
HPV Integration Site Mapping: A Rapid Method of Viral Integration Site (VIS) Analysis and Visualization Using Automated Workflows in CLC Microbial Genomics


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Viral Hybrid Capture (VHC) Analysis and Visualization
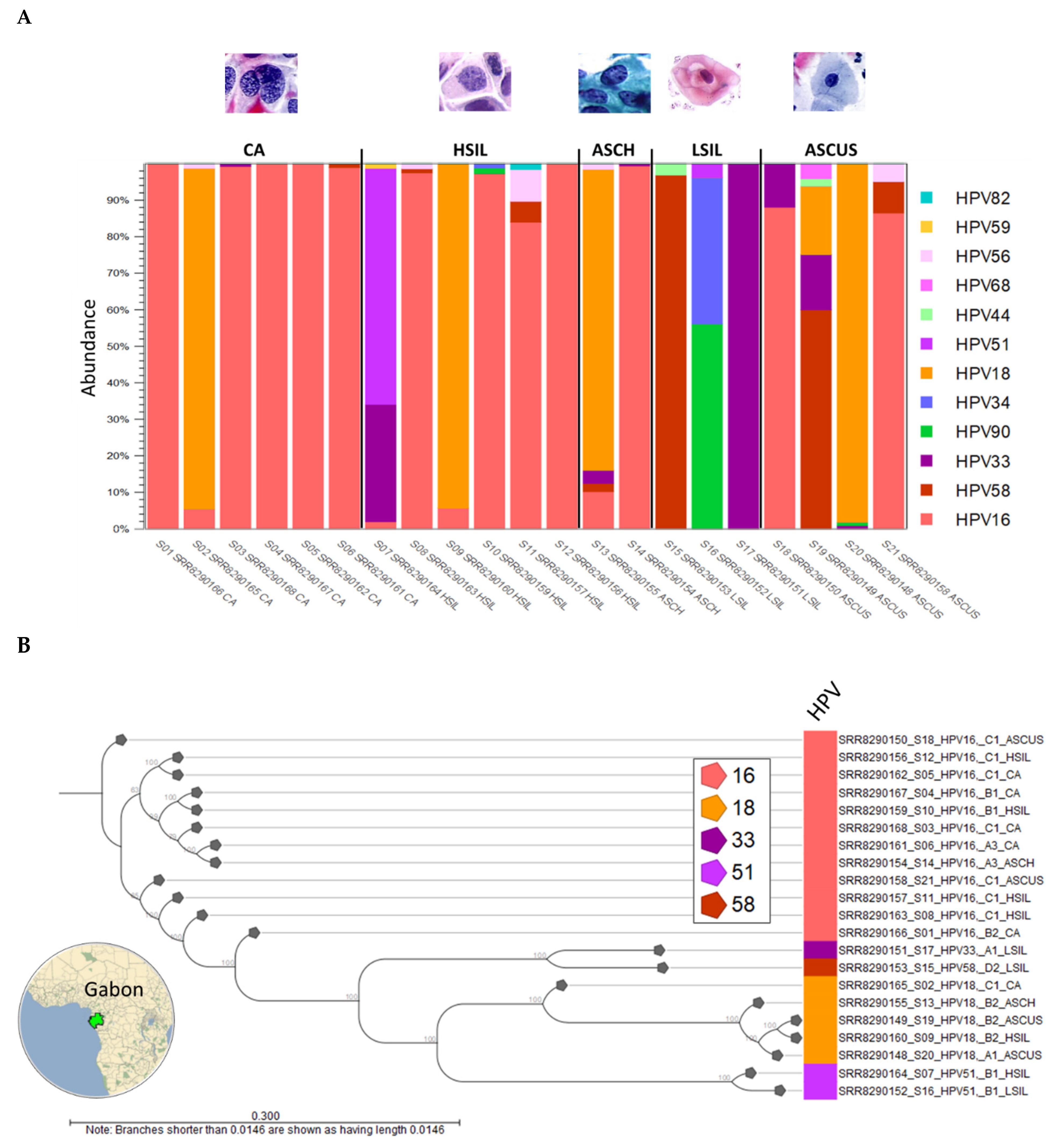
2.1.1. HPV Taxonomic Profiling
2.1.2. HPV Sublineages and Phylogenetic Tree
2.1.3. Viral Hybrid Capture (VHC) Tracklists
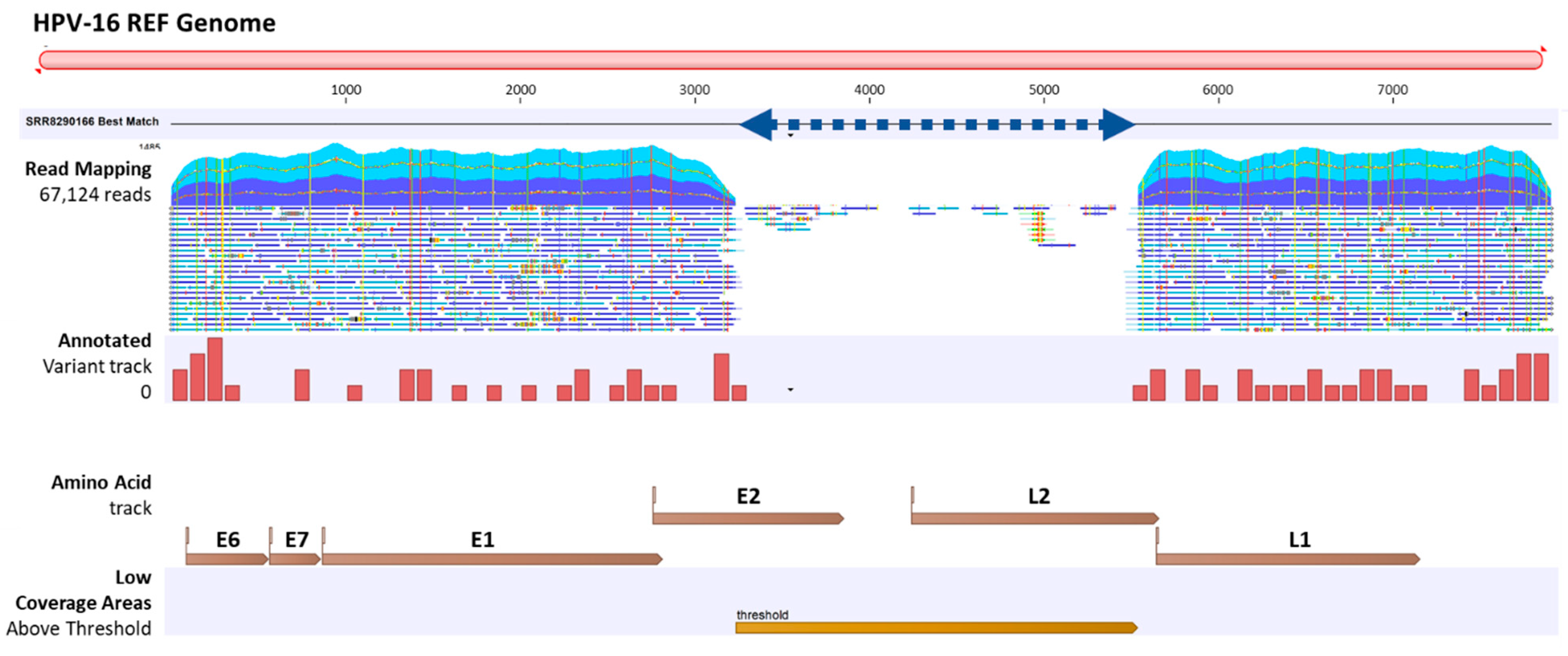
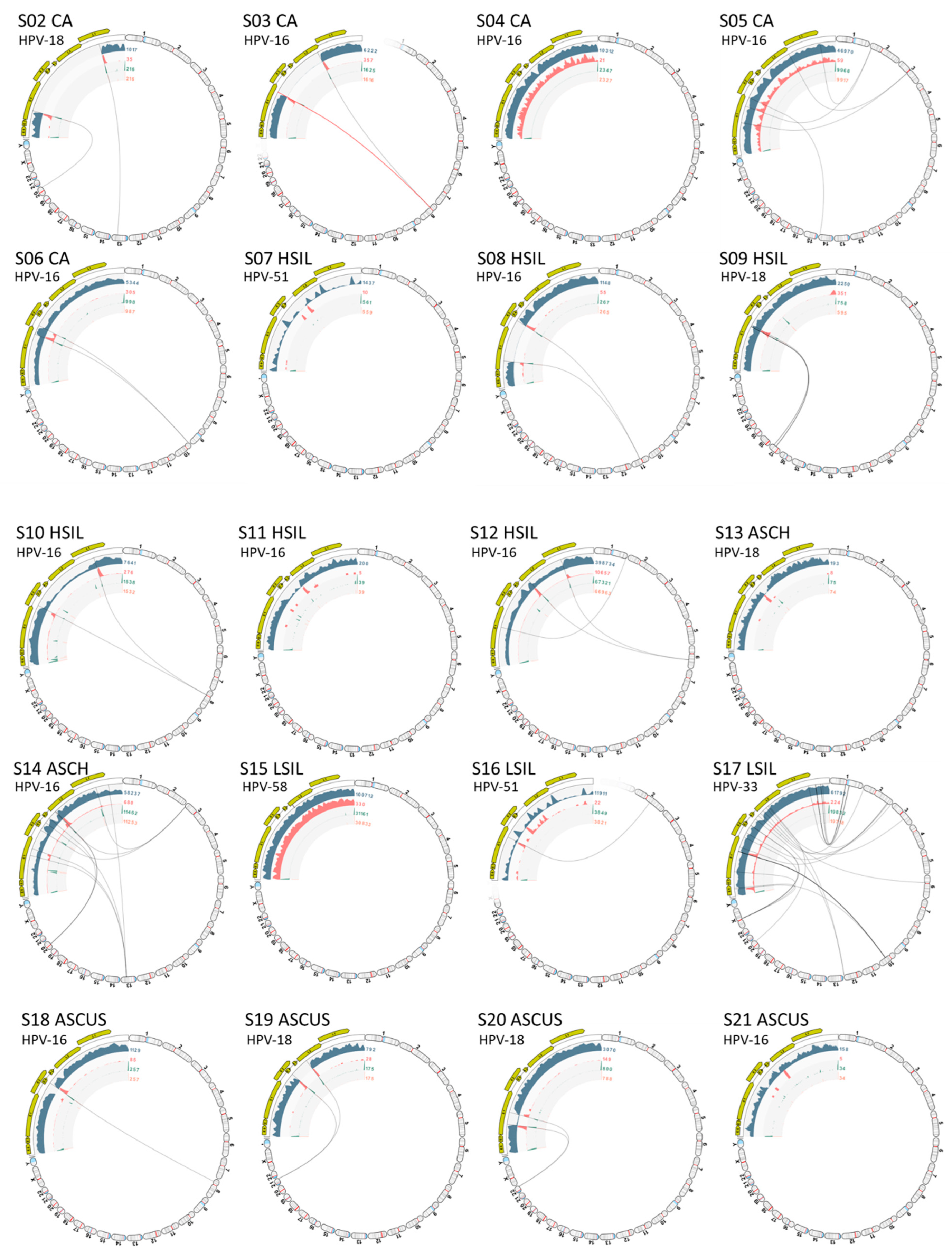
2.2. Viral Integration Site (VIS) Analysis and Visualization
2.3. Workflow Runtimes
3. Discussion
4. Materials and Methods
4.1. NGS Dataset of HPV-Positive Cytology Samples
4.2. Customized HPV Reference Database and Human Reference Genome for CLC Workflows and Tools
4.3. Viral Hybrid Capture Analysis and Workflow
4.4. Viral Integration Site (VIS) Analysis and Workflow
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020; Available online: http://publications.iarc.fr/586 (accessed on 6 June 2022).
- IARC. Cervical Cancer Screening. IARC Handb. Cancer Prev. 2022, 18, 1–456. Available online: https://publications.iarc.fr/604 (accessed on 6 June 2022).
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Hausen, H.Z. Cancers in Humans: A Lifelong Search for Contributions of Infectious Agents, Autobiographic Notes. Annu. Rev. Virol. 2019, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; Hausen, H.Z. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Vinokurova, S.; Doeberitz, M.V.K. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004, 64, 3878–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemsen, A.; Bravo, I.G. Origin and evolution of papillomavirus (onco)genes and genomes. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.A.; Sakakibara, N.; Stepp, W.H.; Jang, M.K. Hitchhiking on host chromatin: How papillomaviruses persist. Biochim. Biophys. Acta 2012, 1819, 820–825. [Google Scholar] [CrossRef] [Green Version]
- Warburton, A.; Della Fera, A.N.; McBride, A.A. Dangerous Liaisons: Long-Term Replication with an Extrachromosomal HPV Genome. Viruses 2021, 13, 1846. [Google Scholar] [CrossRef]
- Coursey, T.L.; McBride, A.A. Hitchhiking of Viral Genomes on Cellular Chromosomes. Annu. Rev. Virol. 2019, 6, 275–296. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakogiannis, D.; Kyriakopoulou, Z.; Ruether, I.G.A.; Amoutzias, G.; Dimitriou, T.; Diamantidou, V.; Kotsovassilis, C.; Markoulatos, P. Determination of human papillomavirus 16 physical status through E1/E6 and E2/E6 ratio analysis. J. Med. Microbiol. 2014, 63 Pt 12, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Kahla, S.; Kochbati, L.; Chanoufi, M.B.; Maalej, M.; Oueslati, R. HPV-16 E2 physical status and molecular evolution in vivo in cervical carcinomas. Int. J. Biol. Markers 2014, 29, e78–e85. [Google Scholar] [CrossRef] [PubMed]
- Della Fera, A.N.; Warburton, A.; Coursey, T.L.; Khurana, S.; McBride, A.A. Persistent Human Papillomavirus Infection. Viruses 2021, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Murthy, S.; Kapoor, A. Evolution of selective-sequencing approaches for virus discovery and virome analysis. Virus Res. 2017, 239, 172–179. [Google Scholar] [CrossRef]
- Fitzpatrick, A.H.; Rupnik, A.; O’Shea, H.; Crispie, F.; Keaveney, S.; Cotter, P. High Throughput Sequencing for the Detection and Characterization of RNA Viruses. Front. Microbiol. 2021, 12, 621719. [Google Scholar] [CrossRef]
- Agilent: Sure Select Custom DNA Target Enrichment Probes. Available online: https://www.agilent.com/en/product/next-generation-sequencing/hybridization-based-next-generation-sequencing-ngs/ngs-custom-target-enrichment-probes/ngs-custom-target-enrichment-probes-232874 (accessed on 4 June 2022).
- Illumina: Target Enrichment. Available online: https://www.illumina.com/techniques/sequencing/dna-sequencing/targeted-resequencing/target-enrichment.html (accessed on 4 June 2022).
- Roche: Target Enrichment. Available online: https://sequencing.roche.com/en-us/products-solutions/by-category/target-enrichment.html (accessed on 4 June 2022).
- Qiagen: QIAseq xHYB Viral STI Panel. Available online: https://www.qiagen.com/us/products/next-generation-sequencing/metagenomics/targeted-metagenomics/qiaseq-xhyb-viral-and-bacterial-panels/ (accessed on 4 June 2022).
- Meyong, A.A.N.; Boundzanga, P.M.; Labouba, I.; Koumakpayi, I.H.; Jeannot, E.; Declère, S.D.; Garau, X.S.; Leroy, E.M.; Belembaogo, E.; Berthet, N. Genome-wide profiling of human papillomavirus DNA integration in liquid-based cytology specimens from a Gabonese female population using HPV capture technology. Sci. Rep. 2019, 9, 1504. [Google Scholar] [CrossRef]
- Bs, L.M.P.; Gu, W.; Wang, Y.; Bs, A.E.; Ms, A.D.B.; Ba, C.V.B.; Carey, T.E.; Mills, R.E.; Brenner, J.C. SearcHPV: A novel approach to identify and assemble human papillomavirus-host genomic integration events in cancer. Cancer 2021, 127, 3531–3540. [Google Scholar] [CrossRef]
- Mainguené, J.; Vacher, S.; Kamal, M.; Hamza, A.; Masliah-Planchon, J.; Baulande, S.; Ibadioune, S.; Borcoman, E.; Cacheux, W.; Calugaru, V.; et al. Human papilloma virus integration sites and genomic signatures in head and neck squamous cell carcinoma. Mol. Oncol. 2022. [Google Scholar] [CrossRef]
- Reynolds, C.J.; Wyatt, J.C. Open source, open standards, and health care information systems. J. Med. Internet Res. 2011, 13, e24. [Google Scholar] [CrossRef]
- Muir, P.; Li, S.; Lou, S.; Wang, D.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 53, Erratum in Genome Biol. 2016, 17, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DoD Issuances: DoDI 8500.01 Cybersecurity. Available online: https://www.esd.whs.mil/Directives/issuances/dodi/ (accessed on 6 June 2022).
- Qiagen Digital Insights. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-microbial-genomics-module/ (accessed on 4 June 2022).
- Gunther, J.S.; Wang, Y.; Lai, Z.; Poage, G.M.; Perez, L.; Huang, T.H.M. Deep sequencing of HPV E6/E7 genes reveals loss of genotypic diversity and gain of clonal dominance in high-grade intraepithelial lesions of the cervix. BMC Genom. 2017, 18, 231. [Google Scholar] [CrossRef] [Green Version]
- International Agency for Research on Cancer: Cytopathology of the Uterine Cervix—Digital Atlas. Available online: https://screening.iarc.fr/atlascyto.php (accessed on 4 June 2022).
- Clifford, G.M.; Tenet, V.; Georges, D.; Alemany, L.; Pavón, M.A.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Bass, S.; et al. Human papillomavirus 16 sub-lineage dispersal and cervical cancer risk worldwide: Whole viral genome sequences from 7116 HPV16-positive women. Papillomavirus Res. 2019, 7, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Harlé, A.; Guillet, J.; Thomas, J.; Demange, J.; Dolivet, G.; Peiffert, D.; Leroux, A.; Garau, X.S. HPV insertional pattern as a personalized tumor marker for the optimized tumor diagnosis and follow-up of patients with HPV-associated carcinomas: A case report. BMC Cancer 2019, 19, 277. [Google Scholar] [CrossRef]
- McEllin, B.; Searle, B.C.; DePledge, L.; Sun, G.; Cobbs, C.; Karimi, M. Detection of Human Papillomavirus Integration in Brain Metastases from Oropharyngeal Tumors by Targeted Sequencing. Viruses 2021, 13, 1536. [Google Scholar] [CrossRef]
- Camarero, S.C.; Segura, P.P. Liquid Biopsy in Head and Neck Cancer: Current Evidence and Future Perspective on Squamous Cell, Salivary Gland, Paranasal Sinus and Nasopharyngeal Cancers. Cancers 2022, 14, 2858. [Google Scholar] [CrossRef]
- European Nucleotide Archive. Available online: https://www.ebi.ac.uk/ena/browser/text-search?query=SUB4880803 (accessed on 4 June 2022).
- Gunther, J.S.; Xia, Q.; Cai, H.; Wang, Y. HPV DeepSeq: An Ultra-Fast Method of NGS Data Analysis and Visualization Using Automated Workflows and a Customized Papillomavirus Database in CLC Genomics Workbench. Pathogens 2021, 10, 1026. [Google Scholar] [CrossRef]
- NIAID PaVE: The Paillomavirus Episteme. Available online: https://pave.niaid.nih.gov/explore/variants/variant_nomenclature (accessed on 4 June 2022).
- CLC Qiagen Digital Insights. Available online: https://digitalinsights.qiagen.com/technical-support/system-requirements/ (accessed on 18 July 2022).
- How to BLAST Guide National Center for Biotechnology Information. Available online: https://ftp.ncbi.nlm.nih.gov/pub/factsheets/HowTo_BLASTGuide.pdf (accessed on 6 June 2022).





 ); (B) VHC workflow steps (1–10) with user-defined parameter settings for Taxonomic Profiling (bold), i.e., HPV reference index and Host genome index; (C) VIS workflow steps (1–4) with selected HPV and Host reference genomes and user-defined search parameters entered for this study.
); (B) VHC workflow steps (1–10) with user-defined parameter settings for Taxonomic Profiling (bold), i.e., HPV reference index and Host genome index; (C) VIS workflow steps (1–4) with selected HPV and Host reference genomes and user-defined search parameters entered for this study.
); (B) VHC workflow steps (1–10) with user-defined parameter settings for Taxonomic Profiling (bold), i.e., HPV reference index and Host genome index; (C) VIS workflow steps (1–4) with selected HPV and Host reference genomes and user-defined search parameters entered for this study.
); (B) VHC workflow steps (1–10) with user-defined parameter settings for Taxonomic Profiling (bold), i.e., HPV reference index and Host genome index; (C) VIS workflow steps (1–4) with selected HPV and Host reference genomes and user-defined search parameters entered for this study.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen-Gunther, J.; Cai, H.; Wang, Y. HPV Integration Site Mapping: A Rapid Method of Viral Integration Site (VIS) Analysis and Visualization Using Automated Workflows in CLC Microbial Genomics. Int. J. Mol. Sci. 2022, 23, 8132. https://doi.org/10.3390/ijms23158132
Shen-Gunther J, Cai H, Wang Y. HPV Integration Site Mapping: A Rapid Method of Viral Integration Site (VIS) Analysis and Visualization Using Automated Workflows in CLC Microbial Genomics. International Journal of Molecular Sciences. 2022; 23(15):8132. https://doi.org/10.3390/ijms23158132
Chicago/Turabian StyleShen-Gunther, Jane, Hong Cai, and Yufeng Wang. 2022. "HPV Integration Site Mapping: A Rapid Method of Viral Integration Site (VIS) Analysis and Visualization Using Automated Workflows in CLC Microbial Genomics" International Journal of Molecular Sciences 23, no. 15: 8132. https://doi.org/10.3390/ijms23158132
APA StyleShen-Gunther, J., Cai, H., & Wang, Y. (2022). HPV Integration Site Mapping: A Rapid Method of Viral Integration Site (VIS) Analysis and Visualization Using Automated Workflows in CLC Microbial Genomics. International Journal of Molecular Sciences, 23(15), 8132. https://doi.org/10.3390/ijms23158132