
Therapeutic Effect of Erythropoietin on Alzheimer’s Disease by Activating the Serotonin Pathway

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. EPO Improves Cognitive Memory Function in Aβ-Treated Mice
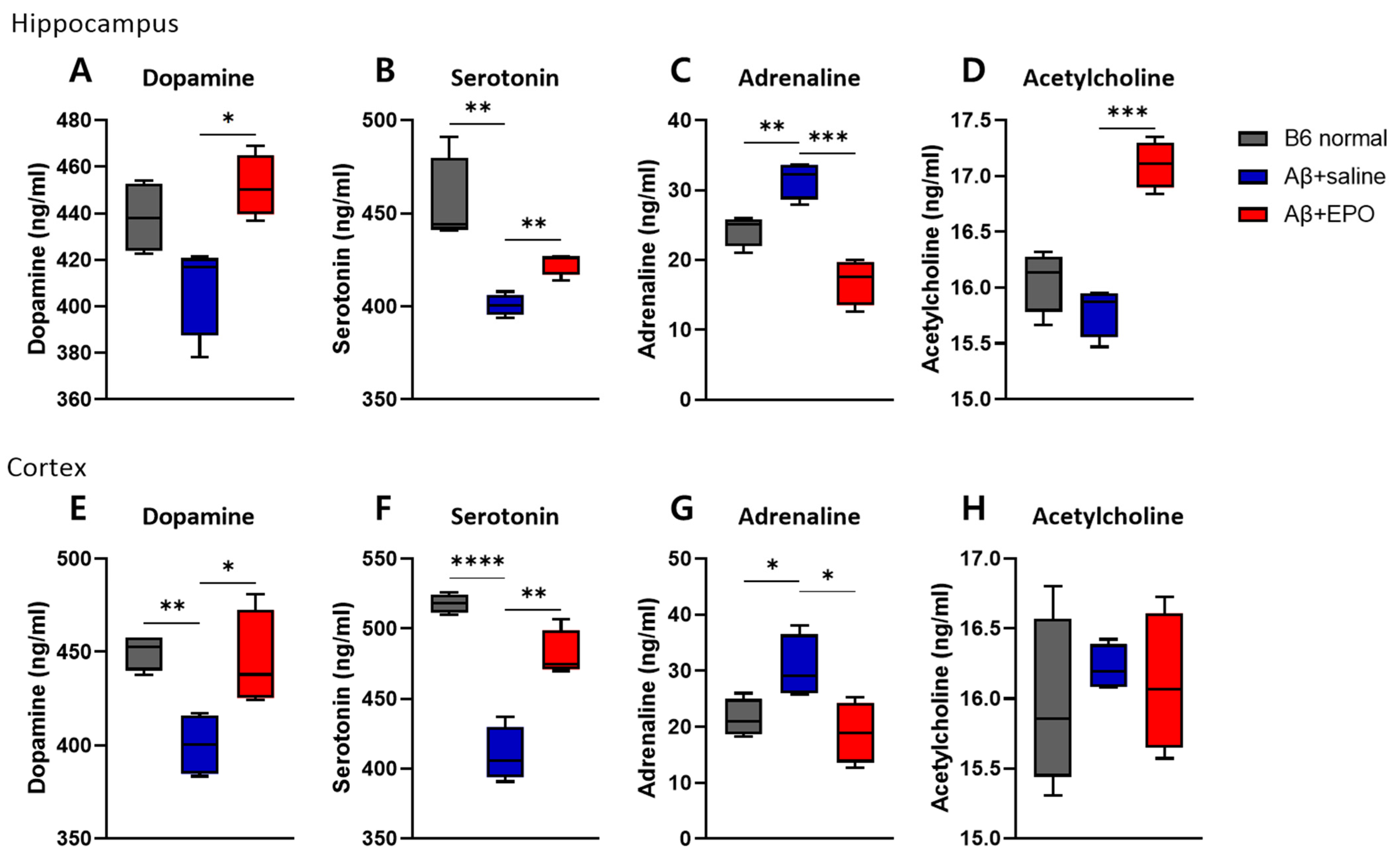
2.2. EPO Regulates Several Neurotransmitter Levels in the Brain, including the Serotonin Pathway
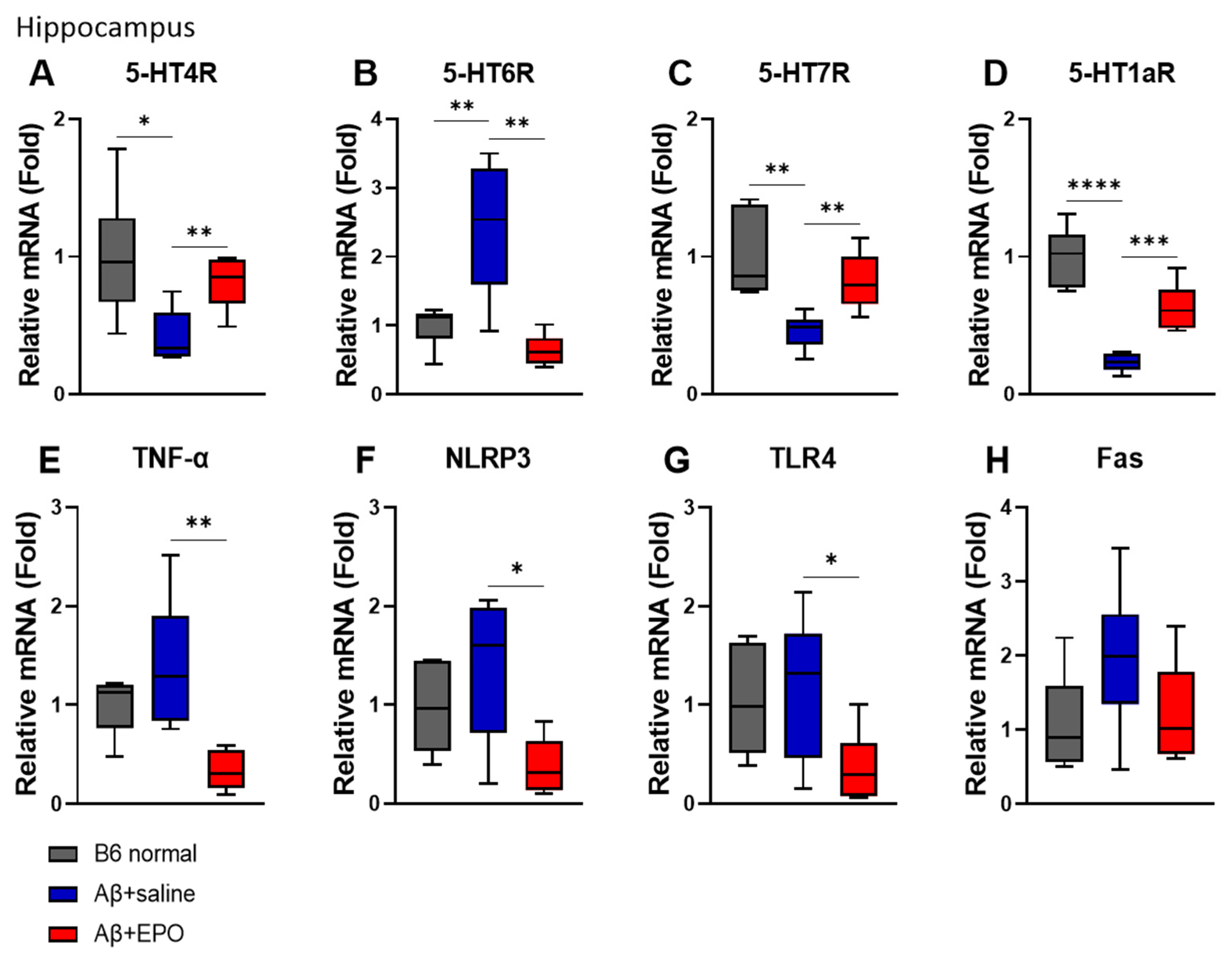
2.3. EPO Recovers Serotonergic Receptor Activities and the Neuroinflammatory Status at mRNA Levels in the Hippocampus
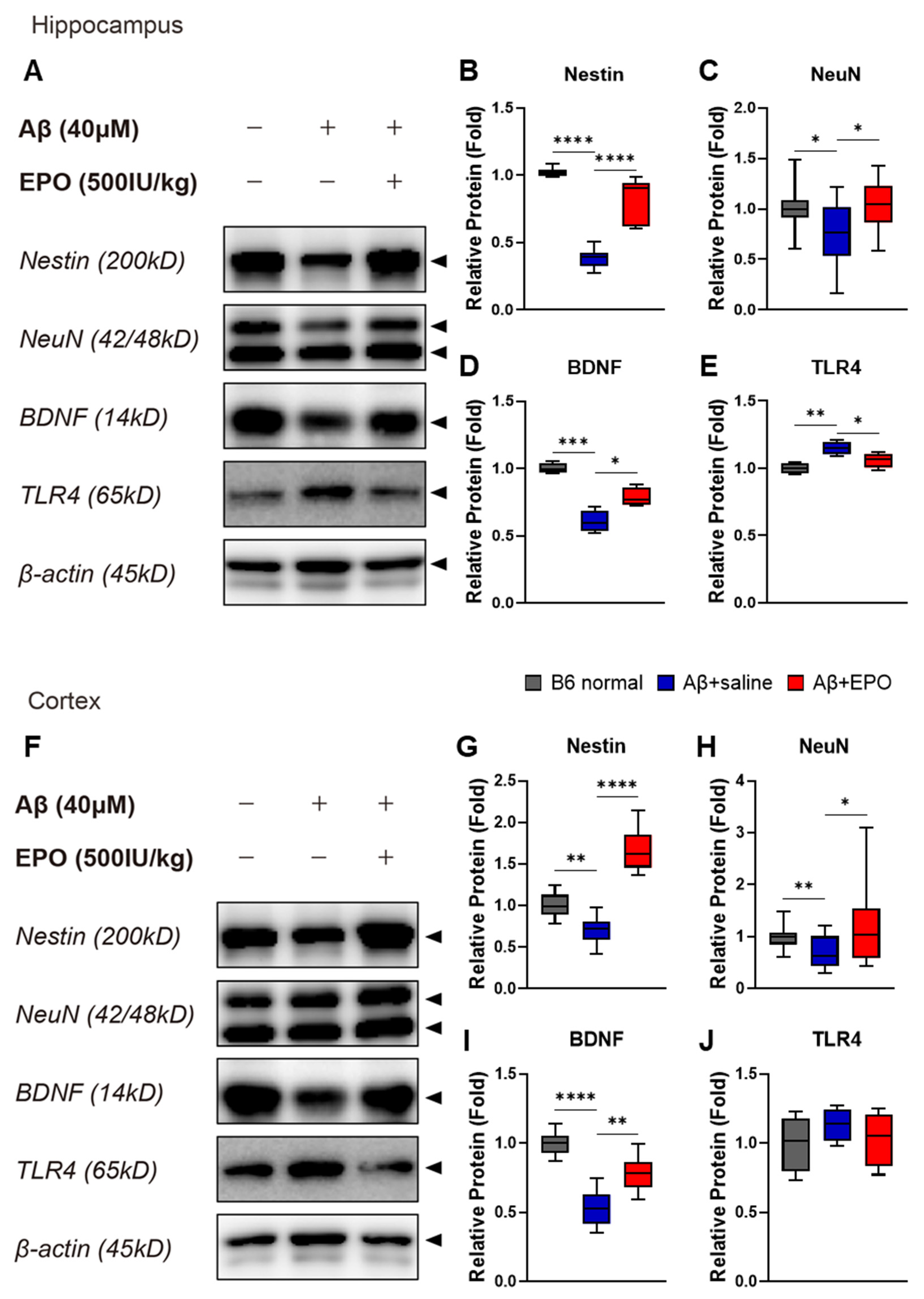
2.4. EPO Promotes Neurogenesis and Suppresses Neuroinflammation at the Protein Level
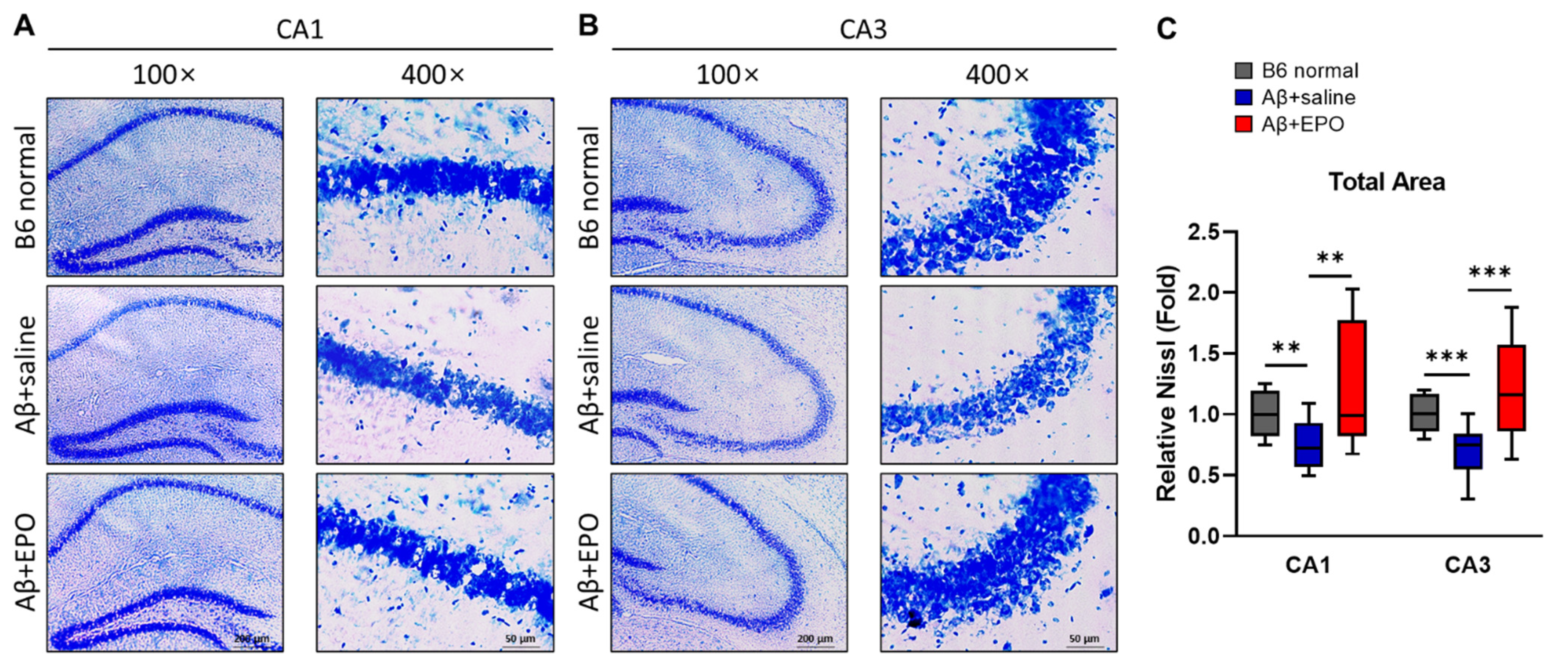
2.5. EPO Stimulates Neurogenesis in the CA1 and CA3 Areas of the Hippocampal Pyramidal Neuron Layer
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Recognition Memory Assessment Using NORT
4.3. Event-Space Memory Assessment Using the Passive Avoidance Task
4.4. Brain Tissue Preparation and ELISA Assay
4.5. Western Blot Analysis
4.6. Quantitative Real-Time PCR
4.7. Tissue Preparation for Cresyl Violet Staining and Photomicrograph
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Chang, R.; Al Maghribi, A.; Vanderpoel, V.; Vasilevko, V.; Cribbs, D.H.; Boado, R.; Pardridge, W.M.; Sumbria, R.K. Brain Penetrating Bifunctional Erythropoietin-Transferrin Receptor Antibody Fusion Protein for Alzheimer’s Disease. Mol. Pharm. 2018, 15, 4963–4973. [Google Scholar] [CrossRef]
- Maurice, T.; Mustafa, M.H.; Desrumaux, C.; Keller, E.; Naert, G.; de la, C.G.-B.M.; Rodriguez Cruz, Y.; Garcia Rodriguez, J.C. Intranasal formulation of erythropoietin (EPO) showed potent protective activity against amyloid toxicity in the Abeta(2)(5)(-)(3)(5) non-transgenic mouse model of Alzheimer’s disease. J. Psychopharmacol. 2013, 27, 1044–1057. [Google Scholar] [CrossRef]
- Arabpoor, Z.; Hamidi, G.; Rashidi, B.; Shabrang, M.; Alaei, H.; Sharifi, M.R.; Salami, M.; Dolatabadi, H.R.; Reisi, P. Erythropoietin improves neuronal proliferation in dentate gyrus of hippocampal formation in an animal model of Alzheimer’s disease. Adv. Biomed. Res. 2012, 1, 50. [Google Scholar] [CrossRef]
- Li, Y.P.; Yang, G.J.; Jin, L.; Yang, H.M.; Chen, J.; Chai, G.S.; Wang, L. Erythropoietin attenuates Alzheimer-like memory impairments and pathological changes induced by amyloid beta42 in mice. Brain Res. 2015, 1618, 159–167. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Park, J.E.; Jung, K.H.; Jeon, D.; Lim, J.Y.; Lee, S.K.; Kim, M.; Roh, J.K. Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-beta burden in Alzheimer’s disease models. J. Neurochem. 2012, 120, 115–124. [Google Scholar] [CrossRef]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassouna, I.; Ott, C.; Wustefeld, L.; Offen, N.; Neher, R.A.; Mitkovski, M.; Winkler, D.; Sperling, S.; Fries, L.; Goebbels, S.; et al. Revisiting adult neurogenesis and the role of erythropoietin for neuronal and oligodendroglial differentiation in the hippocampus. Mol. Psychiatry 2016, 21, 1752–1767. [Google Scholar] [CrossRef] [PubMed]
- Vitellaro-Zuccarello, L.; Mazzetti, S.; Madaschi, L.; Bosisio, P.; Gorio, A.; De Biasi, S. Erythropoietin-mediated preservation of the white matter in rat spinal cord injury. Neuroscience 2007, 144, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Son, H. Effects of serotonin on erythropoietin expression in mouse hippocampus. Exp. Neurobiol. 2013, 22, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Svob Strac, D.; Pivac, N.; Muck-Seler, D. The serotonergic system and cognitive function. Transl. Neurosci. 2016, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Roux, C.M.; Leger, M.; Freret, T. Memory Disorders Related to Hippocampal Function: The Interest of 5-HT4Rs Targeting. Int. J. Mol. Sci. 2021, 22, 12082. [Google Scholar] [CrossRef]
- Perez-Garcia, G.S.; Meneses, A. Effects of the potential 5-HT7 receptor agonist AS 19 in an autoshaping learning task. Behav. Brain Res. 2005, 163, 136–140. [Google Scholar] [CrossRef]
- Radley, J.J.; Jacobs, B.L. 5-HT1A receptor antagonist administration decreases cell proliferation in the dentate gyrus. Brain Res. 2002, 955, 264–267. [Google Scholar] [CrossRef]
- Bali, A.; Singh, S. Serotonergic 5-HT6 Receptor Antagonists: Heterocyclic Chemistry and Potential Therapeutic Significance. Curr. Top. Med. Chem. 2015, 15, 1643–1662. [Google Scholar] [CrossRef]
- Marjanska, M.; Curran, G.L.; Wengenack, T.M.; Henry, P.G.; Bliss, R.L.; Poduslo, J.F.; Jack, C.R.J.; Ugurbil, K.; Garwood, M. Monitoring disease progression in transgenic mouse models of Alzheimer’s disease with proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 11906–11910. [Google Scholar] [CrossRef] [Green Version]
- Haam, J.; Yakel, J.L. Cholinergic modulation of the hippocampal region and memory function. J. Neurochem. 2017, 142, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shohayeb, B.; Diab, M.; Ahmed, M.; Ng, D.C.H. Factors that influence adult neurogenesis as potential therapy. Transl. Neurodegener. 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Claeysen, S.; Bockaert, J.; Giannoni, P. Serotonin: A New Hope in Alzheimer’s Disease? ACS Chem. Neurosci. 2015, 6, 940–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiese, K.; Chong, Z.Z.; Li, F.; Shang, Y.C. Erythropoietin: Elucidating new cellular targets that broaden therapeutic strategies. Prog. Neurobiol. 2008, 85, 194–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1999, 19, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Chu, K.; Sinn, D.I.; Jung, K.H.; Kim, E.H.; Kim, S.J.; Kim, J.M.; Ko, S.Y.; Kim, M.; Roh, J.K. Erythropoietin reduces perihematomal inflammation and cell death with eNOS and STAT3 activations in experimental intracerebral hemorrhage. J. Neurochem. 2006, 96, 1728–1739. [Google Scholar] [CrossRef]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef] [Green Version]
- Robertson, C.S.; Hannay, H.J.; Yamal, J.M.; Gopinath, S.; Goodman, J.C.; Tilley, B.C.; Epo Severe, T.B.I.T.I.; Baldwin, A.; Rivera Lara, L.; Saucedo-Crespo, H.; et al. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: A randomized clinical trial. JAMA 2014, 312, 36–47. [Google Scholar] [CrossRef]
- Sun, J.; Martin, J.M.; Vanderpoel, V.; Sumbria, R.K. The Promises and Challenges of Erythropoietin for Treatment of Alzheimer’s Disease. Neuromol. Med. 2019, 21, 12–24. [Google Scholar] [CrossRef]
- Hooshmandi, E.; Motamedi, F.; Moosavi, M.; Katinger, H.; Zakeri, Z.; Zaringhalam, J.; Maghsoudi, A.; Ghasemi, R.; Maghsoudi, N. CEPO-Fc (An EPO Derivative) Protects Hippocampus Against Abeta-induced Memory Deterioration: A Behavioral and Molecular Study in a Rat Model of Abeta Toxicity. Neuroscience 2018, 388, 405–417. [Google Scholar] [CrossRef]
- Choung, J.S.; Kim, J.M.; Ko, M.H.; Cho, D.S.; Kim, M. Therapeutic efficacy of repetitive transcranial magnetic stimulation in an animal model of Alzheimer’s disease. Sci. Rep. 2021, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Xu, M.; Gan, J.; Yang, X.; Wu, N.; Song, L.; Yuan, W.; Liu, Z. Erythropoietin improves neurobehavior by reducing dopaminergic neuron loss in a 6hydroxydopamineinduced rat model. Int. J. Mol. Med. 2014, 34, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Koshimura, K.; Kawaguchi, M.; Sohmiya, M.; Murakami, Y.; Kato, Y. Stimulating effect of erythropoietin on the release of dopamine and acetylcholine from the rat brain slice. Neurosci. Lett. 2000, 292, 131–133. [Google Scholar] [CrossRef]
- Meltzer, C.C.; Smith, G.; DeKosky, S.T.; Pollock, B.G.; Mathis, C.A.; Moore, R.Y.; Kupfer, D.J.; Reynolds, C.F., 3rd. Serotonin in aging, late-life depression, and Alzheimer’s disease: The emerging role of functional imaging. Neuropsychopharmacology 1998, 18, 407–430. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, G.P.; Mason, S.L.; Meldrum, A.; De Keczer, S.; Parnes, H.; Eglen, R.M.; Wong, E.H. 5-Hydroxytryptamine (5-HT)4 receptors in post mortem human brain tissue: Distribution, pharmacology and effects of neurodegenerative diseases. Br. J. Pharm. 1995, 114, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Bockaert, J.; Claeysen, S.; Compan, V.; Dumuis, A. 5-HT(4) receptors: History, molecular pharmacology and brain functions. Neuropharmacology 2008, 55, 922–931. [Google Scholar] [CrossRef]
- Klempin, F.; Babu, H.; De Pietri Tonelli, D.; Alarcon, E.; Fabel, K.; Kempermann, G. Oppositional effects of serotonin receptors 5-HT1a, 2, and 2c in the regulation of adult hippocampal neurogenesis. Front Mol. Neurosci. 2010, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Choi, J.; Kim, M. Combining Human Umbilical Cord Blood Cells With Erythropoietin Enhances Angiogenesis/Neurogenesis and Behavioral Recovery After Stroke. Front Neurol. 2019, 10, 357. [Google Scholar] [CrossRef]
- Lazarov, O.; Marr, R.A. Neurogenesis and Alzheimer’s disease: At the crossroads. Exp. Neurol. 2010, 223, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Brazo, J.; Castro, E.; Diaz, A.; Valdizan, E.M.; Pilar-Cuellar, F.; Vidal, R.; Treceno, B.; Pazos, A. Modulation of neuroplasticity pathways and antidepressant-like behavioural responses following the short-term (3 and 7 days) administration of the 5-HT(4) receptor agonist RS67333. Int. J. Neuropsychopharmacol. 2012, 15, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugolini, F.; Lana, D.; Nardiello, P.; Nosi, D.; Pantano, D.; Casamenti, F.; Giovannini, M.G. Different Patterns of Neurodegeneration and Glia Activation in CA1 and CA3 Hippocampal Regions of TgCRND8 Mice. Front. Aging Neurosci. 2018, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Gambuzza, M.E.; Sofo, V.; Salmeri, F.M.; Soraci, L.; Marino, S.; Bramanti, P. Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol. Disord. Drug Targets 2014, 13, 1542–1558. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Garcia-Rodriguez, C.; Villalobos, C.; Nunez, L. Role of Toll Like Receptor 4 in Alzheimer’s Disease. Front Immunol. 2020, 11, 1588. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.K.; Chung, B.R.; Kim, H.V.; Kim, Y. Intracerebroventricular Injection of Amyloid-beta Peptides in Normal Mice to Acutely Induce Alzheimer-like Cognitive Deficits. J. Vis. Exp. 2016, 109, 53308. [Google Scholar] [CrossRef] [Green Version]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Processing 2012, 13, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Sun, D. Neurobehavioral Assessments of Focal Cerebral Ischemia: Cognitive Deficit. In Animal Models of Acute Neurological Injuries II; Chen, J., Xu, X.-M., Xu, Z.C., Zhang, J.H., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 157–162. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | ELISA Product | Manufacturer, #Catalog No. |
|---|---|---|
| Dopamine | Mouse Dopamine (Competitive EIA) ELISA Kit | LSBio, Seattle, WA, USA #LS-F28308 |
| Serotonin | Mouse Serotonin (Competitive EIA) ELISA Kit | LSBio, Seattle, WA, USA #LS-F28406 |
| Adrenaline/Epinephrine | Mouse Adrenaline / Epinephrine (Competitive EIA) ELISA Kit | LSBio, Seattle, WA, USA #LS-F28134 |
| Acetylcholine | Mouse Acetylcholine (Competitive EIA) ELISA Kit | LSBio, Seattle, WA, USA #LS-F28199 |
| Target Protein | Name of Antibody | Host Animal | Manufacturer, #Catalog No. | Dilution for Western Blot |
|---|---|---|---|---|
| Nestin | Nestin antibody [2Q178] | Mouse, monoclonal | Abcam, Cambridge, England #ab6142 | 1:1000 |
| NeuN | NeuN antibody | Rabbit, polyclonal | Merck Millipore, Darmstadt, Germany #ABN78 | 1:1000 |
| BDNF | BDNF antibody | Rabbit, polyclonal | Invitrogen, Waltham, MA, USA #PA5-85730 | 1:1000 |
| TLR4 | TLR4 antibody | Rabbit, polyclonal | Novus Biologicals, Centennial, CO, USA #NBP1-78427 | 1:1000 |
| β-actin | β-actin (C4) antibody | Mouse, monoclonal | Santa Cruz, Dallas, TX, USA #sc-47778 | 1:1000 |
| Rabbit | rabbit IgG, HRP-linked antibody | Goat | Cell Signaling, Danvers, MA, USA #7074 | 1:10,000 |
| Mouse | mouse IgG, HRP-linked antibody | Horse | Cell Signaling, Danvers, MA, USA #7076 | 1:20,000 |
| Gene | Gene Bank Number | Primer | Sequence (5′–3′) | Annealing Temperature (°C) | Product Size (bp) |
|---|---|---|---|---|---|
| 5-HT4R | NM_008313 | Forward | TGCTCACGTTCCTTGCAGTGGT | 60 | 134 |
| Reverse | GTCAGCAAAGGCGAGAGACACA | ||||
| 5-HT6R | NM_021358 | Forward | TGCCATCTGCTTCACCTACTGC | 60 | 139 |
| Reverse | CTACTGTCAGCAGACTCCATCC | ||||
| 5-HT7R | NM_008315 | Forward | TCATGACCCTGTGCGTGATCAG | 60 | 119 |
| Reverse | GAGAAGCCAGACCGACAGAATC | ||||
| 5-HT1aR | NM_008308 | Forward | TGCCAACTATCTCATCGGCTCC | 60 | 103 |
| Reverse | CAGAGTCCACTTGTTGAGCACC | ||||
| FAS | NM_007987 | Forward | TATCAAGGAGGCCCATTTTGC | 60 | 195 |
| Reverse | TGTTTCCACCTCTAAACCATGCT | ||||
| NLRP3 | NM_004895 | Forward | GGACTGAAGCACCTGTTGTGCA | 60 | 153 |
| Reverse | TCCTGAGTCTCCCAAGGCATTC | ||||
| TLR4 | NM_138554 | Forward | CCCTGAGGCATTTAGGCAGCTA | 60 | 126 |
| Reverse | AGGTAGAGAGGTGGCTTAGGCT | ||||
| TNF | NM_013693 | Forward | CCAACGGCATGGATCTCAAAGACA | 60 | 141 |
| Reverse | AGATAGCAAATCGGCTGACGGTGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, K.-H.; Ha, S.; Choung, J.S.; Choi, J.I.; Kim, D.Y.; Kim, J.M.; Kim, M. Therapeutic Effect of Erythropoietin on Alzheimer’s Disease by Activating the Serotonin Pathway. Int. J. Mol. Sci. 2022, 23, 8144. https://doi.org/10.3390/ijms23158144
Shim K-H, Ha S, Choung JS, Choi JI, Kim DY, Kim JM, Kim M. Therapeutic Effect of Erythropoietin on Alzheimer’s Disease by Activating the Serotonin Pathway. International Journal of Molecular Sciences. 2022; 23(15):8144. https://doi.org/10.3390/ijms23158144
Chicago/Turabian StyleShim, Kyu-Ho, Sungchan Ha, Jin Seung Choung, Jee In Choi, Daniel Youngsuk Kim, Jong Moon Kim, and MinYoung Kim. 2022. "Therapeutic Effect of Erythropoietin on Alzheimer’s Disease by Activating the Serotonin Pathway" International Journal of Molecular Sciences 23, no. 15: 8144. https://doi.org/10.3390/ijms23158144
APA StyleShim, K.-H., Ha, S., Choung, J. S., Choi, J. I., Kim, D. Y., Kim, J. M., & Kim, M. (2022). Therapeutic Effect of Erythropoietin on Alzheimer’s Disease by Activating the Serotonin Pathway. International Journal of Molecular Sciences, 23(15), 8144. https://doi.org/10.3390/ijms23158144