. https://doi.org/10.3390/ijms23158283
Chicago/Turabian StyleDe Pablo-Moreno, Juan A., Luis Javier Serrano, Luis Revuelta, María José Sánchez, and Antonio Liras. 2022. "The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V" International Journal of Molecular Sciences 23, no. 15: 8283. https://doi.org/10.3390/ijms23158283
APA StyleDe Pablo-Moreno, J. A., Serrano, L. J., Revuelta, L., Sánchez, M. J., & Liras, A. (2022). The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. International Journal of Molecular Sciences, 23(15), 8283. https://doi.org/10.3390/ijms23158283