
Effects of Inhaled Corticosteroids on the Innate Immunological Response to Pseudomonas aeruginosa Infection in Patients with COPD

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
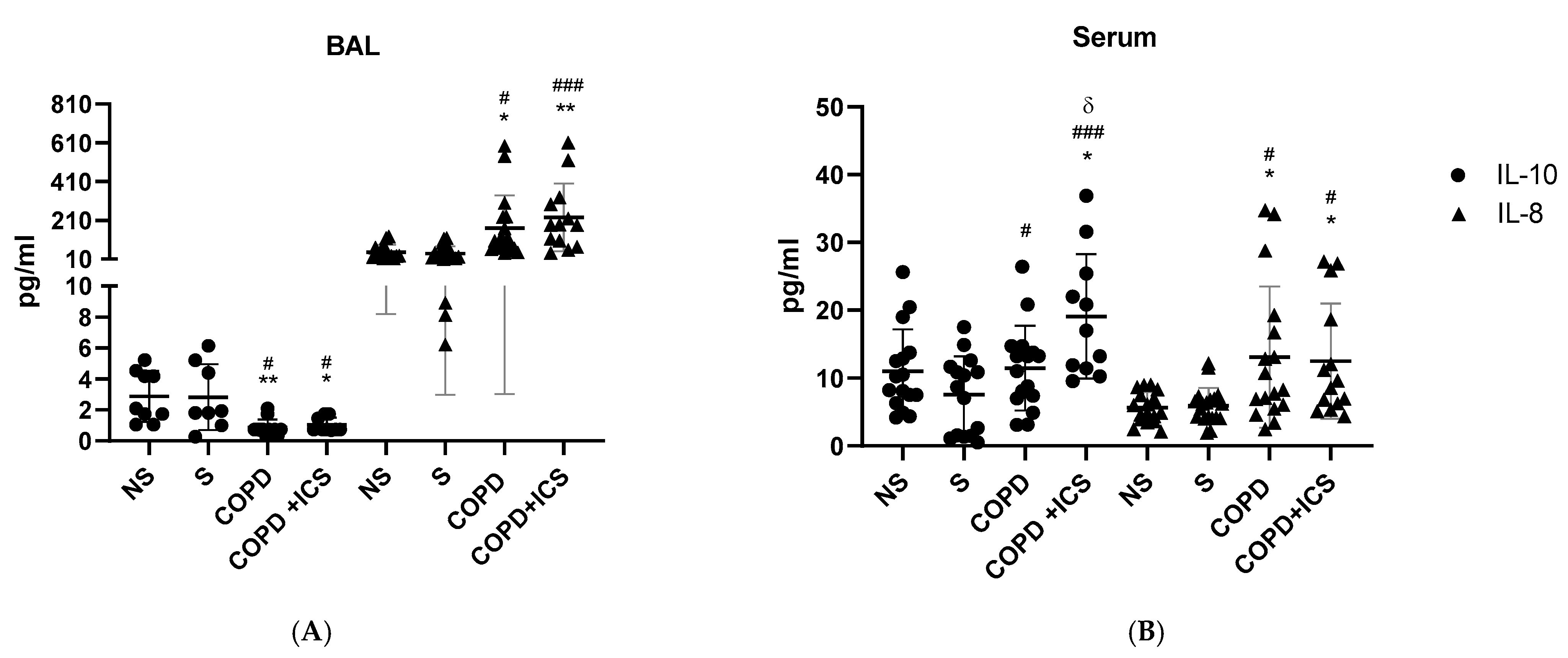
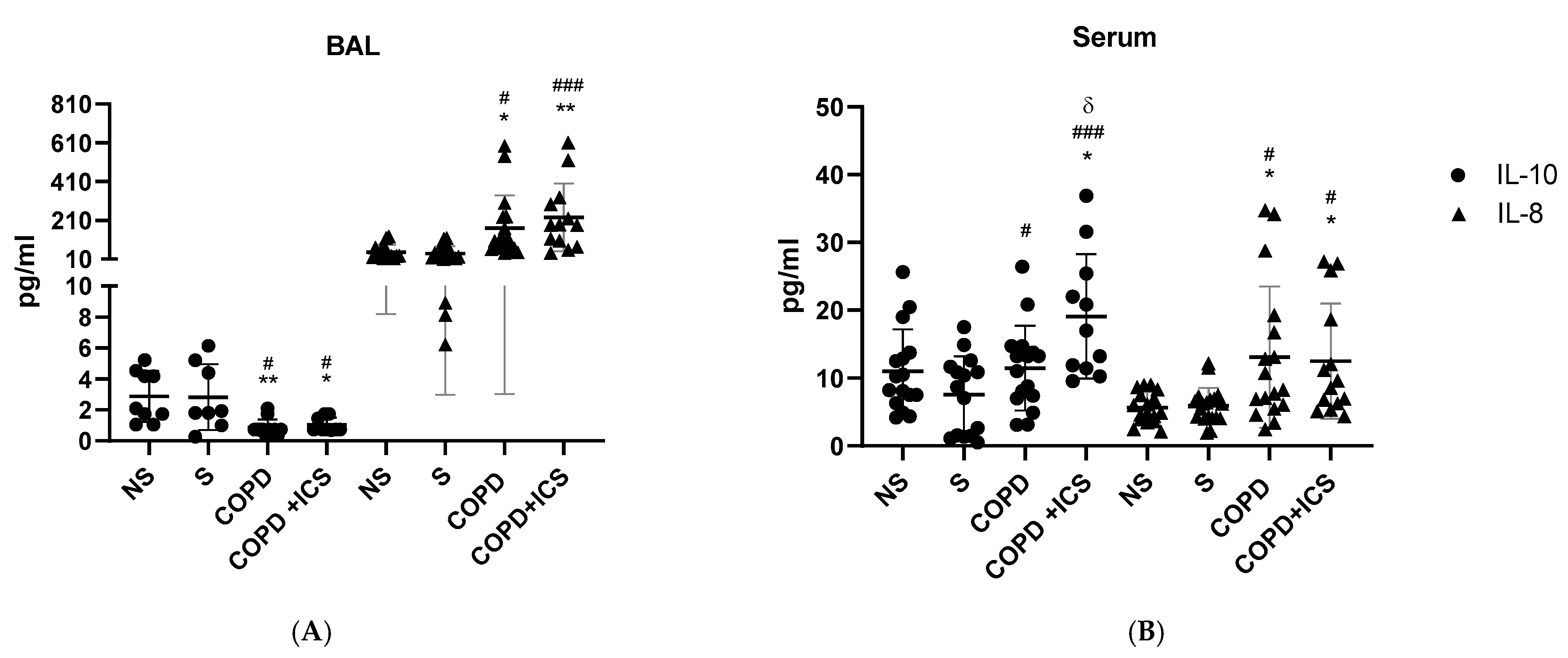
2.2. Inflammatory Pattern in Patients’ BAL and Serum
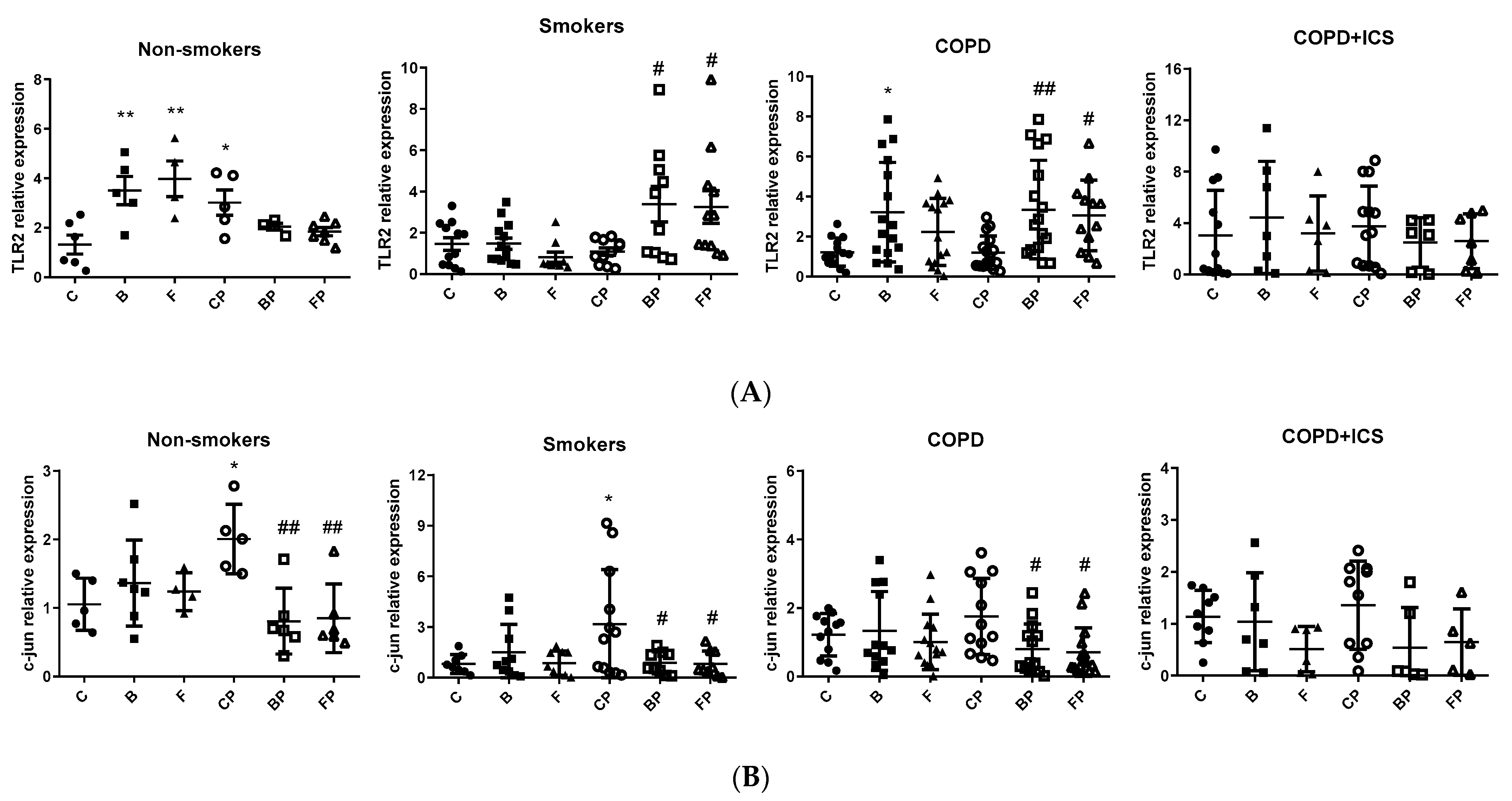
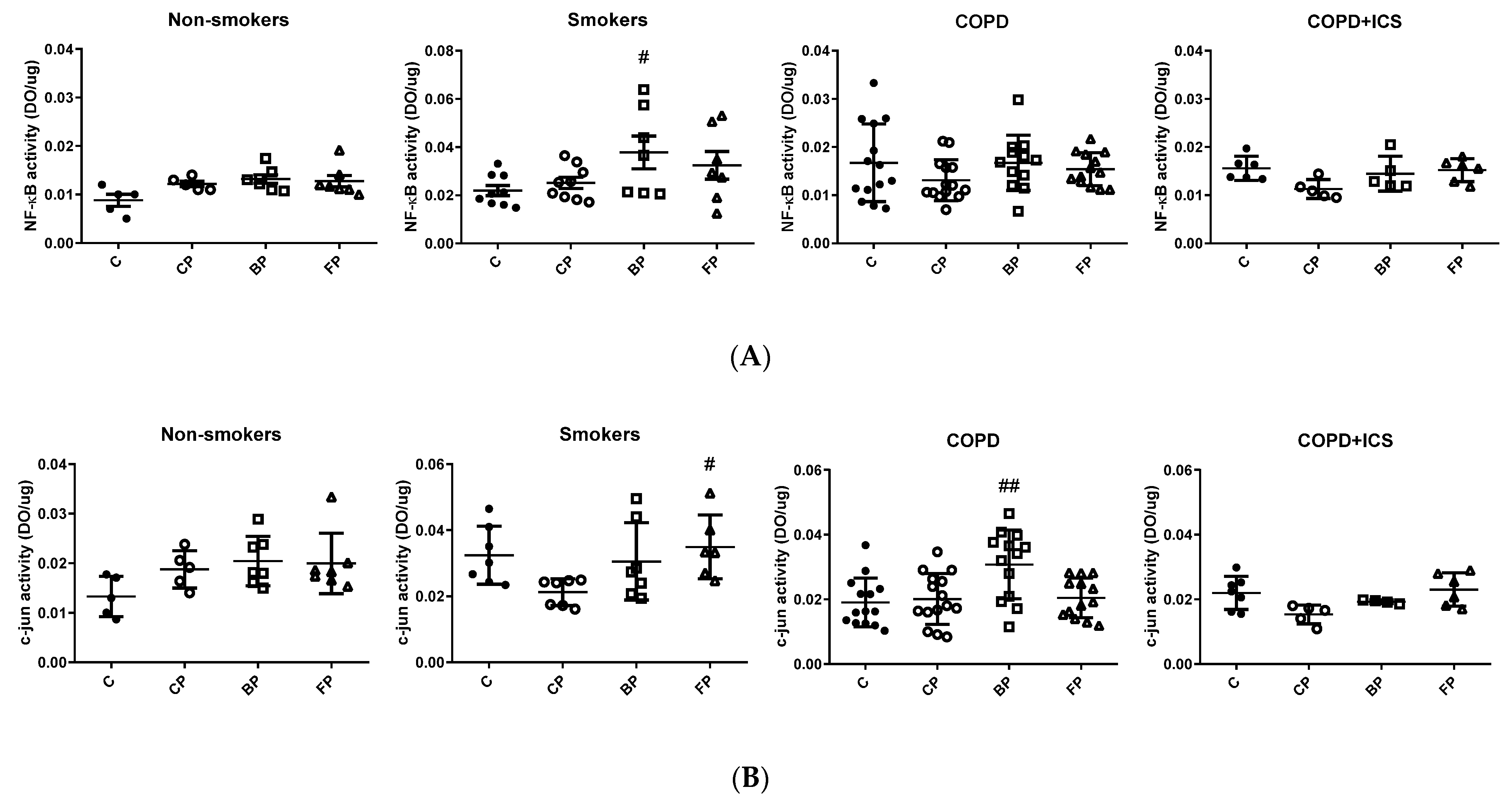
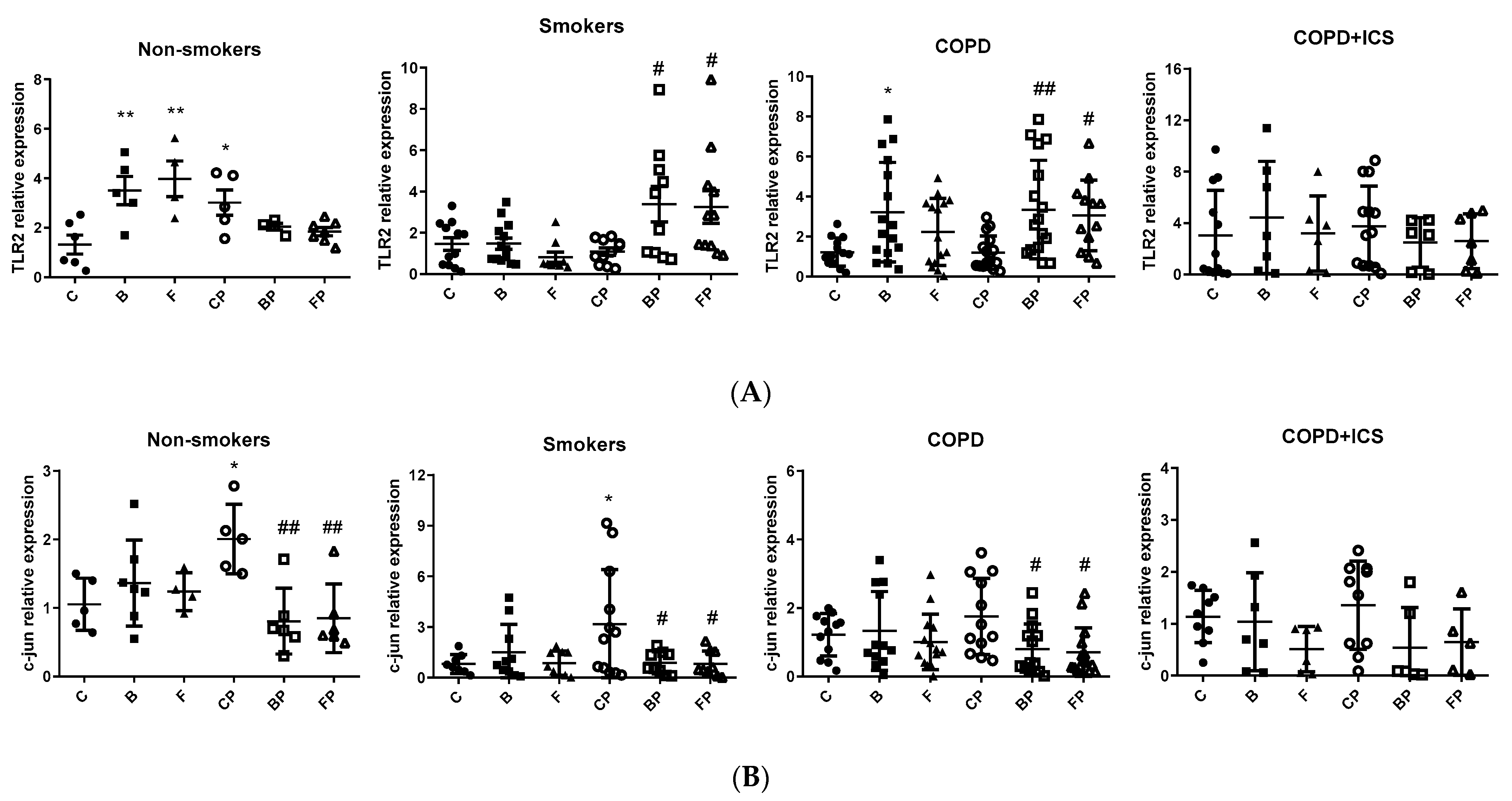
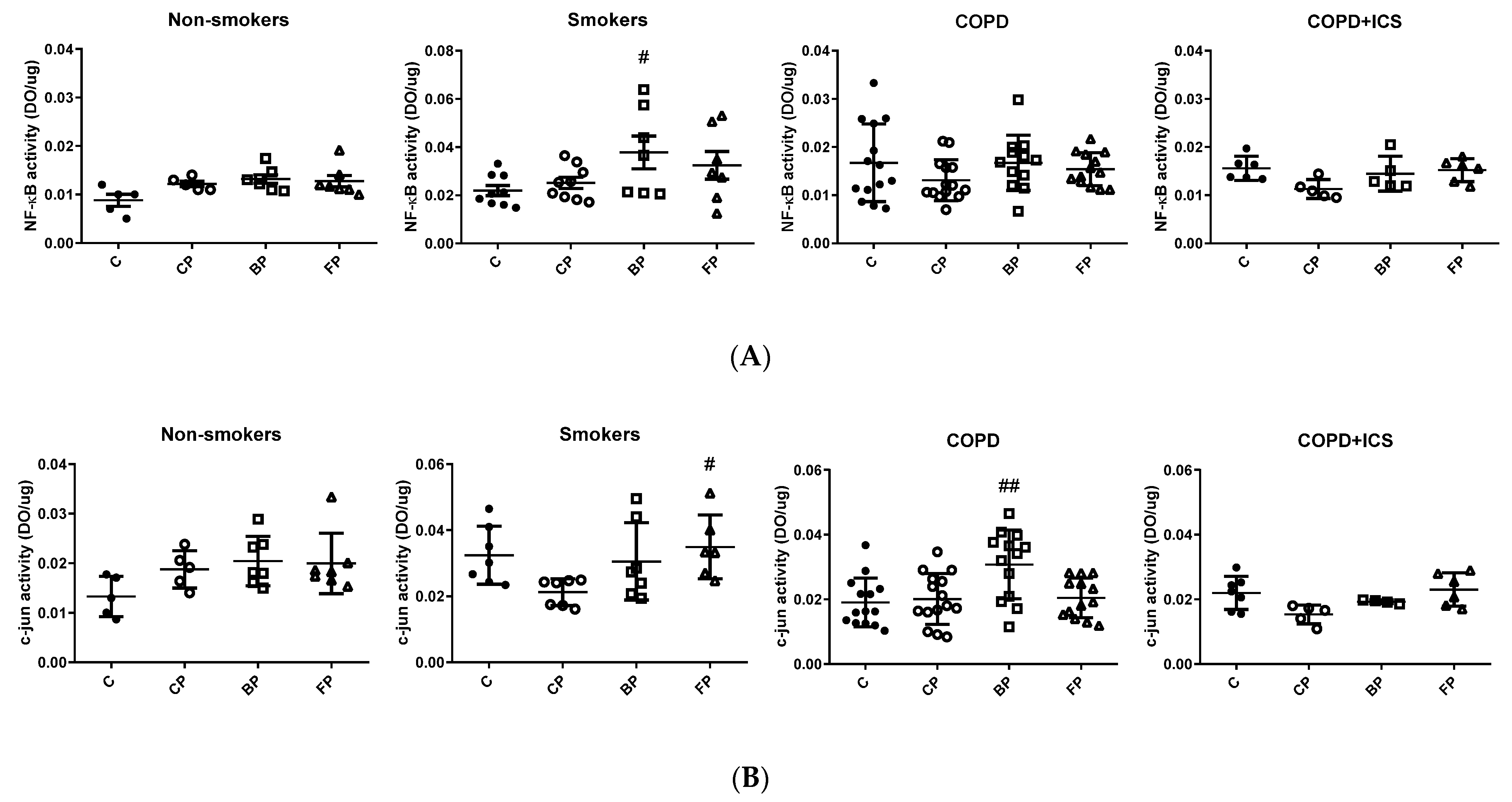
2.3. TLR2 and c-Jun Gene Expression in Primary Human Alveolar Macrophages upon Infection and in Response to ICS Cotreatment
3. Discussion
4. Materials and Methods
4.1. Study Design and Ethics
4.2. Population
4.3. Bacterial Strains and Preparation
4.4. Peripheral Blood Sampling and Processing
4.5. Cytokine Determination
4.6. Cell Culture
4.7. NF-κB and c-Jun Activity
4.8. RT-PCR
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. 2021. Available online: https://goldcopd.org (accessed on 17 May 2022).
- Price, D.; Yawn, B.; Brusselle, G.; Rossi, A. Risk-to-Benefit Ratio of Inhaled Corticosteroids in Patients with COPD. Prim. Care Respir. J. J. Gen. Pract. Airw. Group 2013, 22, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suissa, S.; Patenaude, V.; Lapi, F.; Ernst, P. Inhaled Corticosteroids in COPD and the Risk of Serious Pneumonia. Thorax 2013, 68, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festic, E.; Bansal, V.; Gupta, E.; Scanlon, P.D. Association of Inhaled Corticosteroids with Incident Pneumonia and Mortality in COPD Patients; Systematic Review and Meta-Analysis. COPD J. Chronic Obstr. Pulm. 2016, 13, 312–326. [Google Scholar] [CrossRef]
- Shafiek, H.; Verdú, J.; Iglesias, A.; Ramon-Clar, L.; Toledo-Pons, N.; Lopez-Causape, C.; Juan, C.; Fraile-Ribot, P.; Oliver, A.; Cosio, B.G. Inhaled Corticosteroid Dose Is Associated with Pseudomonas Aeruginosa Infection in Severe COPD. BMJ Open Respir. Res. 2021, 8, e001067. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Goldmann, T.; Rohmann, K.; Rupp, J.; Marwitz, S.; Rotta Detto Loria, J.; Limmer, S.; Zabel, P.; Dalhoff, K.; Drömann, D. Budesonide Inhibits Intracellular Infection with Non-Typeable Haemophilus Influenzae Despite Its Anti-Inflammatory Effects in Respiratory Cells and Human Lung Tissue: A Role for P38 MAP Kinase. Respir. Int. Rev. Thorac. Dis. 2015, 90, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Lessmann, A.; Juárez-Rubio, C.; Plaza-Moral, V. Role of Toll-like Receptors in Respiratory Diseases. Arch. Bronconeumol. 2010, 46, 135–142. [Google Scholar] [CrossRef]
- Cigana, C.; Lorè, N.I.; Bernardini, M.L.; Bragonzi, A. Dampening Host Sensing and Avoiding Recognition in Pseudomonas Aeruginosa Pneumonia. J. Biomed. Biotechnol. 2011, 2011, 852513. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.-S.; Lee, J.-H.; Paek, S.-H.; Jung, Y.W.; Ha, U.-H. Pseudomonas Aeruginosa-Dependent Upregulation of TLR2 Influences Host Responses to a Secondary Staphylococcus Aureus Infection. Pathog. Dis. 2013, 69, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Hotamisligil, G.S. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat. Rev. Drug Discov. 2010, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Chun, W.; Lee, H.J.; Min, J.-H.; Kim, S.-M.; Seo, J.-Y.; Ahn, K.-S.; Oh, S.-R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef]
- Culpitt, S.V.; Rogers, D.F.; Shah, P.; De Matos, C.; Russell, R.E.K.; Donnelly, L.E.; Barnes, P.J. Impaired Inhibition by Dexamethasone of Cytokine Release by Alveolar Macrophages from Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2003, 167, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.A.; Panzica, L.; Kalathil, S.G.; Thanavala, Y. Immune Dysfunction in Patients with Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2015, 12, S169–S175. [Google Scholar] [CrossRef] [PubMed]
- Cosío, B.G.; Jahn, A.; Iglesias, A.; Shafiek, H.; Busquets, X.; Agustí, A. Haemophilus Influenzae Induces Steroid-Resistant Inflammatory Responses in COPD. BMC Pulm. Med. 2015, 15, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eklöf, J.; Ingebrigtsen, T.S.; Sørensen, R.; Saeed, M.I.; Alispahic, I.A.; Sivapalan, P.; Boel, J.B.; Bangsborg, J.; Ostergaard, C.; Dessau, R.B.; et al. Use of Inhaled Corticosteroids and Risk of Acquiring Pseudomonas Aeruginosa in Patients with Chronic Obstructive Pulmonary Disease. Thorax 2022, 77, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Role of Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Von Schéele, I.; Billing, B.; Dahlén, B.; Lantz, A.-S.; Larsson, K.; Palmberg, L. Effects of Budesonide on Toll-like Receptor Expression in Alveolar Macrophages from Smokers with and without COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Higham, A.; Karur, P.; Jackson, N.; Cunoosamy, D.M.; Jansson, P.; Singh, D. Differential Anti-Inflammatory Effects of Budesonide and a P38 MAPK Inhibitor AZD7624 on COPD Pulmonary Cells. Available online: https://www.dovepress.com/differential-anti-inflammatory-effects-of-budesonide-and-a-p38-mapk-in-peer-reviewed-fulltext-article-COPD (accessed on 5 August 2020).
- Peñaloza, H.F.; Nieto, P.A.; Muñoz-Durango, N.; Salazar-Echegarai, F.J.; Torres, J.; Parga, M.J.; Alvarez-Lobos, M.; Riedel, C.A.; Kalergis, A.M.; Bueno, S.M. Interleukin-10 Plays a Key Role in the Modulation of Neutrophils Recruitment and Lung Inflammation during Infection by Streptococcus Pneumoniae. Immunology 2015, 146, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Shan, F.; Zhang, Y.; Jiang, L.; Cheng, Z. Increased Serum IL-17 and Decreased Serum IL-10 and IL-35 Levels Correlate with the Progression of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 2483–2494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zheng, H.; Zhang, H.; Ma, W.; Wang, F.; Liu, C.; He, S. Increased Interleukin (IL)-8 and Decreased IL-17 Production in Chronic Obstructive Pulmonary Disease (COPD) Provoked by Cigarette Smoke. Cytokine 2011, 56, 717–725. [Google Scholar] [CrossRef]
- Nightingale, J.A.; Rogers, D.F.; Chung, K.F.; Barnes, P.J. No Effect of Inhaled Budesonide on the Response to Inhaled Ozone in Normal Subjects. Am. J. Respir. Crit. Care Med. 2000, 161, 479–486. [Google Scholar] [CrossRef]
- Barnes, P.J. Inhaled Corticosteroids Are Not Beneficial in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2000, 161, 342–344, discussion 344. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, R.M.; Lea, S.R.; Metcalfe, H.J.; Singh, D. Mechanisms of Corticosteroid Insensitivity in COPD Alveolar Macrophages Exposed to NTHi. Respir. Res. 2017, 18, 61. [Google Scholar] [CrossRef] [Green Version]
- Freeman, C.M.; Martinez, F.J.; Han, M.K.; Washko, G.R.; McCubbrey, A.L.; Chensue, S.W.; Arenberg, D.A.; Meldrum, C.A.; McCloskey, L.; Curtis, J.L. Lung CD8+ T Cells in COPD Have Increased Expression of Bacterial TLRs. Respir. Res. 2013, 14, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, J.; Sauleda, J.; Regueiro, V.; Santos, C.; López, M.; Ferrer, J.; Agustí, A.G.N.; Bengoechea, J.A. Expression of Toll-like Receptor 2 Is up-Regulated in Monocytes from Patients with Chronic Obstructive Pulmonary Disease. Respir. Res. 2006, 7, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Scheele, I.; Larsson, K.; Palmberg, L. Budesonide Enhances Toll-like Receptor 2 Expression in Activated Bronchial Epithelial Cells. Inhal. Toxicol. 2010, 22, 493–499. [Google Scholar] [CrossRef]
- Simpson, J.L.; McDonald, V.M.; Baines, K.J.; Oreo, K.M.; Wang, F.; Hansbro, P.M.; Gibson, P.G. Influence of Age, Past Smoking, and Disease Severity on TLR2, Neutrophilic Inflammation, and MMP-9 Levels in COPD. Mediat. Inflamm. 2013, 2013, 462934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provost, K.A.; Smith, M.; Miller-Larsson, A.; Gudleski, G.D.; Sethi, S. Bacterial Regulation of Macrophage Bacterial Recognition Receptors in COPD Are Differentially Modified by Budesonide and Fluticasone Propionate. PLoS ONE 2019, 14, e0207675. [Google Scholar] [CrossRef]
- Rossios, C.; To, Y.; To, M.; Ito, M.; Barnes, P.J.; Adcock, I.M.; Johnson, M.; Ito, K. Long-acting fluticasone furoate has a superior pharmacological profile to fluticasone propionate in human respiratory cells. Eur. J. Pharmacol. 2011, 670, 244–251. [Google Scholar] [CrossRef]
- Huszczynski, S.M.; Lam, J.S.; Khursigara, C.M. The Role of Pseudomonas Aeruginosa Lipopolysaccharide in Bacterial Pathogenesis and Physiology. Pathogens 2019, 9, E6. [Google Scholar] [CrossRef] [Green Version]
- Phaybouth, V.; Wang, S.-Z.; Hutt, J.A.; McDonald, J.D.; Harrod, K.S.; Barrett, E.G. Cigarette Smoke Suppresses Th1 Cytokine Production and Increases RSV Expression in a Neonatal Model. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L222–L231. [Google Scholar] [CrossRef]
- Van Zyl-Smit, R.N.; Binder, A.; Meldau, R.; Semple, P.L.; Evans, A.; Smith, P.; Bateman, E.D.; Dheda, K. Cigarette Smoke Impairs Cytokine Responses and BCG Containment in Alveolar Macrophages. Thorax 2014, 69, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette Smoking and Inflammation: Cellular and Molecular Mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lea, S.R.; Reynolds, S.L.; Kaur, M.; Simpson, K.D.; Hall, S.R.; Hessel, E.M.; Singh, D. The Effects of Repeated Toll-like Receptors 2 and 4 Stimulation in COPD Alveolar Macrophages. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinenov, Y.; Rogatsky, I. Glucocorticoids and the Innate Immune System: Crosstalk with the Toll-like Receptor Signaling Network. Mol. Cell. Endocrinol. 2007, 275, 30–42. [Google Scholar] [CrossRef]
- Gagliardo, R.; Chanez, P.; Profita, M.; Bonanno, A.; Albano, G.D.; Montalbano, A.M.; Pompeo, F.; Gagliardo, C.; Merendino, A.M.; Gjomarkaj, M. IκB Kinase-Driven Nuclear Factor-ΚB Activation in Patients with Asthma and Chronic Obstructive Pulmonary Disease. J. Allergy Clin. Immunol. 2011, 128, 635–645.e2. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; Van der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of Spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [Green Version]
- Roca, J.; Sanchis, J.; Agusti-Vidal, A.; Segarra, F.; Navajas, D.; Rodriguez-Roisin, R.; Casan, P.; Sans, S. Spirometric Reference Values from a Mediterranean Population. Bull. Eur. Physiopathol. Respir. 1986, 22, 217–224. [Google Scholar]
- Cosio, B.G.; Tsaprouni, L.; Ito, K.; Jazrawi, E.; Adcock, I.M.; Barnes, P.J. Theophylline Restores Histone Deacetylase Activity and Steroid Responses in COPD Macrophages. J. Exp. Med. 2004, 200, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete Genome Sequence of Pseudomonas Aeruginosa PAO1, an Opportunistic Pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Yuan, Z.; Mehta, H.J.; Mohammed, K.; Nasreen, N.; Roman, R.; Brantly, M.; Sadikot, R.T. TREM-1 Is Induced in Tumor Associated Macrophages by Cyclo-Oxygenase Pathway in Human Non-Small Cell Lung Cancer. PLoS ONE 2014, 9, e94241. [Google Scholar] [CrossRef]
- Esteva-Socias, M.; Gómez-Romano, F.; Carrillo-Ávila, J.A.; Sánchez-Navarro, A.L.; Villena, C. Impact of Different Stabilization Methods on RT-QPCR Results Using Human Lung Tissue Samples. Sci. Rep. 2020, 10, 3579. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Variable | COPD + ICS (n = 11) | COPD (n = 16) | NS (n = 10) | S (n = 12) | Sig. (p) |
|---|---|---|---|---|---|
| Age | 68.27 ± 7.35 | 67.25 ± 8.23 | 61.8 ± 13.89 | 64 ± 9.85 | 0.392 |
| Gender Male/female | 7 (63.6)/4 (36.4) | 13 (81.3)/3 (18.8) | 2 (20)/8 (80) | 8 (66.7)/4 (33.3) | 0.018 * |
| Smoking history Smoker Exsmoker Nonsmoker | 4 (36.4) 7 (63.6) 0 (0) | 9 (56.3) 7 (43.8) 0 (0) | 0 (0) 0 (0) 10 (100) | 8 (66.7) 4 (33.3) 0 (0) | <0.001 * |
| Smoking index | 61 (40–69) | 52.5 (40–60) | 0 (0–0) | 39.5 (23–60) | <0.001 * |
| BMI (kg/m2) | 25.33 ± 7.09 | 28.05 ± 4.02 | 25.53 ± 4.18 | 25.9 ± 4.75 | 0.583 |
| Spirometry | |||||
| FVC (L) | 3.27 ± 0.88 | 3.45 ± 0.84 | 3.05 ± 0.59 | 4.09 ± 1.13 | 0.300 |
| FVC (% predicted) | 86.8 ± 11.44 | 95.43 ± 23.72 | 100.25 ± 2.87 | 100.6 ± 11.76 | 0.404 |
| FEV1 (L) | 1.74 ± 0.69 | 2.08 ± 0.52 | 2.35 ± 0.65 | 3.01 ± 0.85 | 0.014 * |
| FEV1 (% predicted) | 68 (50–72) | 71.25 (60–90) | 99 (94–103) | 102 (98–105) | 0.005 * |
| FEV1/FVC | 56.56 (48–66.88) | 62.26 (55.96–65.52) | 75.32 (70.45–81.3) | 73.4 (70–76) | 0.001 * |
| DLCO | 62 (57–67) | 69 (58–82) | 83 (82.5–85.5) | 83.5 (71.5–95.5) | 0.118 |
| KCO | 69.5 (54–83) | 64 (57.5–77.5) | 88 (87–90.5) | 80 (77–81) | 0.033 * |
| GOLD categories $ | |||||
| GOLD I | 2 (18.2) | 4 (25) | NA | NA | 0.389 |
| GOLD II | 5 (45.5) | 8 (50) | |||
| GOLD III | 1 (9.1) | 1 (6.25) | |||
| GOLD IV | 2 (18.2) | 0 (0) | |||
| Gene | Sequence |
|---|---|
| TLR2 | F: GGACTTCTCCCATTTCCGTCT |
| R: CTCCAGGTAGGTCTTGGTGTTC | |
| c-jun | F: AAAGGATAGTGCGATGTTTC |
| R: TAAAATCTGCCACCAATTCC | |
| B2M | F: ACCCCCACTGAAAAAGATGAG |
| R: ATCTTCAAACCTCCATGATGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerón-Pisa, N.; Shafiek, H.; Martín-Medina, A.; Verdú, J.; Jordana-Lluch, E.; Escobar-Salom, M.; Barceló, I.M.; López-Causapé, C.; Oliver, A.; Juan, C.; et al. Effects of Inhaled Corticosteroids on the Innate Immunological Response to Pseudomonas aeruginosa Infection in Patients with COPD. Int. J. Mol. Sci. 2022, 23, 8127. https://doi.org/10.3390/ijms23158127
Cerón-Pisa N, Shafiek H, Martín-Medina A, Verdú J, Jordana-Lluch E, Escobar-Salom M, Barceló IM, López-Causapé C, Oliver A, Juan C, et al. Effects of Inhaled Corticosteroids on the Innate Immunological Response to Pseudomonas aeruginosa Infection in Patients with COPD. International Journal of Molecular Sciences. 2022; 23(15):8127. https://doi.org/10.3390/ijms23158127
Chicago/Turabian StyleCerón-Pisa, Noemi, Hanaa Shafiek, Aina Martín-Medina, Javier Verdú, Elena Jordana-Lluch, Maria Escobar-Salom, Isabel M. Barceló, Carla López-Causapé, Antonio Oliver, Carlos Juan, and et al. 2022. "Effects of Inhaled Corticosteroids on the Innate Immunological Response to Pseudomonas aeruginosa Infection in Patients with COPD" International Journal of Molecular Sciences 23, no. 15: 8127. https://doi.org/10.3390/ijms23158127
APA StyleCerón-Pisa, N., Shafiek, H., Martín-Medina, A., Verdú, J., Jordana-Lluch, E., Escobar-Salom, M., Barceló, I. M., López-Causapé, C., Oliver, A., Juan, C., Iglesias, A., & Cosío, B. G. (2022). Effects of Inhaled Corticosteroids on the Innate Immunological Response to Pseudomonas aeruginosa Infection in Patients with COPD. International Journal of Molecular Sciences, 23(15), 8127. https://doi.org/10.3390/ijms23158127