
Circ-LocNet: A Computational Framework for Circular RNA Sub-Cellular Localization Prediction

 ,
, 
Abstract
:1. Introduction
- Development of a benchmark circRNA sub-cellular localization prediction dataset using public RNALocate database [34] which is comprised of 1,205 circRNA sequences annotated against 8 different sub-cellular localities.
- A comprehensive performance analysis of residue frequency, residue order and frequency, and residue physicochemical property-based sequence descriptors is performed to find an appropriate standalone sequence descriptor for circular RNA sub-cellular localization prediction.
- A detailed performance impact of K-order sequence descriptor fusion is performed by ensembling similar as well dissimilar genres of statistical representation learning approaches to reap the combined benefits in order to investigate whether sequence descriptor fusion significantly optimizes the statistical representation of circRNA sequences and which K-order sequence descriptor fusion manages to capture more discriminative residue distribution important for sub-cellular localization prediction.
- Extensive empirical evaluation of 5 different generative, discriminative, and tree-based classifiers using various sequence descriptors is performed to investigate which type of classifier extracts a residue correlation that is important to accurately predict circular RNA sub-cellular localization.
- An end-to-end computational framework (Circ-LocNet) that explores the performance of seven different standalone sequence descriptors, second-order sequence descriptor fusion, third-order sequence descriptor fusion, and so on all the way up to seventh-order sequence descriptor fusion with the five most widely used machine learning classifiers in order to determine an appropriate combination of sequence descriptor and machine learning classifier for circular RNA sub-cellular localization prediction.
- Development and deployment of the very first interactive and user-friendly circRNAs sub-cellular localization prediction platform.
2. Results
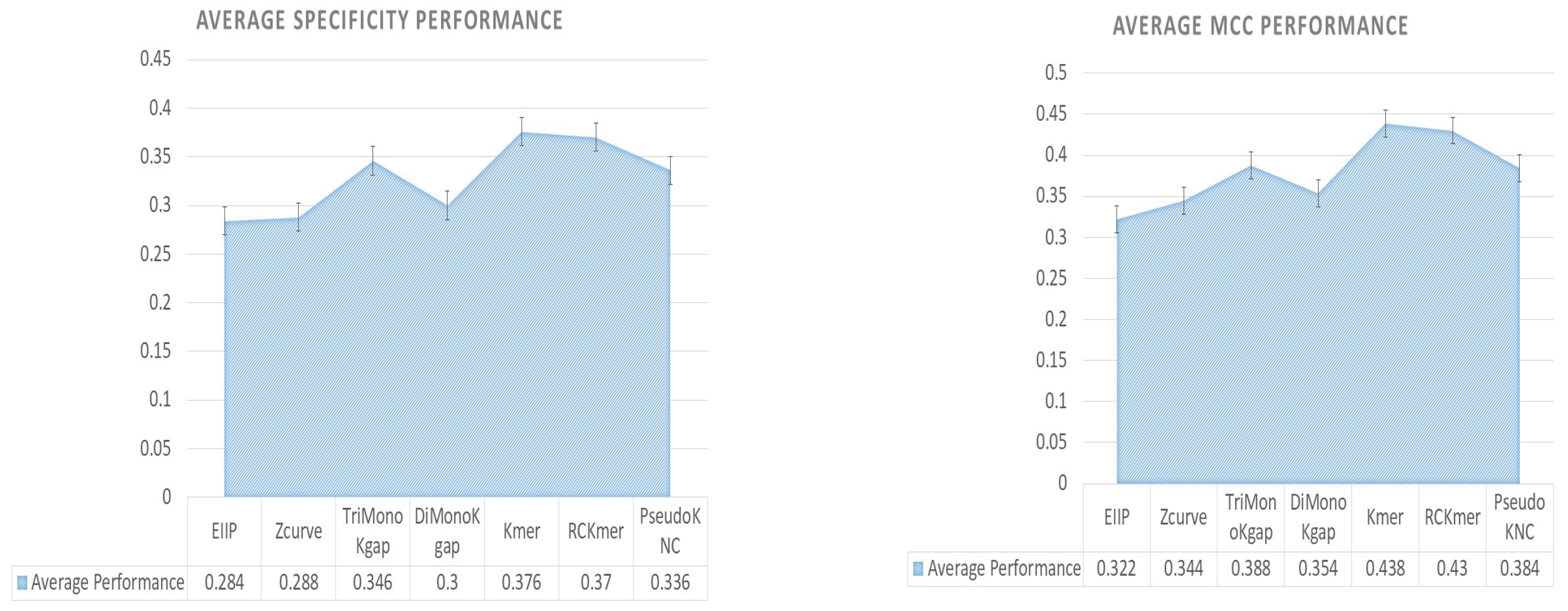
2.1. Performance Assessment of Different Standalone Sequence Descriptors Using Distinct Genre Machine Learning Classifiers
2.2. Performance Assessment of K-Order Sequence Descriptors Fusion Using Distinct Genre Machine Learning Classifiers
3. Discussion
Interactive and User-Friendly Circ-Locnet Web Server
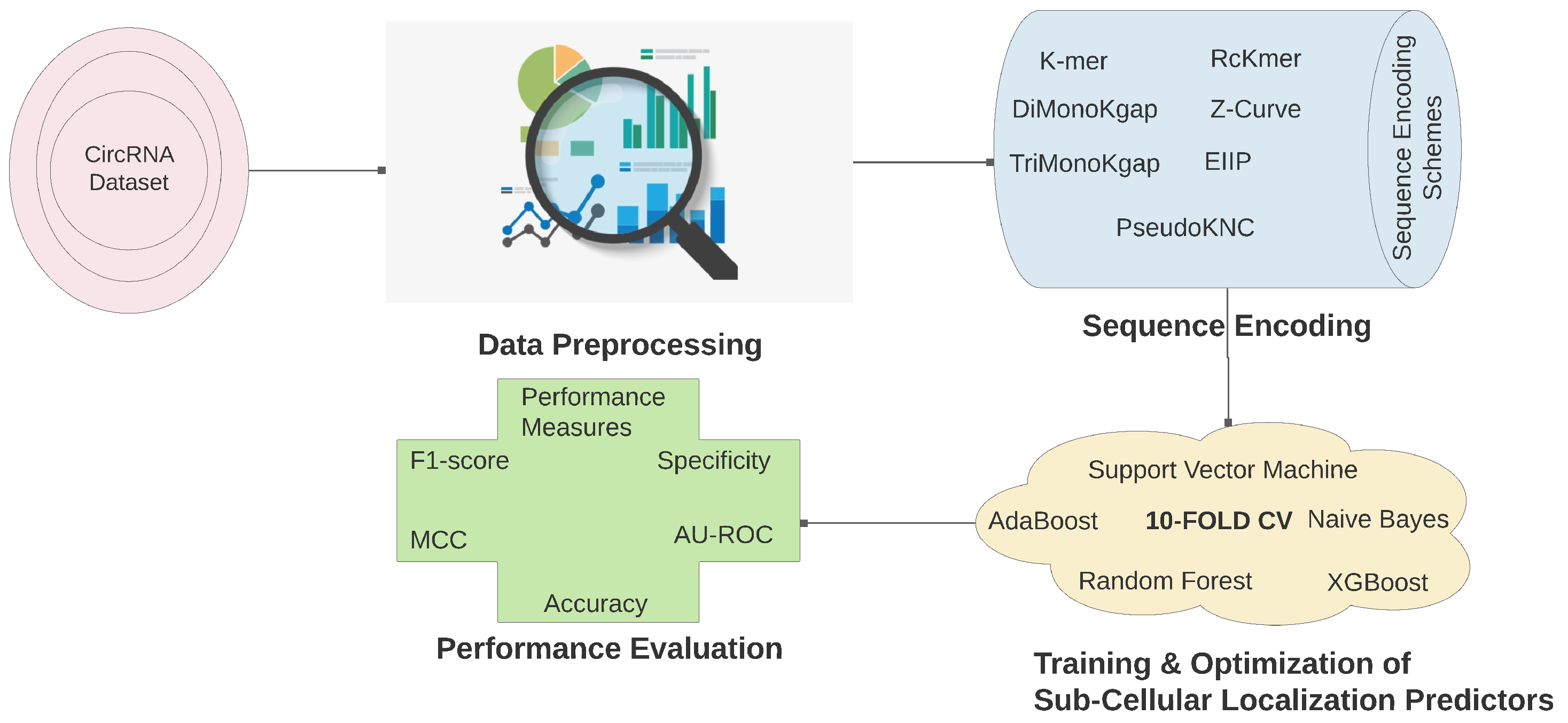
4. Materials and Methods
4.1. Circ-Locnet
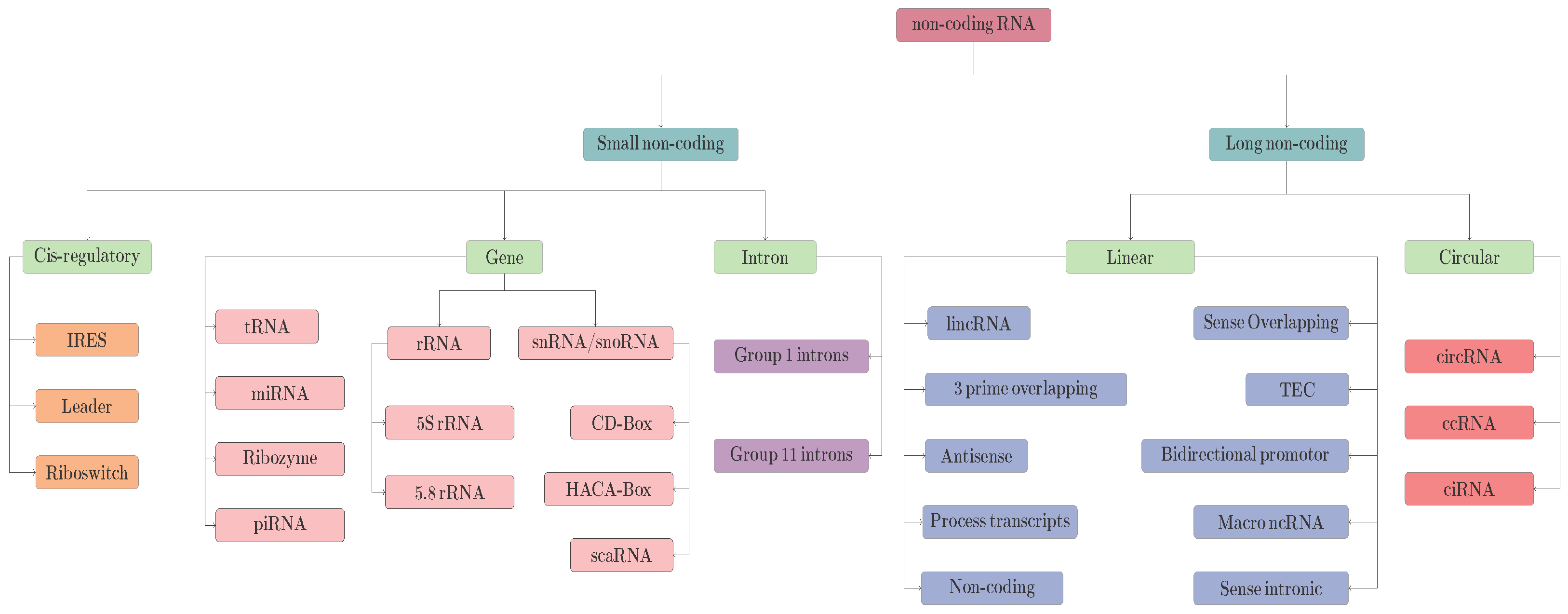
4.1.1. Sequence Encoding Approaches
4.1.2. K-Mer
4.1.3. Reverse Compliment Kmer
4.1.4. Psuedoknc
4.1.5. Z-Curve
4.1.6. Electron–Ion Interaction Pseudopotentials of Trinucleotide (Eiip)
4.1.7. Xxkgap
4.1.8. Circular RNA Sub-Cellular Localization Predictors
4.2. Circular RNA Sub-Cellular Localization Dataset
4.3. Evaluation Metrics
4.4. Experimental Setup
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Leung, K.S. Quantification of non-coding RNA target localization diversity and its application in cancers. J. Mol. Cell Biol. 2018, 10, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Frías-Lasserre, D.; Villagra, C.A. The importance of ncRNAs as epigenetic mechanisms in phenotypic variation and organic evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Li, X.; Zhang, P.; Wang, J.; Zhou, Y.; Chen, M. Circular RNA: An emerging key player in RNA world. Briefings Bioinform. 2017, 18, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Cocquerelle, C.; Daubersies, P.; Majerus, M.A.; Kerckaert, J.P.; Bailleul, B. Splicing with inverted order of exons occurs proximal to large introns. EMBO J. 1992, 11, 1095–1098. [Google Scholar] [CrossRef]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [Green Version]
- Cocquerelle, C.; Mascrez, B.; Hétuin, D.; Bailleul, B. Mis-splicing yields circular RNA molecules. FASEB J. 1993, 7, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Zaphiropoulos, P.G. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: Correlation with exon skipping. Proc. Natl. Acad. Sci. USA 1996, 93, 6536–6541. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PloS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Geng, X.; Jia, Y.; Zhang, Y.; Shi, L.; Li, Q.; Zang, A.; Wang, H. Circular RNA: Biogenesis, degradation, functions and potential roles in mediating resistance to anticarcinogens. Epigenomics 2020, 12, 267–283. [Google Scholar] [CrossRef]
- Liu, J.; Yang, L.; Fu, Q.; Liu, S. Emerging roles and potential biological value of circRNA in osteosarcoma. Front. Oncol. 2020, 10, 552236. [Google Scholar] [CrossRef]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell. Mol. Life Sci. 2018, 75, 1071–1098. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhang, L.; Li, W.; Deng, J.; Zheng, J.; An, M.; Lu, J.; Zhou, Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/β-catenin pathway. Oncotarget 2015, 6, 6001. [Google Scholar] [CrossRef] [Green Version]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis and normal human tissues. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Li, P.; Chen, S.; Chen, H.; Mo, X.; Li, T.; Shao, Y.; Xiao, B.; Guo, J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin. Chim. Acta 2015, 444, 132–136. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Holdt, L.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.; Nicolaou, A.; et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat. Commun. 2016, 7, 12429. [Google Scholar]
- Tang, W.; Ji, M.; He, G.; Yang, L.; Niu, Z.; Jian, M.; Wei, Y.; Ren, L.; Xu, J. Silencing CDR1as inhibits colorectal cancer progression through regulating microRNA-7. Oncotargets Ther. 2017, 10, 2045. [Google Scholar] [CrossRef] [Green Version]
- Du, W.W.; Yang, W.; Chen, Y.; Wu, Z.K.; Foster, F.S.; Yang, Z.; Li, X.; Yang, B.B. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur. Heart J. 2017, 38, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, X.; Li, C.; Yue, L.; Ding, N.; Riordan, T.; Yang, L.; Li, Y.; Jen, C.; Lin, S.; et al. Circular RNA profiling provides insights into their subcellular distribution and molecular characteristics in HepG2 cells. RNA Biol. 2019, 16, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Cha, D.J.; Franklin, J.L.; Higginbotham, J.N.; Jeppesen, D.K.; Weaver, A.M.; Prasad, N.; Levy, S.; Coffey, R.J.; Patton, J.G.; et al. Circular RNAs are down-regulated in KRAS mutant colon cancer cells and can be transferred to exosomes. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.L.; Wang, Y.; et al. Extensive translation of circular RNAs driven by N 6-methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Bramham, C.R.; Wells, D.G. Dendritic mRNA: Transport, translation and function. Nat. Rev. Neurosci. 2007, 8, 776. [Google Scholar] [CrossRef]
- Lécuyer, E.; Yoshida, H.; Parthasarathy, N.; Alm, C.; Babak, T.; Cerovina, T.; Hughes, T.R.; Tomancak, P.; Krause, H.M. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 2007, 131, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Chin, A.; Lecuyer, E. RNA localization: Making its way to the center stage. Biochim. Biophys. Acta-(Bba)-Gen. Subj. 2017, 1861, 2956–2970. [Google Scholar] [CrossRef]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest–and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, K.N.; Lindebjerg, J.; Kalmár, A.; Molnár, B.; Sørensen, F.B.; Hansen, T.F.; Nielsen, B.S. miR-21 expression analysis in budding colon cancer cells by confocal slide scanning microscopy. Clin. Exp. Metastasis 2018, 35, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Kallen, A.N.; Zhou, X.B.; Xu, J.; Qiao, C.; Ma, J.; Yan, L.; Lu, L.; Liu, C.; Yi, J.S.; Zhang, H.; et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 2013, 52, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wei, Y.; Yu, L.; Xiao, Y. Identification of altered circular RNA expression in serum exosomes from patients with papillary thyroid carcinoma by high-throughput sequencing. Med. Sci. Monit. Int. Med J. Exp. Clin. Res. 2019, 25, 2785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 2016, 45, D135–D138. [Google Scholar]
- Aken, B.L.; Achuthan, P.; Akanni, W.; Amode, M.R.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2017, 45, D635–D642. [Google Scholar] [CrossRef]
- Bernstein, B.; Birney, E.; Dunham, I.; Green, E.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Meher, P.K.; Satpathy, S.; Rao, A.R. miRNALoc: Predicting miRNA subcellular localizations based on principal component scores of physico-chemical properties and pseudo compositions of di-nucleotides. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Asim, M.N.; Dengel, A.; Ahmed, S. A convNet based multi-label microRNA subcellular location predictor, by incorporating k-mer positional encoding. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Gudenas, B.L.; Wang, L. Prediction of lncRNA subcellular localization with deep learning from sequence features. Sci. Rep. 2018, 8, 16385. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.D.; Huang, Y.; Zhang, Z.Y.; Zhao, Y.W.; Wang, D.; Chen, W.; Chou, K.C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef]
- Cao, Z.; Pan, X.; Yang, Y.; Huang, Y.; Shen, H.B. The lncLocator: A subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics 2018, 34, 2185–2194. [Google Scholar] [CrossRef]
- Xiao, Y.; Cai, J.; Yang, Y.; Zhao, H.; Shen, H. Prediction of MicroRNA Subcellular Localization by Using a Sequence-to-Sequence Model. In Proceedings of the IEEE International Conference on Data Mining (ICDM), Singapore, 17–20 November 2018; pp. 1332–1337. [Google Scholar]
- Asim, M.N.; Malik, M.I.; Zehe, C.; Trygg, J.; Dengel, A.; Ahmed, S. MirLocPredictor: A ConvNet-Based Multi-Label MicroRNA Subcellular Localization Predictor by Incorporating k-Mer Positional Information. Genes 2020, 11, 1475. [Google Scholar] [CrossRef]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2019, 21, 1–16. [Google Scholar] [CrossRef]
- Rafiee, A.; Riazi-Rad, F.; Havaskary, M.; Nuri, F. Long noncoding RNAs: Regulation, function and cancer. Biotechnol. Genet. Eng. Rev. 2018, 34, 153–180. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Fu, L.; Wang, Q.; Fu, G.; Lu, C.; Feng, J.; Cong, B.; Li, S. Circular RNA as a potential biomarker for forensic age prediction using multiple machine learning models: A preliminary study. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, L.L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef]
- Asim, M.N.; Ibrahim, M.A.; Imran Malik, M.; Dengel, A.; Ahmed, S. Advances in computational methodologies for classification and sub-cellular locality prediction of non-coding RNAs. Int. J. Mol. Sci. 2021, 22, 8719. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, C.; Li, F.; Xiang, D.; Chen, Y.Z.; Akutsu, T.; Daly, R.J.; Webb, G.I.; Zhao, Q.; et al. iLearnPlus: A comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021, 49, e60. [Google Scholar] [CrossRef]
- Monga, I.; Banerjee, I. Computational identification of piRNAs using features based on rna sequence, structure, thermodynamic and physicochemical properties. Curr. Genom. 2019, 20, 508–518. [Google Scholar] [CrossRef]
- Stricker, M.; Asim, M.N.; Dengel, A.; Ahmed, S. CircNet: An encoder—Decoder-based convolution neural network (CNN) for circular RNA identification. Neural Comput. Appl. 2021, 34, 11441–11452. [Google Scholar] [CrossRef]
- Niu, M.; Zhang, J.; Li, Y.; Wang, C.; Liu, Z.; Ding, H.; Zou, Q.; Ma, Q. CirRNAPL: A web server for the identification of circRNA based on extreme learning machine. Comput. Struct. Biotechnol. J. 2020, 18, 834–842. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, Z.M.; Li, S.H.; Tan, J.X.; Chen, W.; Lin, H. Evaluation of different computational methods on 5-methylcytosine sites identification. Briefings Bioinform. 2020, 21, 982–995. [Google Scholar] [CrossRef]
- Lee, D.; Karchin, R.; Beer, M.A. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011, 21, 2167–2180. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Dennis, J.; Thurman, R.E.; Kingston, R.; Stamatoyannopoulos, J.A.; Noble, W.S. Predicting human nucleosome occupancy from primary sequence. PLoS Comput. Biol. 2008, 4, e1000134. [Google Scholar]
- Noble, W.S.; Kuehn, S.; Thurman, R.; Yu, M.; Stamatoyannopoulos, J. Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics 2005, 21, i338–i343. [Google Scholar] [CrossRef]
- Fletez-Brant, C.; Lee, D.; McCallion, A.S.; Beer, M.A. kmer-SVM: A web server for identifying predictive regulatory sequence features in genomic data sets. Nucleic Acids Res. 2013, 41, W544–W556. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Zhou, H.; Yue, Z. ProRice: An Ensemble Learning Approach for Predicting Promoters in Rice. In Proceedings of the 4th International Conference on Computer Science and Application Engineering, Sanya, China, 20–22 October 2020; pp. 1–5. [Google Scholar]
- Xu, H.; Jia, P.; Zhao, Z. Deep4mC: Systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning. Briefings Bioinform. 2020, 22, bbaa099. [Google Scholar] [CrossRef]
- Wang, H.; Ding, Y.; Tang, J.; Zou, Q.; Guo, F. Multi-label learning for identi cation of RNA-associated subcellular localizations. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007, 248, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.D.; Huang, J.H.; Liang, R.P.; Lu, X.Q. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: An approach from discrete wavelet transform. Anal. Biochem. 2009, 390, 68–73. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A. Genetic programming for creating Chou’s pseudo amino acid based features for submitochondria localization. Amino Acids 2008, 34, 653–660. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Zou, Q.; Dong, Q.; Chen, Q. Protein remote homology detection by combining Chou’s pseudo amino acid composition and profile-based protein representation. Mol. Inform. 2013, 32, 775–782. [Google Scholar] [CrossRef]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014, 30, 1522–1529. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.R.; Xiao, X.; Chou, K.C. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int. J. Mol. Sci. 2014, 15, 1746–1766. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, Z.; Dai, Z.; Zou, X. Predicting promoters by pseudo-trinucleotide compositions based on discrete wavelets transform. J. Theor. Biol. 2013, 319, 1–7. [Google Scholar] [CrossRef]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2019, 111, 96–102. [Google Scholar] [CrossRef]
- Lin, H.; Deng, E.Z.; Ding, H.; Chen, W.; Chou, K.C. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961–12972. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, C.T. Z curves, an intutive tool for visualizing and analyzing the DNA sequences. J. Biomol. Struct. Dyn. 1994, 11, 767–782. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Liao, Q.; Liu, B. iEsGene-ZCPseKNC: Identify Essential Genes Based on Z Curve Pseudo k-Tuple Nucleotide Composition. IEEE Access 2019, 7, 165241–165247. [Google Scholar] [CrossRef]
- Song, K. Recognition of prokaryotic promoters based on a novel variable-window Z-curve method. Nucleic Acids Res. 2012, 40, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, C.T. Identification of replication origins in archaeal genomes based on the Z-curve method. Archaea 2005, 1, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.B.; Ou, H.Y.; Zhang, C.T. ZCURVE: A new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 2003, 31, 1780–1789. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Pei, Z.; Liu, J.; Qin, S.; Cai, L. Prediction of nucleosome DNA formation potential and nucleosome positioning using increment of diversity combined with quadratic discriminant analysis. Chromosome Res. 2010, 18, 777–785. [Google Scholar] [CrossRef]
- Nair, A.S.; Sreenadhan, S.P. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation 2006, 1, 197. [Google Scholar]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Briefings Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef]
- Muhammod, R.; Ahmed, S.; Md Farid, D.; Shatabda, S.; Sharma, A.; Dehzangi, A. PyFeat: A Python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics 2019, 35, 3831–3833. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Li, X.; Ding, H.; Xu, L.; Xiang, H. Prediction of m5C Modifications in RNA Sequences by Combining Multiple Sequence Features. Mol.-Ther.-Nucleic Acids 2020, 21, 332–342. [Google Scholar] [CrossRef]
- Jia, C.; Yang, Q.; Zou, Q. NucPosPred: Predicting species-specific genomic nucleosome positioning via four different modes of general PseKNC. J. Theor. Biol. 2018, 450, 15–21. [Google Scholar] [CrossRef]
- He, W.; Jia, C.; Zou, Q. 4mCPred: Machine learning methods for DNA N4-methylcytosine sites prediction. Bioinformatics 2019, 35, 593–601. [Google Scholar] [CrossRef]
- Mabrouk, M.S.; Solouma, N.H.; Youssef, A.B.M.; Kadah, Y.M. Eukaryotic Gene Prediction by an Investigation of Nonlinear Dynamical Modeling Techniques On EIIP Coded Sequences 2008. Available online: http://dspace.must.edu.eg/handle/123456789/194 (accessed on 15 June 2022).
- Naeem, S.M.; Mabrouk, M.S.; Eldosoky, M.A.; Sayed, A.Y. Moment invariants for cancer classification based on electron–ion interaction pseudo potentials (EIIP). Netw. Model. Anal. Health Inform. Bioinform. 2020, 9, 1–5. [Google Scholar] [CrossRef]
- El-Badawy, I.M.; Gasser, S.; Aziz, A.M.; Khedr, M.E. On the use of pseudo-EIIP mapping scheme for identifying exons locations in DNA sequences. In Proceedings of the International Conference on Signal and Image Processing Applications (ICSIPA), Kuala Lumpur, Malaysia, 19–21 October 2015; pp. 244–247. [Google Scholar]
- Tang, Q.; Nie, F.; Kang, J.; Chen, W. ncPro-ML: An integrated computational tool for identifying non-coding RNA promoters in multiple species. Comput. Struct. Biotechnol. J. 2020, 18, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.Y.; Khanal, J.; Tayara, H.; Chong, K.T. iEnhancer-RF: Identifying enhancers and their strength by enhanced feature representation using random forest. Chemom. Intell. Lab. Syst. 2021, 212, 104284. [Google Scholar] [CrossRef]
- Yu, T.; Chen, M.; Wang, C. An Improved Method for Identification of Pre-miRNA in Drosophila. IEEE Access 2020, 8, 52173–52180. [Google Scholar] [CrossRef]
- Gu, S. Applying Machine Learning Algorithms for the Analysis of Biological Sequences and Medical Records. Master’s Thesis, South Dakota State University, Brookings, SD, USA, 2019. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Kowsari, K.; Jafari Meimandi, K.; Heidarysafa, M.; Mendu, S.; Barnes, L.; Brown, D. Text classification algorithms: A survey. Information 2019, 10, 150. [Google Scholar] [CrossRef] [Green Version]
- Bağiröz, B.; Doruk, E.; Yildiz, O. Machine Learning In Bioinformatics: Gene Expression And Microarray Studies. In Proceedings of the Medical Technologies Congress (TIPTEKNO), Antalya, Turkey, 19–20 November 2020; pp. 1–4. [Google Scholar]
- John, G.; Langley, P. Estimating Continuous Distributions in Bayesian Classifiers. In Proceedings of the Eleventh Conference on Uncertainty in Artificial Intelligence, Montréal, QC, Canada, 18–20 August 1995. [Google Scholar]
- Saritas, M.M.; Yasar, A. Performance analysis of ANN and Naive Bayes classification algorithm for data classification. Int. J. Intell. Syst. Appl. Eng. 2019, 7, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.; Shpilrain, V.; Najarian, K.; Kahrobaei, D. Private naive bayes classification of personal biomedical data: Application in cancer data analysis. Comput. Biol. Med. 2019, 105, 144–150. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees; CRC: Boca Raton, FL, USA, 1984. [Google Scholar]
- Quinlan, J.R. C4. 5: Programs for Machine Learning; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Sammut, C.; Webb, G.I. Encyclopedia of Machine Learning and Data Mining; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Barman, R.K.; Saha, S.; Das, S. Prediction of interactions between viral and host proteins using supervised machine learning methods. PLoS ONE 2014, 9, e112034. [Google Scholar]
- Eid, F.E.; ElHefnawi, M.; Heath, L.S. DeNovo: Virus-host sequence-based protein–protein interaction prediction. Bioinformatics 2016, 32, 1144–1150. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, Y.; Li, F.; Akutsu, T.; Rawlings, N.D.; Webb, G.I.; Chou, K.C. iProt-Sub: A comprehensive package for accurately mapping and predicting protease-specific substrates and cleavage sites. Briefings Bioinform. 2019, 20, 638–658. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Shekar, B.; Dagnew, G. Grid search-based hyperparameter tuning and classification of microarray cancer data. In Proceedings of the Second International Conference on Advanced Computational and Communication Paradigms (ICACCP), Gangtok, India, 25–28 February 2019; pp. 1–8. [Google Scholar]
- Le, N.Q.K.; Yapp, E.K.Y.; Ho, Q.T.; Nagasundaram, N.; Ou, Y.Y.; Yeh, H.Y. iEnhancer-5Step: Identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal. Biochem. 2019, 571, 53–61. [Google Scholar] [CrossRef]
- Asim, M.N.; Ibrahim, M.A.; Malik, M.I.; Dengel, A.; Ahmed, S. Enhancer-DSNet: A Supervisedly Prepared Enriched Sequence Representation for the Identification of Enhancers and Their Strength. In Proceedings of the International Conference on Neural Information Processing, Bangkok, Thailand, 18–22 November 2020; pp. 38–48. [Google Scholar]



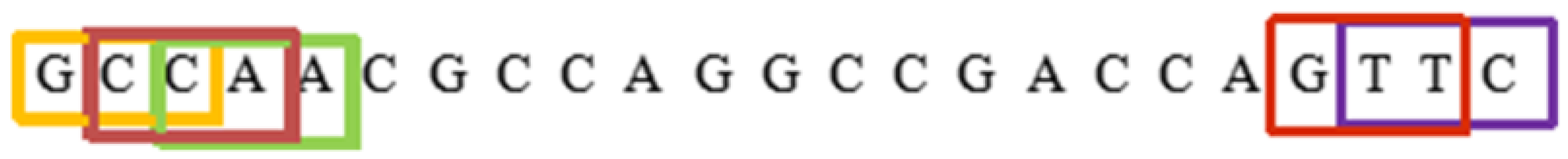


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Descriptors | RandomForest | Xgboost | Naive Bayes | SVM | Adaboost |
|---|---|---|---|---|---|
| EIIP | 0.675 | 0.663 | 0.092 | 0.403 | 0.523 |
| zCurve | 0.621 | 0.612 | 0.270 | 0.405 | 0.526 |
| triMonoKGap | 0.688 | 0.678 | 0.225 | 0.463 | 0.559 |
| diMonoKGap | 0.643 | 0.665 | 0.214 | 0.405 | 0.557 |
| Kmer | 0.685 | 0.670 | 0.606 | 0.469 | 0.490 |
| RCkmer | 0.676 | 0.651 | 0.591 | 0.489 | 0.521 |
| pseudoKNC | 0.685 | 0.658 | 0.249 | 0.446 | 0.564 |
| Sequence Descriptor | RandomForest | Xgboost | Naive Bayes | SVM | Adaboost |
|---|---|---|---|---|---|
| EIIP | 0.619 | 0.613 | 0.109 | 0.232 | 0.532 |
| zCurve | 0.585 | 0.585 | 0.304 | 0.236 | 0.525 |
| triMonoKGap | 0.634 | 0.635 | 0.260 | 0.334 | 0.561 |
| diMonoKGap | 0.602 | 0.624 | 0.248 | 0.235 | 0.562 |
| Kmer | 0.623 | 0.622 | 0.589 | 0.351 | 0.493 |
| RCkmer | 0.617 | 0.601 | 0.582 | 0.385 | 0.521 |
| pseudoKNC | 0.630 | 0.613 | 0.282 | 0.308 | 0.565 |
| Machine Learning Classifier | Best Performing K-Order Sequence Descriptor Fusion | |||||
|---|---|---|---|---|---|---|
| 2nd Order | 3rd Order | 4th Order | 5th Order | 6th Order | 7th Order | |
| Random Forest | TriMonoKGap+ PseudoKNC | RCKmer+ zCurve+ Kmer | diMonoKGap+ RCKmer+ triMonoKGap+ pseudoKNC | diMonoKGap+ RCKmer+ triMonoKGap+ pseudoKNC+ zCurve | diMonoKGap+ EIIP+ triMonoKGap+ pseudoKNC+ zCurve+Kmer | diMonoKGap, RCKmer, EIIP+ triMonoKGap+pseudoKNC+ zCurve+Kmer |
| Xgboost | Kmer+ diMonoKGap | pseudoKNC+ diMonoKGap+ RCKmer | Kmer, triMonoKGap+ zCurve+ RCKmer | Kmer+triMonoKGap+ zCurve+ diMonoKGap+ RCKmer | pseudoKNC+triMonoKGap+ EIIP+zCurve+ diMonoKGap+RCKmer | diMonoKGap+EIIP+RCKmer+ triMonoKGap+pseudoKNC + zCurve+Kmer |
| Naive Bayes | RCKmer+ Kmer | RCKmer+Kmer +pseudoKNC | RCKmer+pseudoKNC+ ZCurve+Kmer | diMonoKGap+RCKmer+ pseudoKNC+ zCurve+Kmer | diMonoKGap+RCKmer+ triMonoKGap+pseudoKNC +zCurve+Kmer | diMonoKGap+EIIP+RCKmer+ triMonoKGap+pseudoKNC + zCurve+Kmer |
| SVM | diMonoKGap+ EIIP | diMonoKGap+EIIP+ zCurve | diMonoKGap+ EIIP+RCKmer +triMonoKGap | diMonoKGap+EIIP+RCKmer +triMonoKGap+pseudoKNC | diMonoKGap+EIIP+ RCKmer+triMonoKGap+ pseudoKNC+zCurve | diMonoKGap+EIIP+RCKmer+ triMonoKGap+pseudoKNC + zCurve+Kmer |
| AdaBoost | diMonoKGap+ pseudoKNC | diMonoKGap+ pseudoKNC +zCurve | diMonoKGap+RCKmer+ triMonoKGap+zCurve | diMonoKGap+RCKmer+ pseudoKNC+ zCurve+Kmer | diMonoKGap+RCKmer+ triMonoKGap+pseudoKNC +zCurve+Kmer | diMonoKGap+EIIP+RCKmer+ triMonoKGap+pseudoKNC + zCurve+Kmer |
| Encoder Fusion | RandomForest | Xgboost | Naive Bayes | SVM | Adaboost |
|---|---|---|---|---|---|
| 2nd-order | 0.695 | 0.689 | 0.605 | 0.681 | 0.564 |
| 3rd-order | 0.693 | 0.689 | 0.249 | 0.683 | 0.576 |
| 4th-order | 0.694 | 0.688 | 0.249 | 0.682 | 0.571 |
| 5th-order | 0.687 | 0.686 | 0.247 | 0.681 | 0.562 |
| 6th-order | 0.692 | 0.684 | 0.239 | 0.674 | 0.551 |
| 7th-order | 0.683 | 0.678 | 0.221 | 0.673 | 0.531 |
| Encoder Fusion | RandomForest | Xgboost | Naive Bayes | SVM | Adaboost |
|---|---|---|---|---|---|
| 2nd-order | 0.643 | 0.637 | 0.587 | 0.621 | 0.566 |
| 3rd-order | 0.632 | 0.641 | 0.282 | 0.624 | 0.575 |
| 4th-order | 0.641 | 0.638 | 0.282 | 0.622 | 0.571 |
| 5th-order | 0.633 | 0.637 | 0.278 | 0.621 | 0.564 |
| 6th-order | 0.637 | 0.634 | 0.271 | 0.613 | 0.551 |
| 7th-order | 0.634 | 0.629 | 0.251 | 0.613 | 0.531 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asim, M.N.; Ibrahim, M.A.; Imran Malik, M.; Dengel, A.; Ahmed, S. Circ-LocNet: A Computational Framework for Circular RNA Sub-Cellular Localization Prediction. Int. J. Mol. Sci. 2022, 23, 8221. https://doi.org/10.3390/ijms23158221
Asim MN, Ibrahim MA, Imran Malik M, Dengel A, Ahmed S. Circ-LocNet: A Computational Framework for Circular RNA Sub-Cellular Localization Prediction. International Journal of Molecular Sciences. 2022; 23(15):8221. https://doi.org/10.3390/ijms23158221
Chicago/Turabian StyleAsim, Muhammad Nabeel, Muhammad Ali Ibrahim, Muhammad Imran Malik, Andreas Dengel, and Sheraz Ahmed. 2022. "Circ-LocNet: A Computational Framework for Circular RNA Sub-Cellular Localization Prediction" International Journal of Molecular Sciences 23, no. 15: 8221. https://doi.org/10.3390/ijms23158221
APA StyleAsim, M. N., Ibrahim, M. A., Imran Malik, M., Dengel, A., & Ahmed, S. (2022). Circ-LocNet: A Computational Framework for Circular RNA Sub-Cellular Localization Prediction. International Journal of Molecular Sciences, 23(15), 8221. https://doi.org/10.3390/ijms23158221