
Structure–Activity Relationship of New Chimeric Analogs of Mastoparan from the Wasp Venom Paravespula lewisii

 , , and
, , and
Abstract
:1. Introduction
2. Results
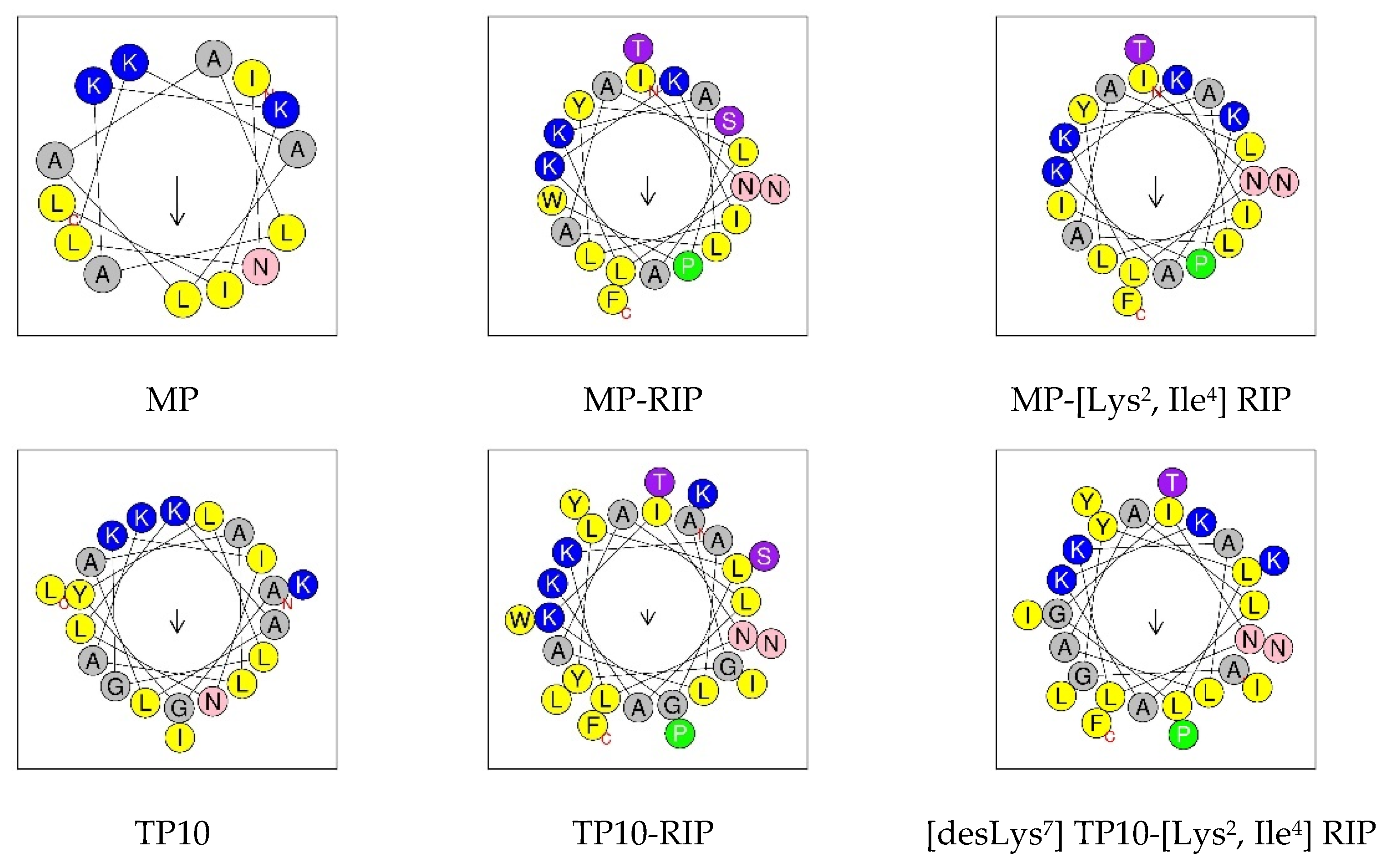
2.1. Design and Synthesis of Peptides
2.2. Antimicrobial Activity Study
2.3. Circular Dichroism Study
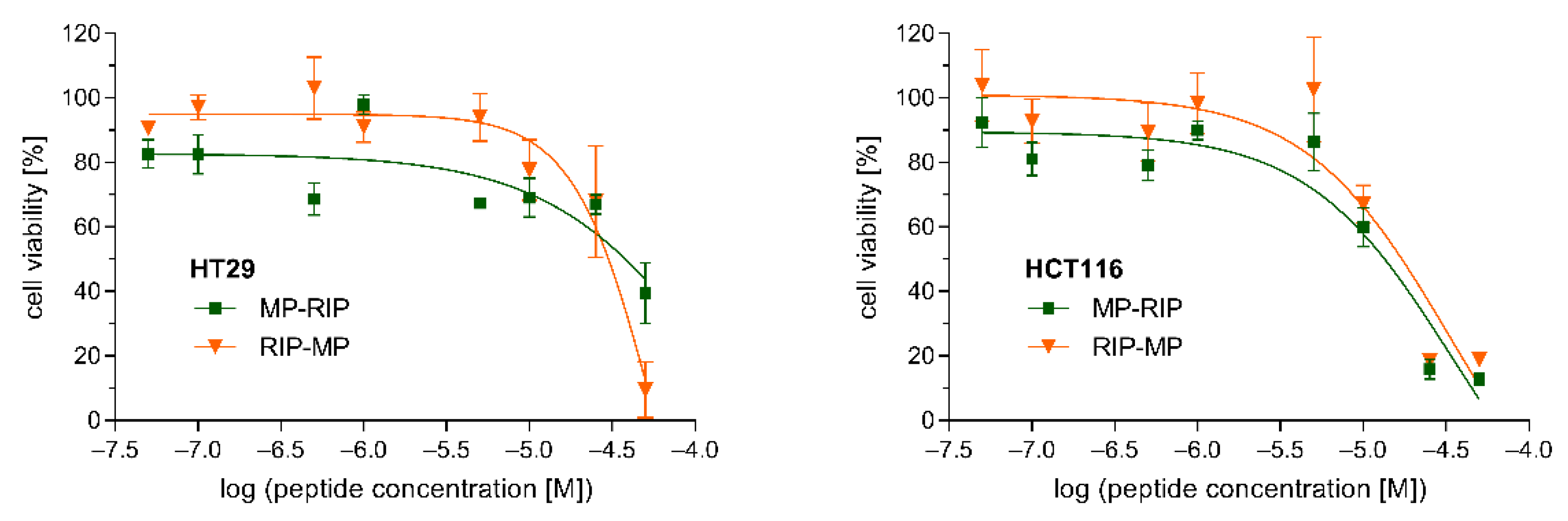
2.4. Cytotoxicity Study
3. Discussion
4. Materials and Methods
4.1. Reagents
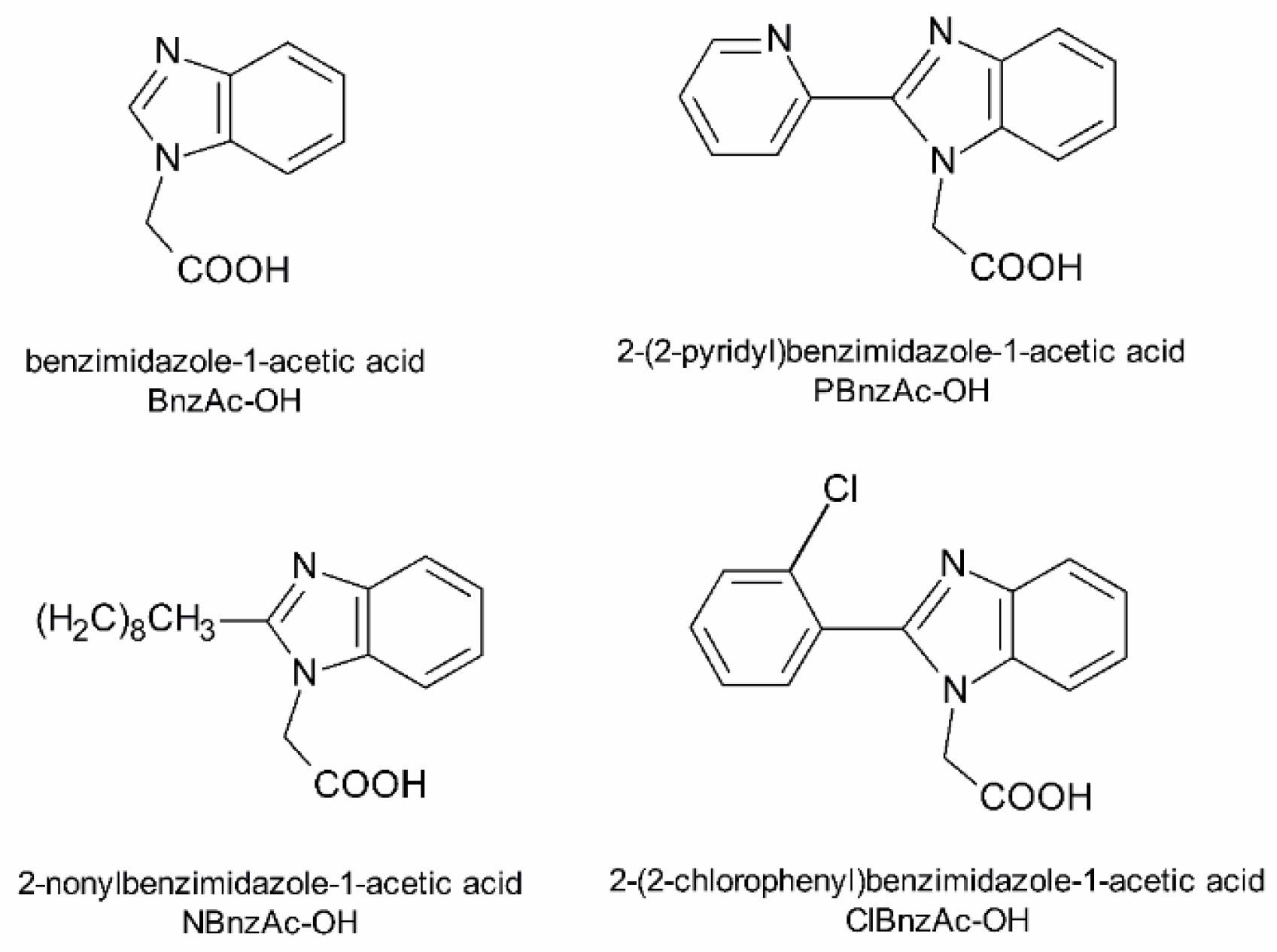
4.2. Preparation of Benzimidazole Derivatives
4.3. Peptide Synthesis and Purification
4.4. Antimicrobial Assay
4.5. Circular Dichroism Spectroscopy
4.6. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPs | antimicrobial peptides |
| BnzAc | benzimidazole-1-acetyl |
| CD | circular dichroism |
| ClBnzAc | 2-(2-chlorophenyl) benzimidazole-1-acetyl |
| CPP | cell-penetrating peptide |
| FBS | fetal bovine serum |
| fH | relative α-helix fraction |
| Gal | galanin |
| Galp | galparan |
| H | mean hydrophobicity |
| HPLC | high-performance liquid chromatography |
| LD50 | peptide concentration sufficient to kill 50% of the peptide-treated cells |
| μH | mean hydrophobic moment |
| MIC | minimal inhibitory concentration |
| MitP | mitoparan |
| MP | mastoparan |
| MTT | 3-(4, 5-dimethylthazol-2-yl)-2, 5-diphenyl tetrazolium bromide |
| NBnzAc | 2-nonylbenzimidazole-1-acetyl |
| PBnzAc | 2-(2-pyridyl) benzimidazole-1-acetyl |
| RIP | RNA III inhibiting peptide |
| SDS | sodium dodecylsulfate |
| TFE | 2,2,2-trifluoroethanol |
| TP | transportan |
| TP10 | transportan 10 |
| Tris | tris (hydroxymethyl) aminomethane |
References
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M. Antimicrobial Peptides: From Design to Clinical Application. Antibiotics 2022, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Boparai, J.K.; Sharma, P.K. Mini Review on Antimicrobial Peptides, Sources, Mechanism and Recent Applications. Protein Pept. Lett. 2020, 27, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Yan, Z.B.; Meng, Y.M.; Hong, X.Y.; Shao, G.; Ma, J.J.; Cheng, X.R.; Liu, J.; Kang, J.; Fu, C.Y. Antimicrobial peptides: Mechanism of action, activity and clinical potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Wu, Y.; Wang, L.; Ma, C.; Xi, X.; Bininda-Emonds, O.R.P.; Shaw, C.; Chen, T.; Zhou, M. Evaluation of the bioactivity of a mastoparan peptide from wasp venom and of its analogues designed through targeted engineering. Int. J. Biol. Sci. 2018, 14, 599–607. [Google Scholar] [CrossRef] [PubMed]
- de la Salud Bea, R.; North, L.J.; Horiuchi, S.; Frawley, E.R.; Shen, Q. Antimicrobial Activity and Toxicity of Analogs of Wasp Venom EMP Peptides. Potential Influence of Oxidized Methionine. Antibiotics 2021, 10, 1208. [Google Scholar] [CrossRef] [PubMed]
- Silva, O.N.; Torres, M.D.T.; Cao, J.; Alves, E.S.F.; Rodrigues, L.V.; Resende, J.M.; Lião, L.M.; Porto, W.F.; Fensterseifer, I.C.M.; Lu, T.K.; et al. Repurposing a peptide toxin from wasp venom into antiinfectives with dual antimicrobial and immunomodulatory properties. Proc. Natl. Acad. Sci. USA 2020, 117, 26936–26945. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Gongpan, P.; Meng, Y.; Nieh, J.C.; Yuan, H.; Tan, K. Functional characterization, antimicrobial effects, and potential antibacterial mechanisms of new mastoparan peptides from hornet venom (Vespa ducalis, Vespa mandarinia, and Vespa affinis). Toxicon 2021, 200, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Vila-Farrés, X.; López-Rojas, R.; Pachón-Ibáñez, M.E.; Teixidó, M.; Pachón, J.; Vila, J.; Giralt, E. Sequence-activity relationship, and mechanism of action of mastoparan analogues against extended-drug resistant Acinetobacter baumannii. Eur. J. Med. Chem. 2015, 101, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, J.R.; Etzerodt, T.; Gjetting, T.; Andresen, T.L. Side chain hydrophobicity modulates therapeutic activity and membrane selectivity of antimicrobial peptide mastoparan-X. PLoS ONE 2014, 9, e91007. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos Cabrera, M.P.; De Souza, B.M.; Fontana, R.; Konno, K.; Palma, M.M.; De Azevedo, W.F., Jr.; Ruggiero Neto, J. Conformation and lytic activity of eumenine mastoparan: A new antimicrobial peptide from wasp venom. J. Pept. Res. 2004, 64, 95–103. [Google Scholar] [CrossRef]
- Dos Santos Cabrera, M.P.; Rangel, M.; Ruggiero Neto, J.; Konno, K. Chemical and Biological Characteristics of Antimicrobial α-Helical Peptides Found in Solitary Wasp Venoms and Their Interactions with Model Membranes. Toxins 2019, 11, 559. [Google Scholar] [CrossRef] [Green Version]
- Howl, J.; Howl, L.; Jones, S. The cationic tetradecapeptide mastoparan as a privileged structure for drug discovery: Enhanced antimicrobial properties of mitoparan analogues modified at position-14. Peptides 2018, 101, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Avram, S.; Buiu, C.; Borcan, F.; Milac, A.-L. More effective antimicrobial mastoparan derivatives, generated by 3D-QSAR-Almond and computational mutagenesis. Mol. Biosyst. 2012, 8, 587–594. [Google Scholar] [CrossRef]
- Xu, X.; Yang, H.; Yu, J.; Li, J.; Lai, R. The mastoparanogen from wasp. Peptides 2006, 27, 3053–3057. [Google Scholar] [CrossRef]
- Moreno, M.; Giralt, E. Three Valuable Peptides from Bee and Wasp Venoms for Therapeutic and Biotechnological Use: Melittin, Apamin and Mastoparan. Toxins 2015, 7, 1126–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Howl, J. Enantiomer-specific bioactivities of peptidomimetic analogues of mastoparan and mitoparan: Characterization of inverso mastoparan as a highly efficient cell penetrating peptide. Bioconjug. Chem. 2012, 23, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Monson de Souza, B.; Dos Santos Cabrera, M.P.; Gomes, P.C.; Dias, N.B.; Stabeli, R.G.; Leite, N.B.; Neto, J.R.; Palma, M.S. Structure-activity relationship of mastoparan analogs: Effects of the number and positioning of Lys residues on secondary structure, interaction with membrane-mimetic systems and biological activity. Peptides 2015, 72, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Howl, J. Charge delocalisation and the design of novel mastoparan analogues: Enhanced cytotoxicity and secretory efficacy of [Lys5, Lys8, Aib10]MP. Regul. Pept. 2004, 121, 121–128. [Google Scholar] [CrossRef]
- Rungsa, P.; Peigneur, S.; Jangpromma, N.; Klaynongsruang, S.; Tytgat, J.; Daduang, S. In Silico and In Vitro Structure–Activity Relationship of Mastoparan and Its Analogs. Molecules 2022, 27, 561. [Google Scholar] [CrossRef]
- Irazazabal, L.N.; Porto, W.F.; Ribeiro, S.M.; Casale, S.; Humblot, V.; Ladram, A.; Franco, O.L. Selective amino acid substitution reduces cytotoxicity of the antimicrobial peptide mastoparan. Biochim. Biophys. Acta 2016, 1858, 2699–2708. [Google Scholar] [CrossRef]
- Ruczyński, J.; Wierzbicki, P.M.; Kogut-Wierzbicka, M.; Mucha, P.; Siedlecka-Kroplewska, K.; Rekowski, P. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem. Cytobiol. 2014, 52, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Ruczyński, J.; Rusiecka, I.; Turecka, K.; Kozłowska, A.; Alenowicz, M.; Gągało, I.; Kawiak, A.; Rekowski, P.; Waleron, K.; Kocić, I. Transportan 10 improves the pharmacokinetics and pharmacodynamics of vancomycin. Sci. Rep. 2019, 9, 3247. [Google Scholar] [CrossRef] [Green Version]
- Rusiecka, I.; Ruczyński, J.; Kozłowska, A.; Backtrog, E.; Mucha, P.; Kocić, I.; Rekowski, P. TP10-dopamine conjugate as a potential therapeutic agent in the treatment of Parkinson’s disease. Bioconjug. Chem. 2019, 30, 760–774. [Google Scholar] [CrossRef]
- Wierzbicki, P.M.; Kogut-Wierzbicka, M.; Ruczyński, J.; Siedlecka-Kroplewska, K.; Kaszubowska, L.; Rybarczyk, A.; Alenowicz, M.; Rekowski, P.; Kmieć, Z. Protein and siRNA delivery by transportan and transportan 10 into colorectal cancer cell lines. Folia Histochem. Cytobiol. 2014, 52, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Zhang, M.; Zhang, F.; Wang, Y.; Ouyang, J.; Luo, X.; Yang, H.; Zhang, D.; Chen, Y.; Yu, H.; et al. Design of a Sea Snake Antimicrobial Peptide Derivative with Therapeutic Potential against Drug-Resistant Bacterial Infection. ACS Infect. Dis. 2020, 6, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.; Maurya, I.K.; Khan, S.I.; Jacob, M.R.; Kumar, V.; Tikoo, K.; Jain, R. Discovery of a Membrane-Active, Ring-Modified Histidine Containing Ultrashort Amphiphilic Peptide That Exhibits Potent Inhibition of Cryptococcus neoformans. J. Med. Chem. 2017, 60, 6607–6621. [Google Scholar] [CrossRef] [PubMed]
- Bellavita, R.; Casciaro, B.; Di Maro, S.; Brancaccio, D.; Carotenuto, A.; Falanga, A.; Cappiello, F.; Buommino, E.; Galdiero, S.; Novellino, E.; et al. First-in-Class Cyclic Temporin L Analogue: Design, Synthesis, and Antimicrobial Assessment. J. Med. Chem. 2021, 64, 11675–11694. [Google Scholar] [CrossRef] [PubMed]
- Gov, Y.; Bitler, A.; Dell’Acqua, G.; Torres, J.V.; Balaban, N. RNAIII inhibiting peptide (RIP), a global inhibitor of Staphylococcus aureus pathogenesis: Structure and function analysis. Peptides 2001, 22, 1609–1620. [Google Scholar] [CrossRef]
- Giacometti, A.; Cirioni, O.; Gov, Y.; Ghiselli, R.; Del Prete, M.S.; Mocchegiani, F.; Saba, V.; Orlando, F.; Scalise, G.; Balaban, N.; et al. RNA III inhibiting peptide inhibits in vivo biofilm formation by drug-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2003, 47, 1979–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Acqua, G.; Giacometti, A.; Cirioni, O.; Ghiselli, R.; Saba, V.; Scalise, G.; Gov, Y.; Balaban, N. Suppression of drug-resistant Staphylococcal Infections by the quorum-sensing inhibitor RNAIII-inhibiting peptide. J. Infect. Dis. 2004, 190, 318–320. [Google Scholar] [CrossRef] [Green Version]
- Giacometti, A.; Cirioni, O.; Ghiselli, R.; Dell’Acqua, G.; Orlando, F.; D’Amato, G.; Mocchegiani, F.; Silvestri, C.; Del Prete, M.S.; Rocchi, M.; et al. RNAIII-inhibiting peptide improves efficacy of clinically used antibiotics in a murine model of staphylococcal sepsis. Peptides 2005, 26, 169–175. [Google Scholar] [CrossRef]
- Ciulla, M.; Di Stefano, A.; Marinelli, L.; Cacciatore, I.; Di Biase, G. RNAIII Inhibiting Peptide (RIP) and Derivatives as Potential Tools for the Treatment of S. aureus Biofilm Infections. Curr. Top. Med. Chem. 2018, 18, 2068–2079. [Google Scholar] [CrossRef]
- Brishty, S.R.; Hossain, M.J.; Khandaker, M.U.; Faruque, M.R.I.; Osman, H.; Rahman, S.M.A. A Comprehensive Account on Recent Progress in Pharmacological Activities of Benzimidazole Derivatives. Front. Pharmacol. 2021, 12, 762807. [Google Scholar] [CrossRef]
- Marinescu, M. Synthesis of Antimicrobial Benzimidazole-Pyrazole Compounds and Their Biological Activities. Antibiotics 2021, 10, 1002. [Google Scholar] [CrossRef]
- Hernández-López, H.; Tejada-Rodríguez, C.J.; Leyva-Ramos, S. A Panoramic Review of Benzimidazole Derivatives and Their Potential Biological Activity. Mini Rev. Med. Chem. 2022, 22, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Kaur, M.; Bansal, B. Antimicrobial Potential of Benzimidazole Derived Molecules. Mini Rev. Med. Chem. 2019, 19, 624–646. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, S.; Kumar, S.; Narasimhan, B. Pharmacological significance of heterocyclic 1H-benzimidazole scaffolds: A review. BMC Chem. 2019, 13, 101. [Google Scholar] [CrossRef] [Green Version]
- Vasava, M.S.; Bhoi, M.N.; Rathwa, S.K.; Jethava, D.J.; Acharya, P.T.; Patel, D.B.; Patel, H.D. Benzimidazole: A Milestone in the Field of Medicinal Chemistry. Mini Rev. Med. Chem. 2020, 20, 532–565. [Google Scholar] [CrossRef]
- Fauchère, J.; Pliška, V. Hydrophobic parameters Π of amino-acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The helical hydrophobic moment: A measure of the amphiphilicity of a helix. Nature 1982, 299, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C.; Wilcox, W. Hydrophobic moments and protein structure. Faraday Symp. Chem. Soc. 1982, 17, 109–120. [Google Scholar] [CrossRef]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Rohl, C.A.; Baldwin, R.L. Deciphering rules of helix stability in peptides. Methods Enzymol. 1998, 295, 1–26. [Google Scholar] [CrossRef]
- Konno, K.; Hisada, M.; Fontana, R.; Lorenzi, C.C.; Naoki, H.; Itagaki, Y.; Miwa, A.; Kawai, N.; Nakata, Y.; Yasuhara, T.; et al. Anoplin, a novel antimicrobial peptide from the venom of the solitary wasp Anoplius samariensis. Biochim. Biophys. Acta 2001, 1550, 70–80. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence |
|---|---|
| MP | INLKALAALAKKIL-NH2 |
| retroMP | LIKKALAALAKLNI-NH2 |
| MP-retroMP | INLKALAALAKKILLIKKALAALAKLNI-NH2 |
| RIP | YSPWTNF-NH2 |
| MP-RIP | INLKALAALAKKILYSPWTNF-NH2 |
| RIP-MP | YSPWTNFINLKALAALAKKIL-NH2 |
| MP (4–14) | KALAALAKKIL-NH2 |
| MP (4–14)-RIP | KALAALAKKILYSPWTNF-NH2 |
| [Lys2, Ile4] RIP | YKPITNF-NH2 |
| MP (4–14)-[Lys2, Ile4] RIP | KALAALAKKILYKPITNF-NH2 |
| MP-[Lys2, Ile4] RIP | INLKALAALAKKILYKPITNF-NH2 |
| [Lys2, Ile4] RIP-MP | YKPITNFINLKALAALAKKIL-NH2 |
| Gal (1–13)-MP (Galp) | GWTLNSAGYLLGPINLKALAALAKKIL-NH2 |
| Gal (1–12)-Lys-MP (TP) | GWTLNSAGYLLGKINLKALAALAKKIL-NH2 |
| Gal (7–12)-Lys-MP (TP10) | AGYLLGKINLKALAALAKKIL-NH2 |
| TP10-RIP | AGYLLGKINLKALAALAKKILYSPWTNF-NH2 |
| TP10-[Lys2, Ile4] RIP | AGYLLGKINLKALAALAKKILYKPITNF-NH2 |
| [desLys7] TP10-[Lys2, Ile4] RIP | AGYLLGINLKALAALAKKILYKPITNF-NH2 |
| [Lys7 (BnzAc)] TP10 | AGYLLGK (BnzAc) INLKALAALAKKIL-NH2 |
| [Lys7 (NBnzAc)] TP10 | AGYLLGK (NBnzAc) INLKALAALAKKIL-NH2 |
| [Lys7 (PBnzAc)] TP10 | AGYLLGK (PBnzAc) INLKALAALAKKIL-NH2 |
| [Lys7 (ClBnzAc)] TP10 | AGYLLGK (ClBnzAc) INLKALAALAKKIL-NH2 |
| Peptide | MW | N | Q | %H | H | µH | Ins |
|---|---|---|---|---|---|---|---|
| MP | 1478.91 | 14 | +4 | 71.43 | 0.576 | 0.398 | 10.91 |
| retroMP | 1478.91 | 14 | +4 | 71.43 | 0.576 | 0.398 | 28.54 |
| MP-retroMP | 2940.81 | 28 | +7 | 71.43 | 0.576 | 0.241 | 20.09 |
| RIP | 912.98 | 7 | +1 | 57.14 | 0.763 | nd | 2.81 |
| MP-RIP | 2374.88 | 21 | +4 | 66.67 | 0.639 | 0.292 | 8.69 |
| RIP-MP | 2374.88 | 21 | +4 | 66.67 | 0.639 | 0.093 | 8.69 |
| MP (4–14) | 1138.49 | 11 | +4 | 72.73 | 0.470 | 0.598 | 18.88 |
| MP (4–14)-RIP | 2034.46 | 18 | +4 | 66.67 | 0.584 | 0.359 | 13.19 |
| [Lys2, Ile4] RIP | 881.03 | 7 | +2 | 57.14 | 0.563 | nd | −45.14 |
| MP (4–14)-[Lys2, Ile4] RIP | 2002.50 | 18 | +5 | 66.67 | 0.506 | 0.379 | −5.46 |
| MP-[Lys2, Ile4] RIP | 2342.92 | 21 | +5 | 66.67 | 0.572 | 0.312 | −7.30 |
| [Lys2, Ile4] RIP-MP | 2342.92 | 21 | +5 | 66.67 | 0.572 | 0.090 | −7.30 |
| Galp | 2809.42 | 27 | +4 | 62.96 | 0.631 | 0.134 | 6.33 |
| TP | 2840.48 | 27 | +5 | 59.26 | 0.567 | 0.173 | 0.04 |
| TP10 | 2181.76 | 21 | +5 | 66.67 | 0.560 | 0.235 | −1.52 |
| TP10-RIP | 3077.73 | 28 | +5 | 64.29 | 0.610 | 0.152 | −0.08 |
| TP10-[Lys2, Ile4] RIP | 3045.78 | 28 | +6 | 64.29 | 0.560 | 0.165 | −12.07 |
| [desLys7] TP10-[Lys2, Ile4] RIP | 2917.60 | 27 | +5 | 66.67 | 0.618 | 0.249 | −9.74 |
| [Lys7 (BnzAc)] TP10 | 2339.76 | 21 | +4 | nd | nd | nd | nd |
| [Lys7 (NBnzAc)] TP10 | 2466.76 | 21 | +4 | nd | nd | nd | nd |
| [Lys7 (PBnzAc)] TP10 | 2417.36 | 21 | +4 | nd | nd | nd | nd |
| [Lys7 (ClBnzAc)] TP10 | 2450.36 | 21 | +4 | nd | nd | nd | nd |
| Peptide | MIC | ||||
|---|---|---|---|---|---|
| S. aureus | E. coli | P. aeruginosae | |||
| [μg/mL] | [μM] | [μg/mL] | [μM] | [μg/mL] | |
| MP | 8 | 5.4 | 32 | 21.6 | 256 |
| retroMP | >256 | >172.8 | 256 | 172.8 | 256 |
| MP-retroMP | >256 | >87.5 | >256 | 87.5 | >256 |
| RIP | >256 | >280.4 | >256 | >280.4 | >256 |
| MP-RIP | 16 | 6.7 | 256 | 107.2 | >256 |
| RIP-MP | 32 | 13.4 | 128 | 53.6 | >256 |
| MP (4–14) | >256 | >224.4 | >256 | >224.4 | >256 |
| MP (4–14)-RIP | >256 | >125.8 | >256 | >125.8 | >256 |
| [Lys2, Ile4] RIP | >256 | >290.6 | >256 | >290.6 | >256 |
| MP (4–14)-[Lys2, Ile4] RIP | 256 | 127.8 | >256 | >127.8 | >256 |
| MP-[Lys2, Ile4] RIP | 64 | 27.8 | 64 | 27.8 | >256 |
| [Lys2, Ile4] RIP-MP | 64 | 27.8 | 256 | 109.3 | >256 |
| Galp | 128 | 45.5 | >256 | >91.1 | >256 |
| TP | 128 | 45.1 | >256 | >90.1 | >256 |
| TP10 | 16 | 7.3 | 128 | 58.7 | 256 |
| TP10-RIP | 128 | 41.6 | >256 | >83.2 | >256 |
| TP10-[Lys2, Ile4] RIP | 128 | 42.0 | >256 | >84,0 | >256 |
| [desLys7] TP10-[Lys2, Ile4] RIP | 64 | 21.9 | 256 | 87.6 | >256 |
| [Lys7 (BnzAc)] TP10 | 256 | 109.4 | >256 | >109.4 | >256 |
| [Lys7 (NBnzAc)] TP10 | >256 | >103.8 | >256 | >103.8 | >256 |
| [Lys7 (PBnzAc)] TP10 | >256 | >105.3 | >256 | >105.3 | >256 |
| [Lys7 (ClBnzAc)] TP10 | >256 | >104.5 | >256 | >104.5 | >256 |
| Gentamicin | 0.5 | 1.1 | 1.0 | 2.1 | 0.25 |
| Peptide | Relative α-Helix Fraction (fH) | ||
|---|---|---|---|
| Water | 50% TFE | 0.1 M Tris + 50 mM SDS | |
| MP | 0.14 | 0.38 | 0.50 |
| retroMP | 0.19 | 0,27 | 0.16 |
| MP-retroMP | 0.04 | 0.24 | 0.17 |
| MP-RIP | 0.11 | 0.34 | 0.18 |
| RIP-MP | 0.15 | 0.39 | 0.23 |
| MP (4–14) | 0.02 | 0.31 | 0.64 |
| MP (4–14)-RIP | 0.05 | 0.23 | 0.23 |
| MP (4–14)-[Lys2, Ile4] RIP | 0.04 | 0.23 | 0.18 |
| MP-[Lys2, Ile4] RIP | 0.09 | 0.27 | 0.32 |
| [Lys2, Ile4] RIP-MP | 0.10 | 0.35 | 0.31 |
| Galp | 0.13 | 0.34 | 0.09 |
| TP | 0.12 | 0.34 | 0.10 |
| TP10 | 0.10 | 0.34 | 0.32 |
| TP10-RIP | 0.08 | 0.27 | 0.25 |
| TP10-[Lys2, Ile4] RIP | 0.08 | 0.29 | 0.31 |
| [desLys7] TP10-[Lys2, Ile4] RIP | 0.13 | 0.34 | 0.25 |
| [Lys7 (PBnzAc)] TP10 | 0.15 | 0.51 | 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruczyński, J.; Parfianowicz, B.; Mucha, P.; Wiśniewska, K.; Piechowicz, L.; Rekowski, P. Structure–Activity Relationship of New Chimeric Analogs of Mastoparan from the Wasp Venom Paravespula lewisii. Int. J. Mol. Sci. 2022, 23, 8269. https://doi.org/10.3390/ijms23158269
Ruczyński J, Parfianowicz B, Mucha P, Wiśniewska K, Piechowicz L, Rekowski P. Structure–Activity Relationship of New Chimeric Analogs of Mastoparan from the Wasp Venom Paravespula lewisii. International Journal of Molecular Sciences. 2022; 23(15):8269. https://doi.org/10.3390/ijms23158269
Chicago/Turabian StyleRuczyński, Jarosław, Brygida Parfianowicz, Piotr Mucha, Katarzyna Wiśniewska, Lidia Piechowicz, and Piotr Rekowski. 2022. "Structure–Activity Relationship of New Chimeric Analogs of Mastoparan from the Wasp Venom Paravespula lewisii" International Journal of Molecular Sciences 23, no. 15: 8269. https://doi.org/10.3390/ijms23158269
APA StyleRuczyński, J., Parfianowicz, B., Mucha, P., Wiśniewska, K., Piechowicz, L., & Rekowski, P. (2022). Structure–Activity Relationship of New Chimeric Analogs of Mastoparan from the Wasp Venom Paravespula lewisii. International Journal of Molecular Sciences, 23(15), 8269. https://doi.org/10.3390/ijms23158269