
An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds

 ,
, 
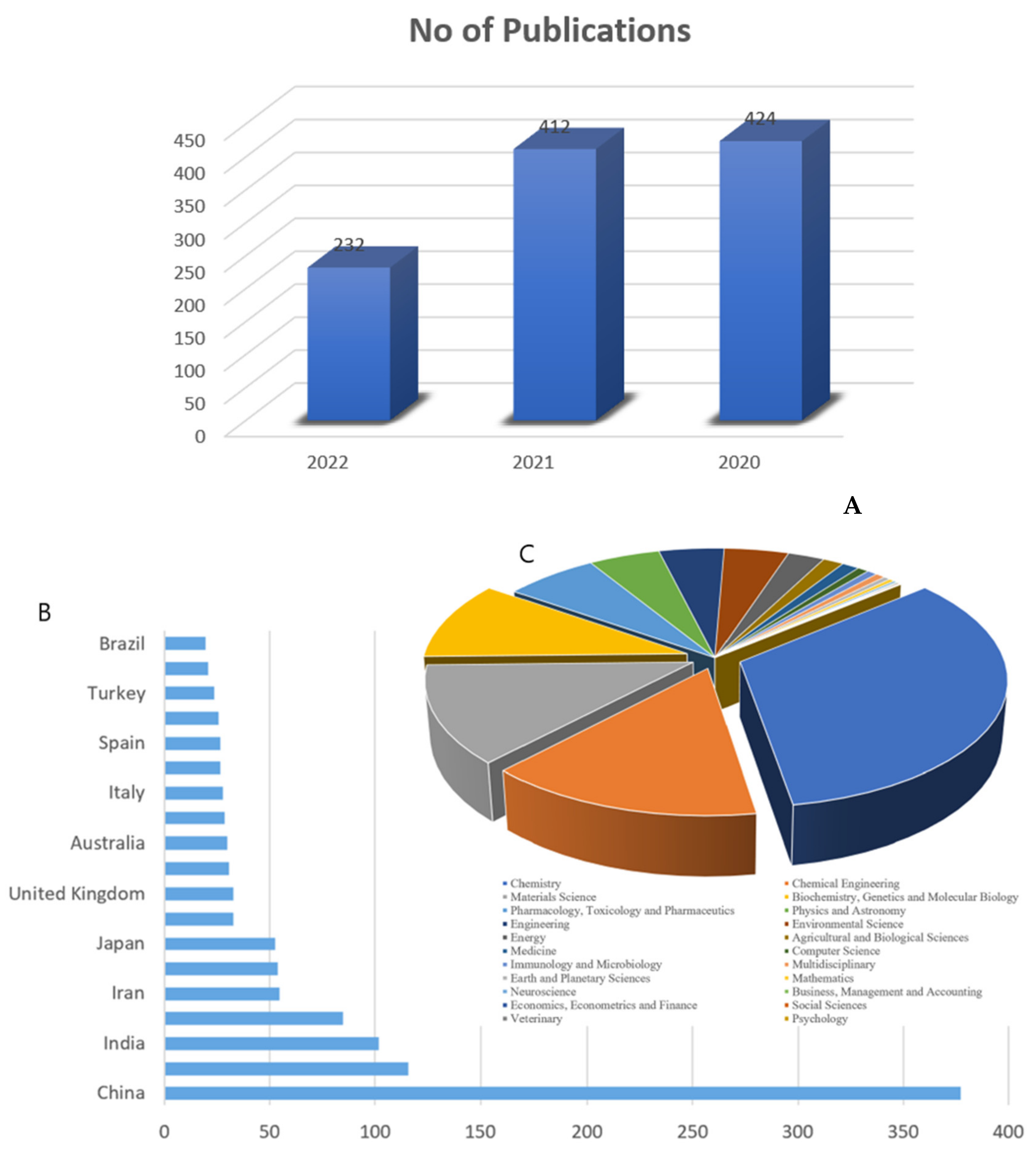{kind=link}
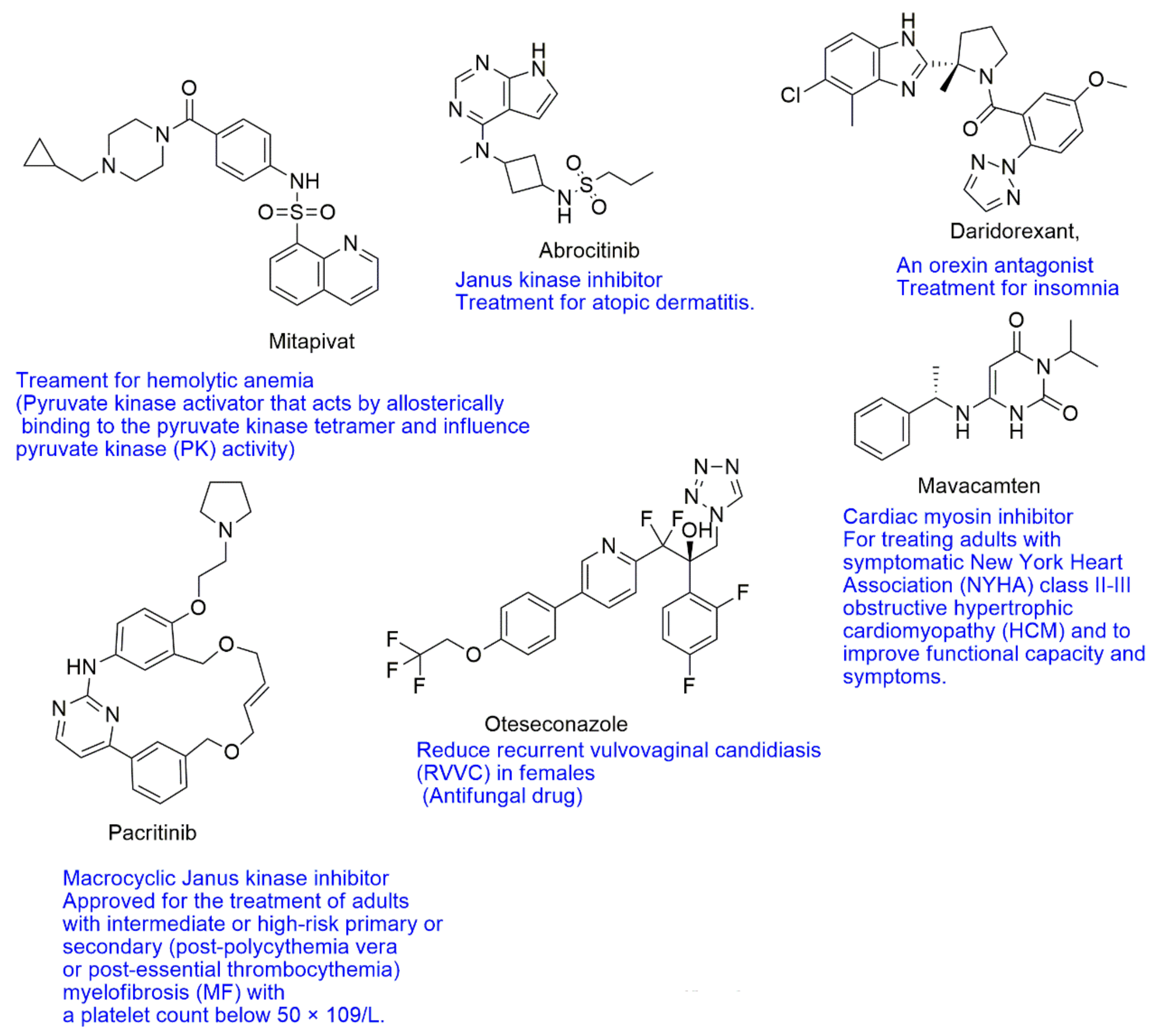{kind=link}
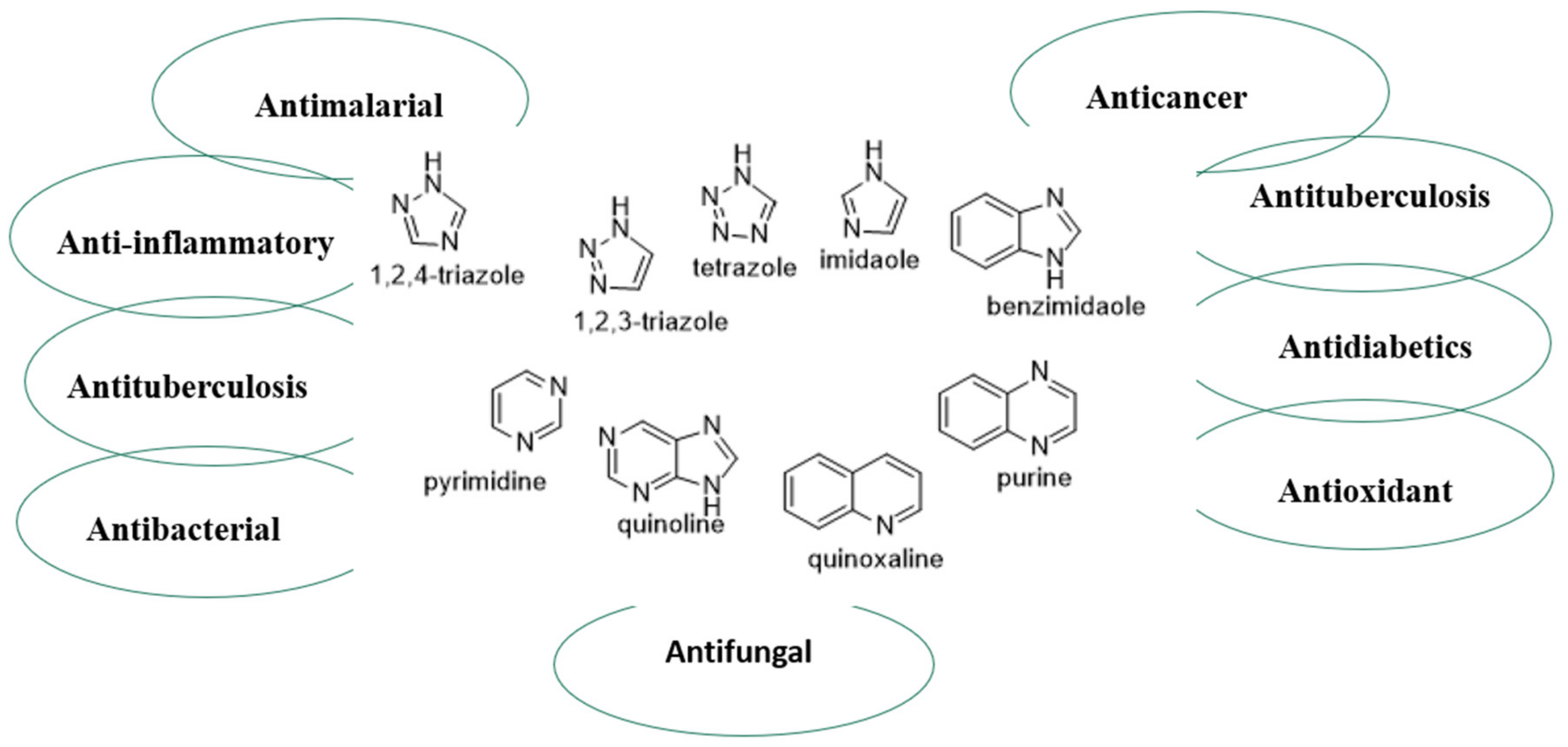{kind=link}
{kind=link}
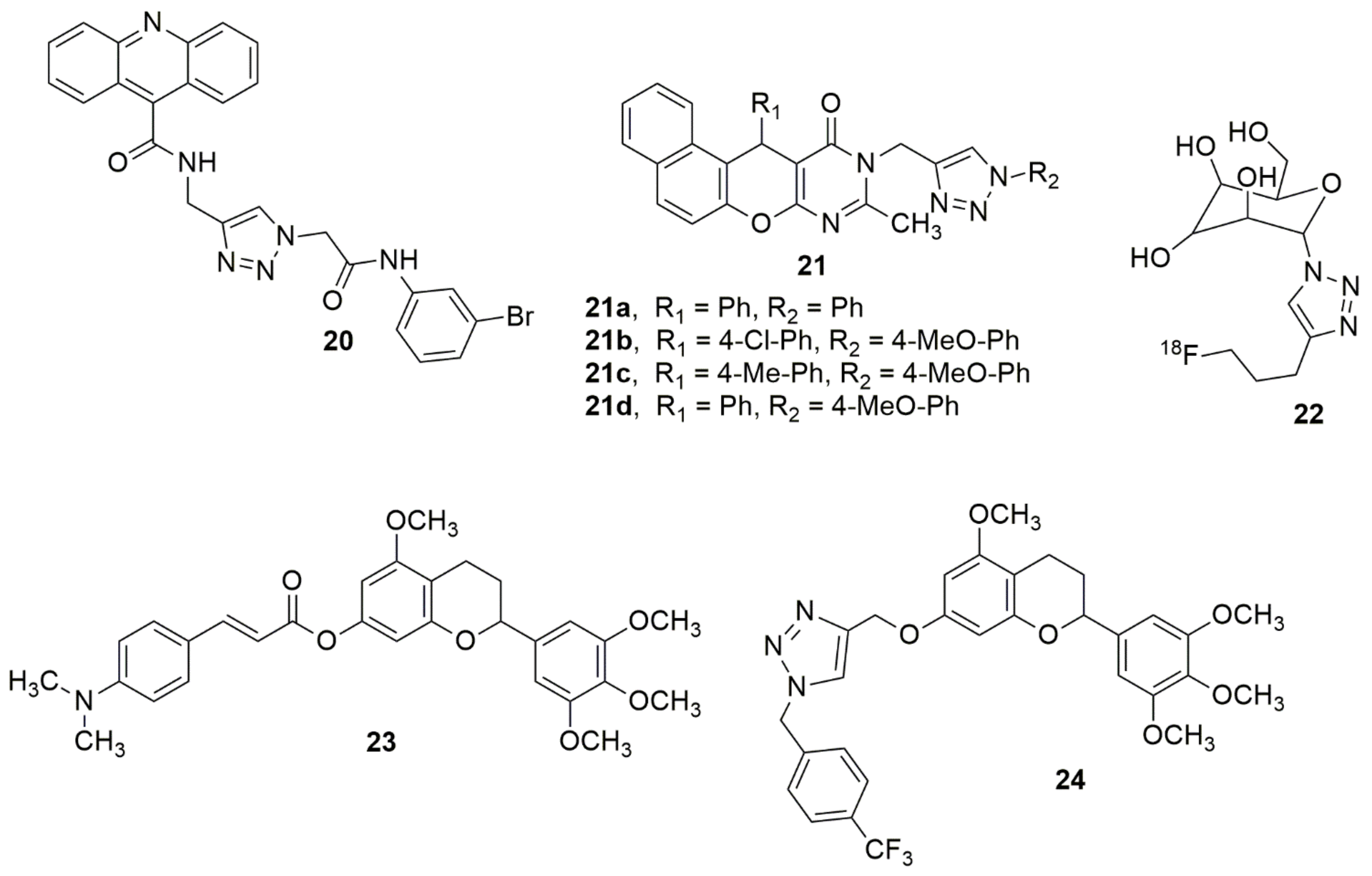{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
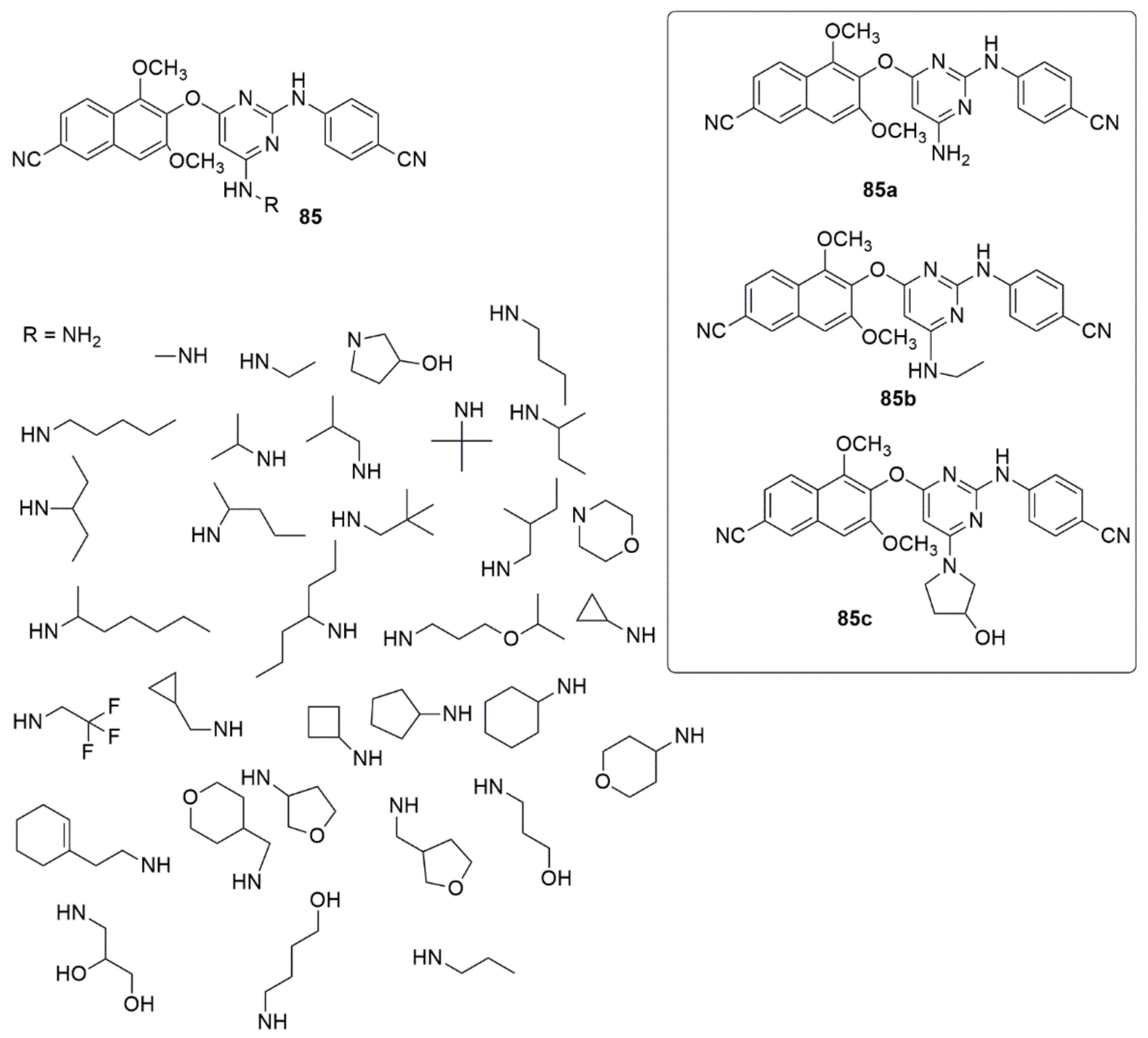{kind=link}
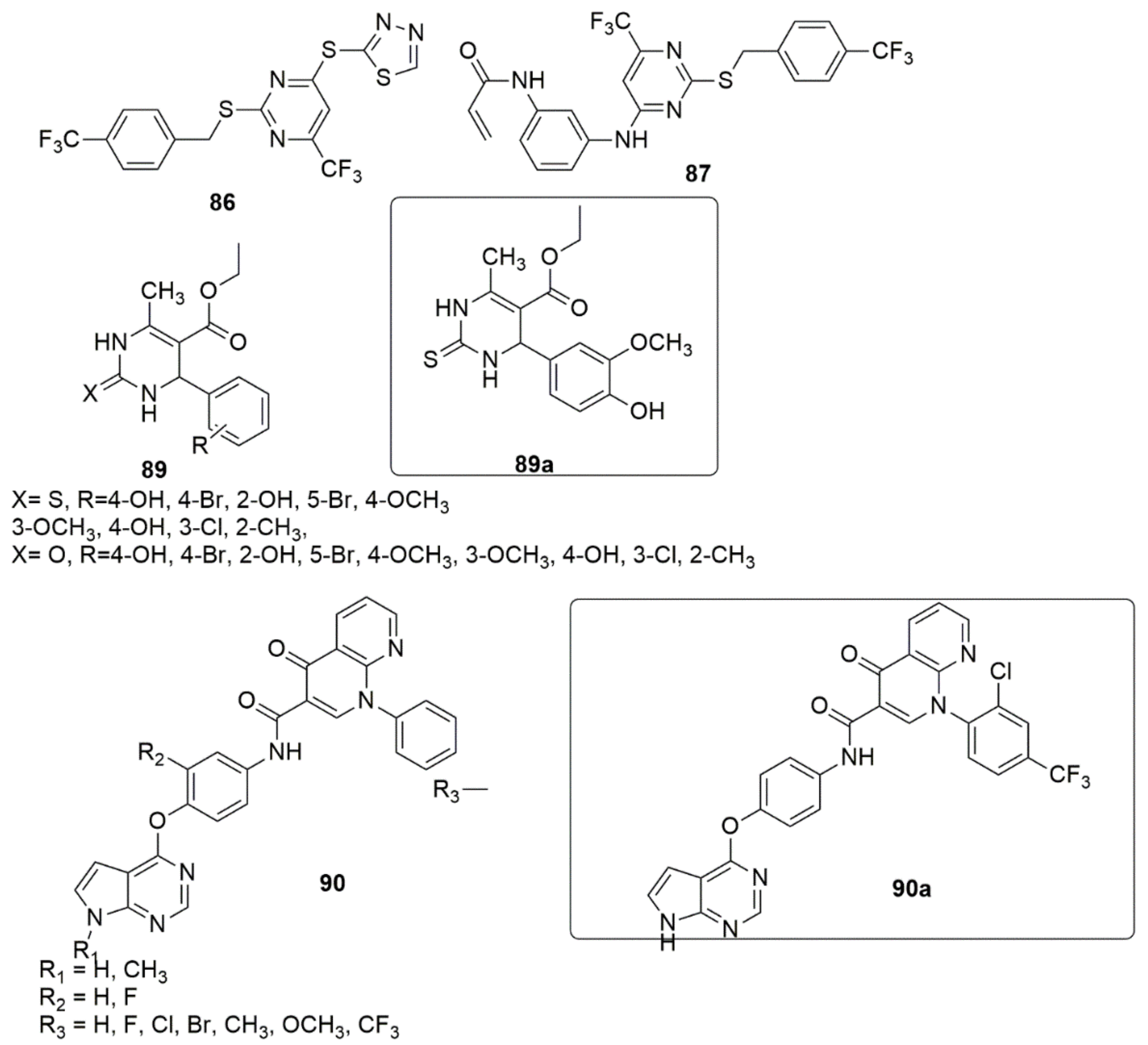{kind=link}
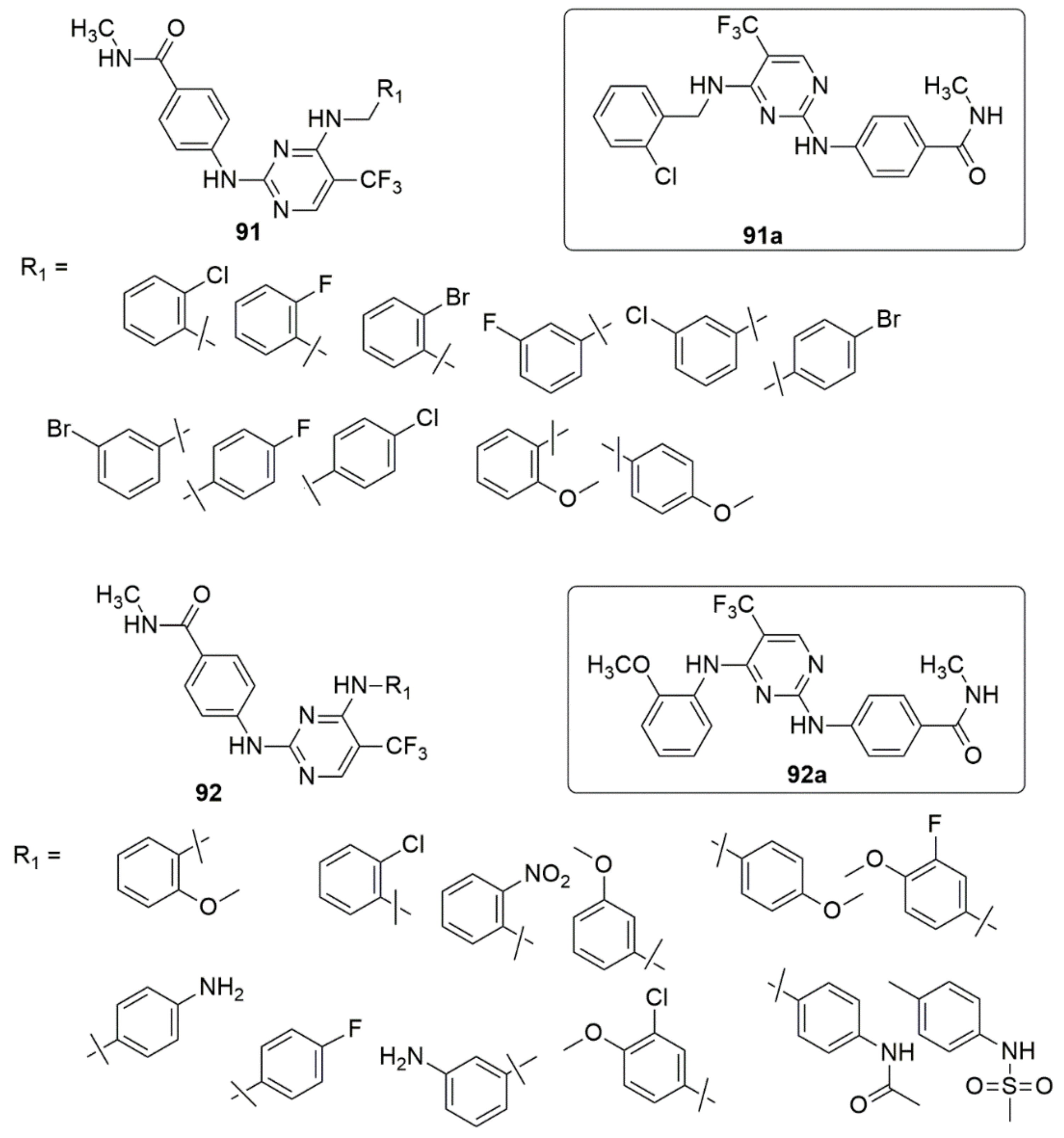{kind=link}
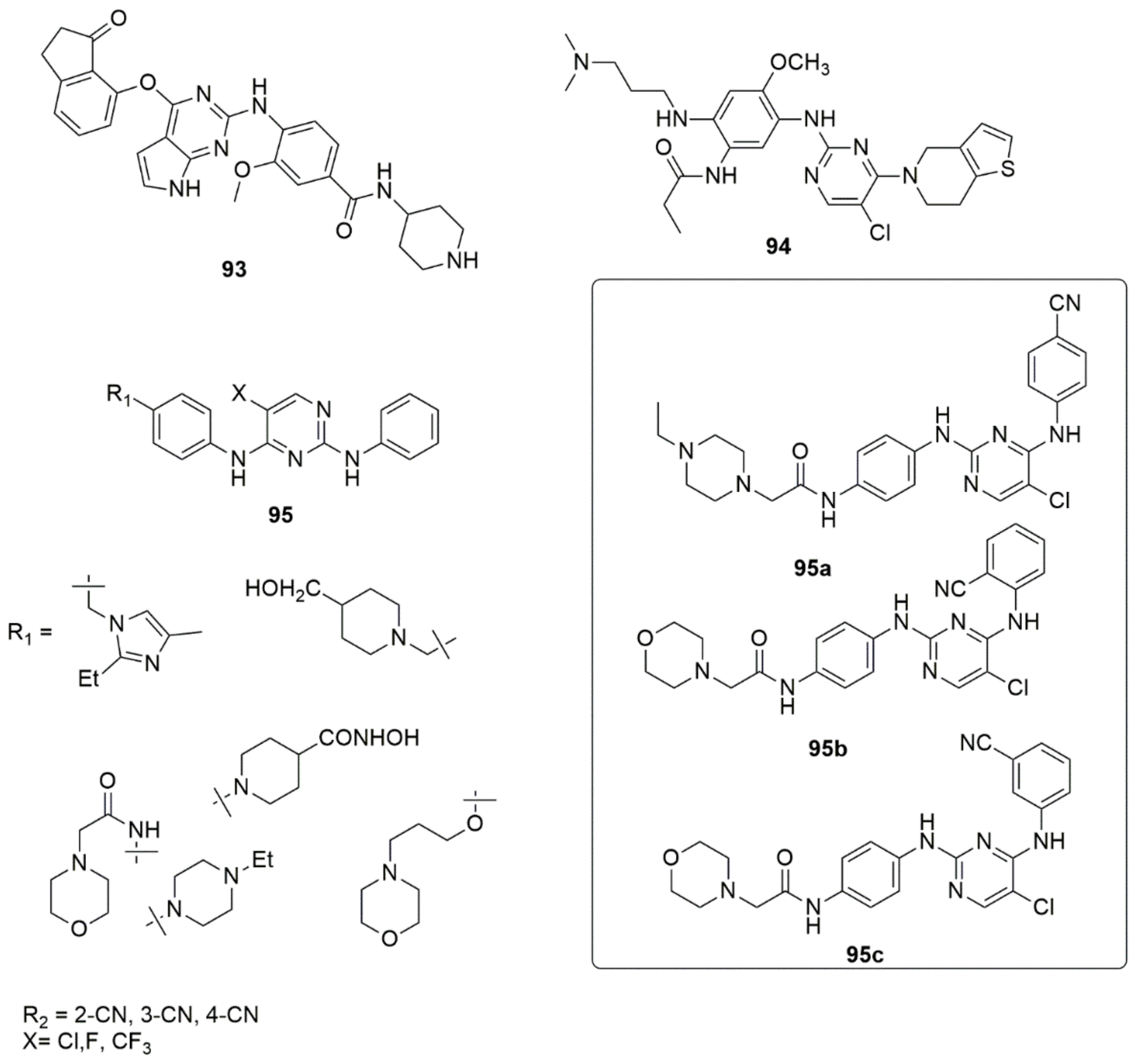{kind=link}
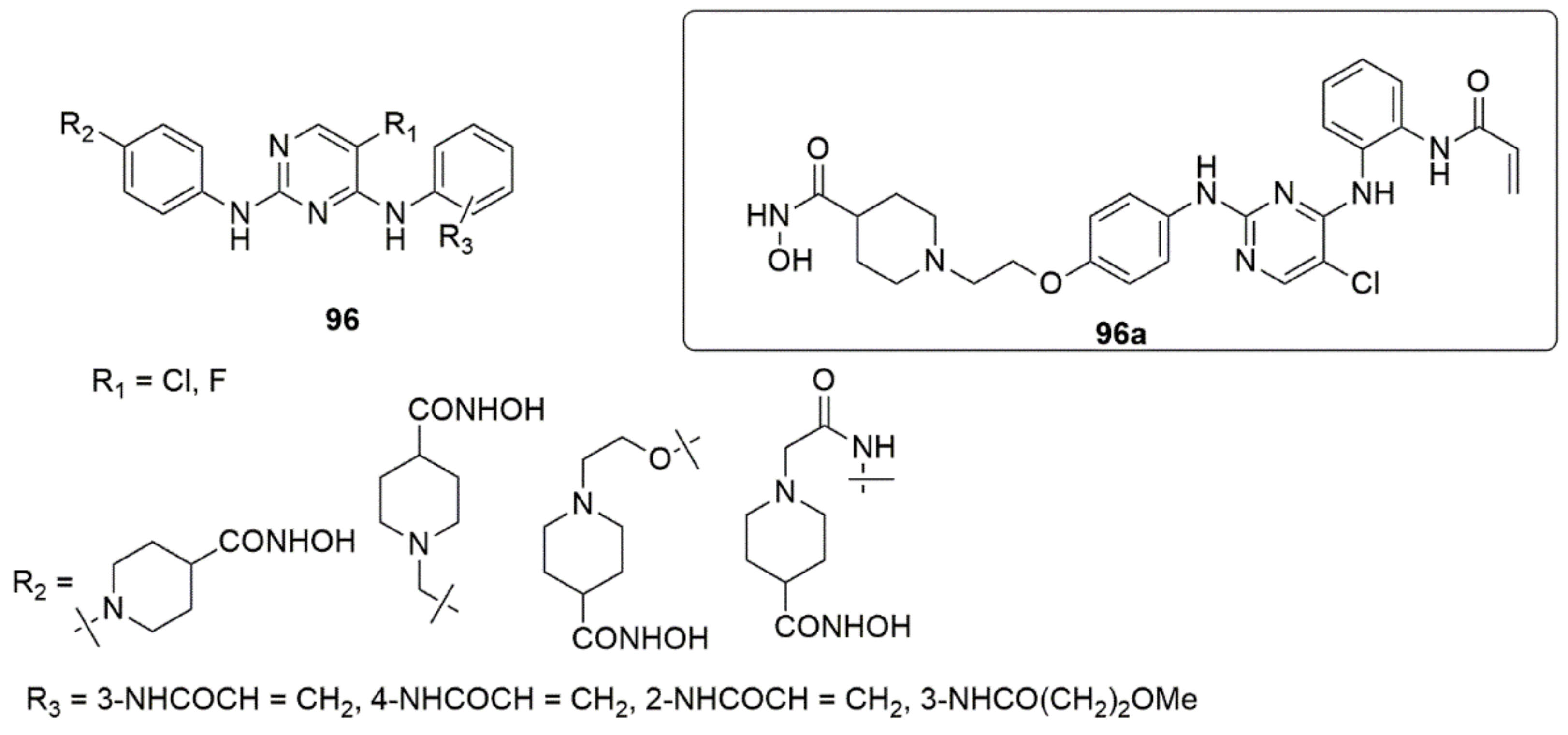{kind=link}
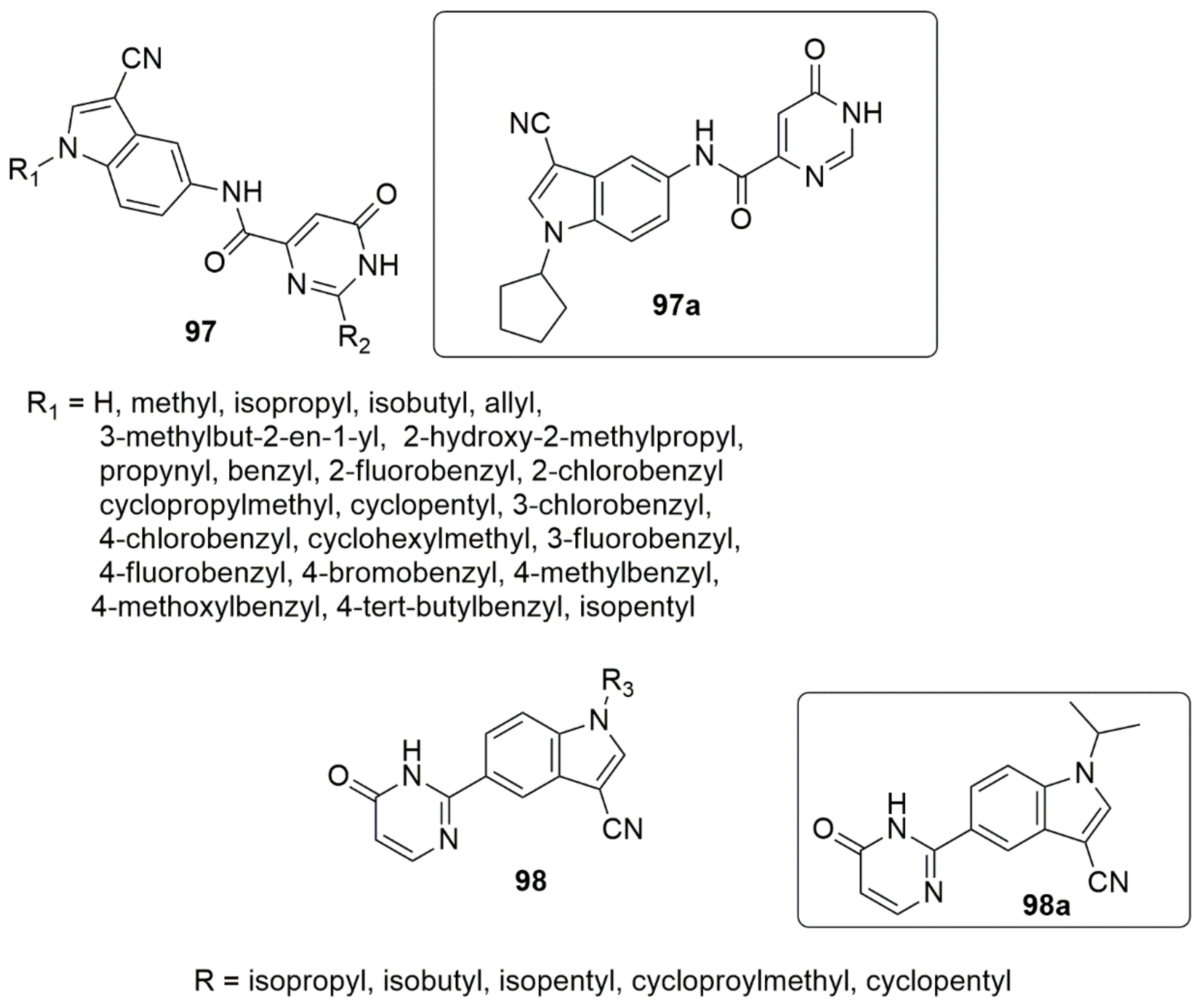{kind=link}
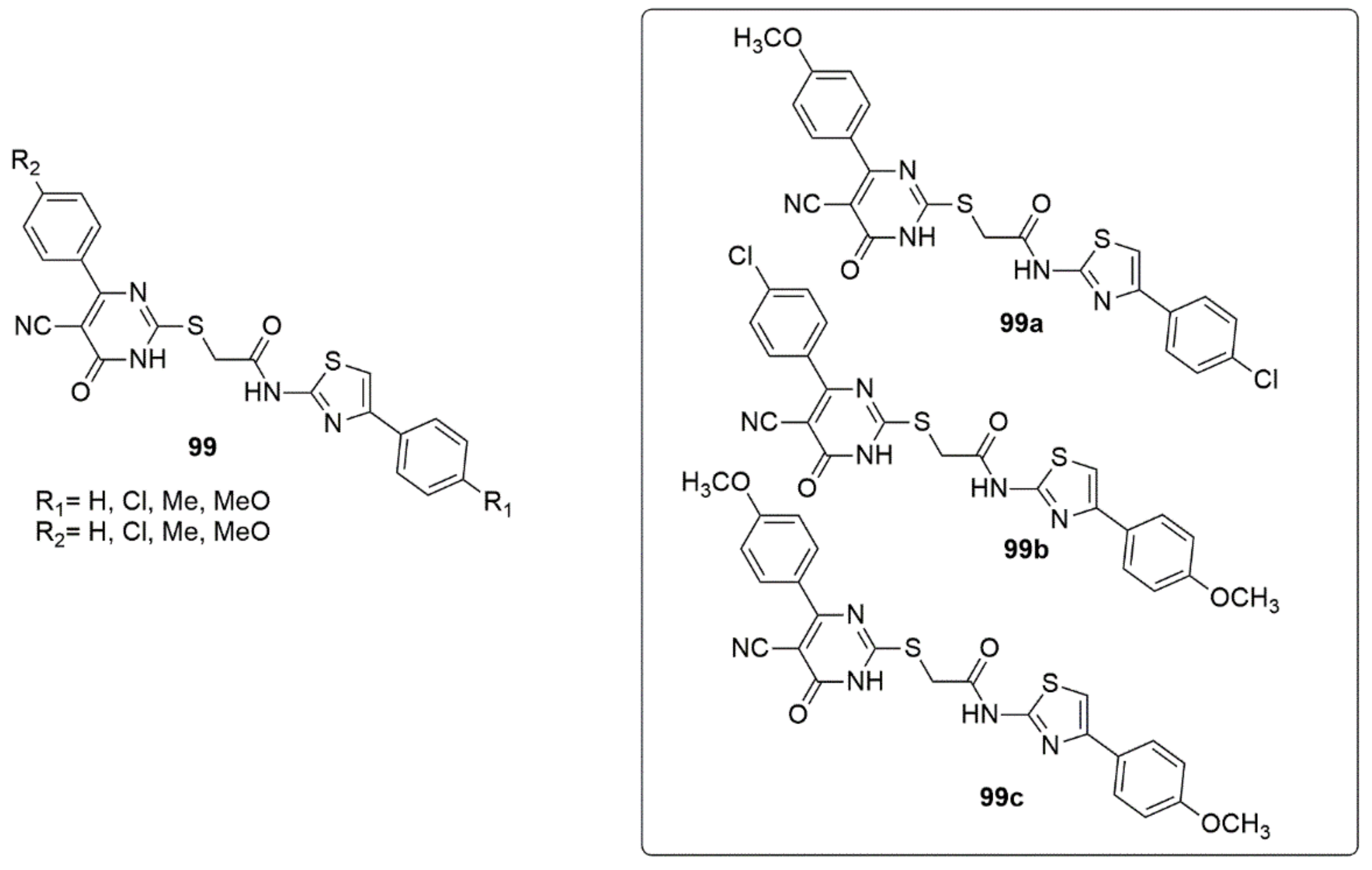{kind=link}
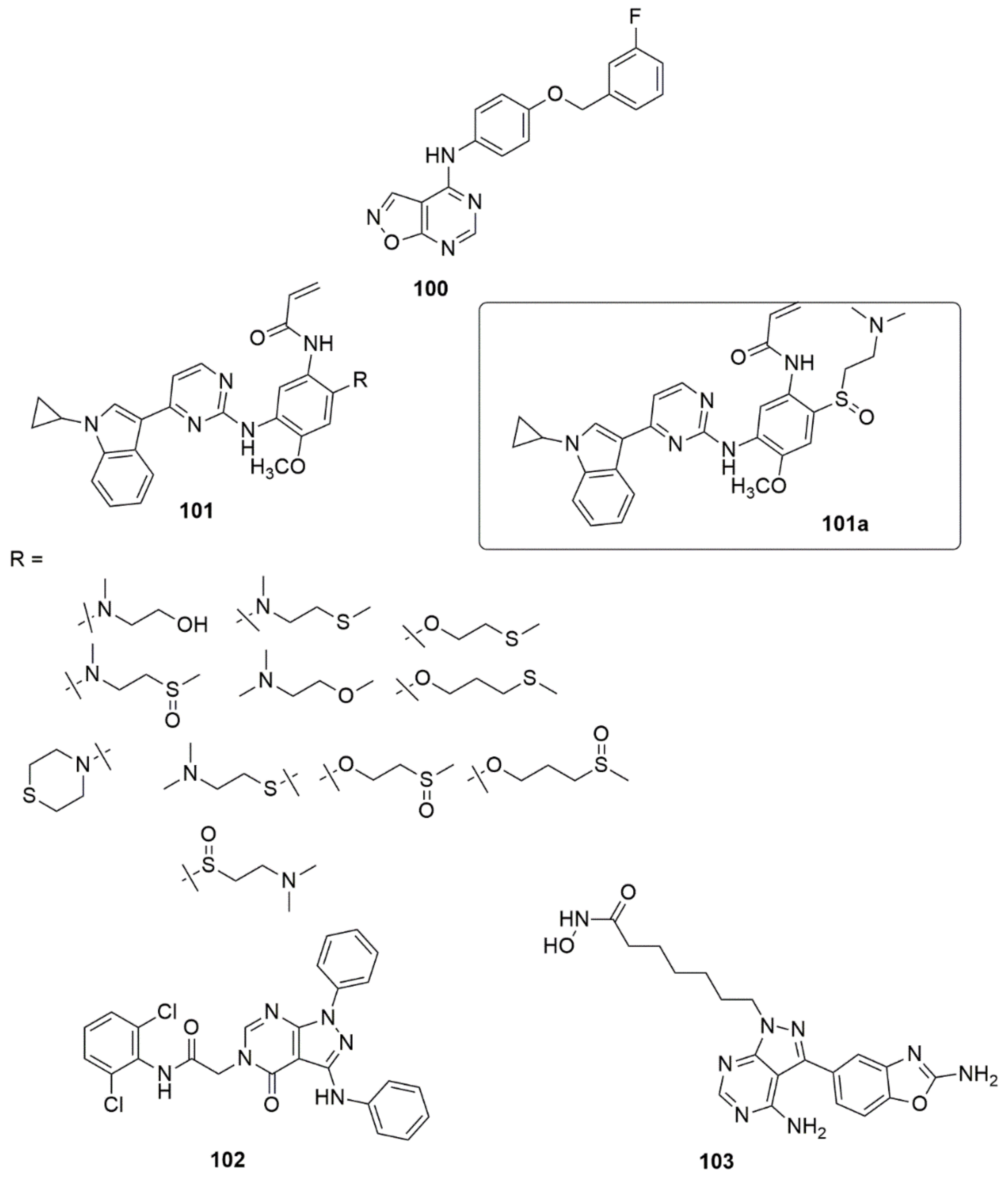{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Five-membered heterocyclic compounds
- Triazole (1,2,3-triazole and 1,2,4-triazole)
- Tetrazole
- Imiazole/Benimidazole
- Six-membered heterocyclic compounds
- Pyrimidine
- Quinoline
- Quinoxaline
- Purine

2. Five-Membered Heterocyclic Compounds
2.1. Triazole
2.2. 1,2,3-Triazole
2.3. 1,2,4-Triazole
2.4. Tetrazole
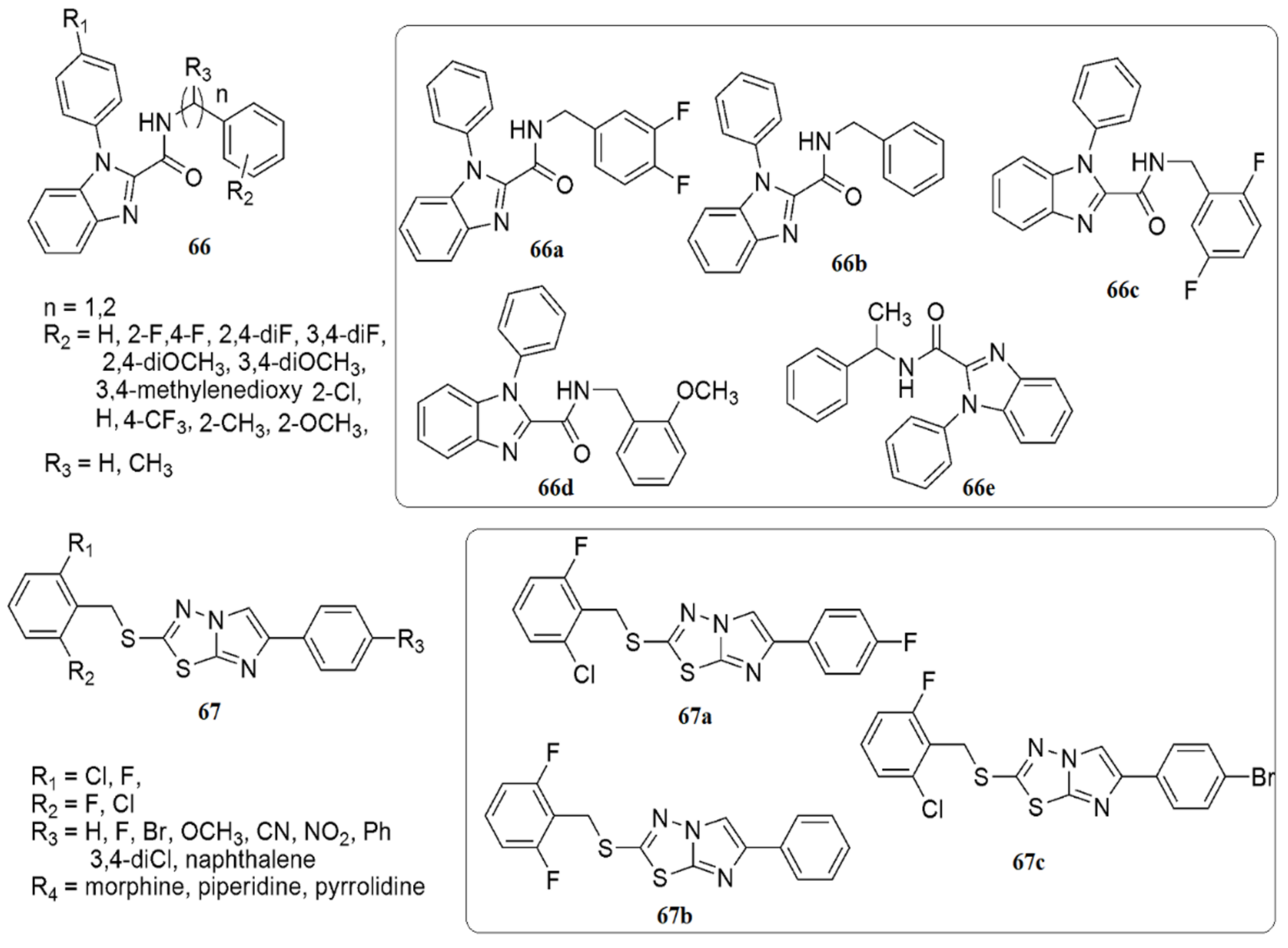
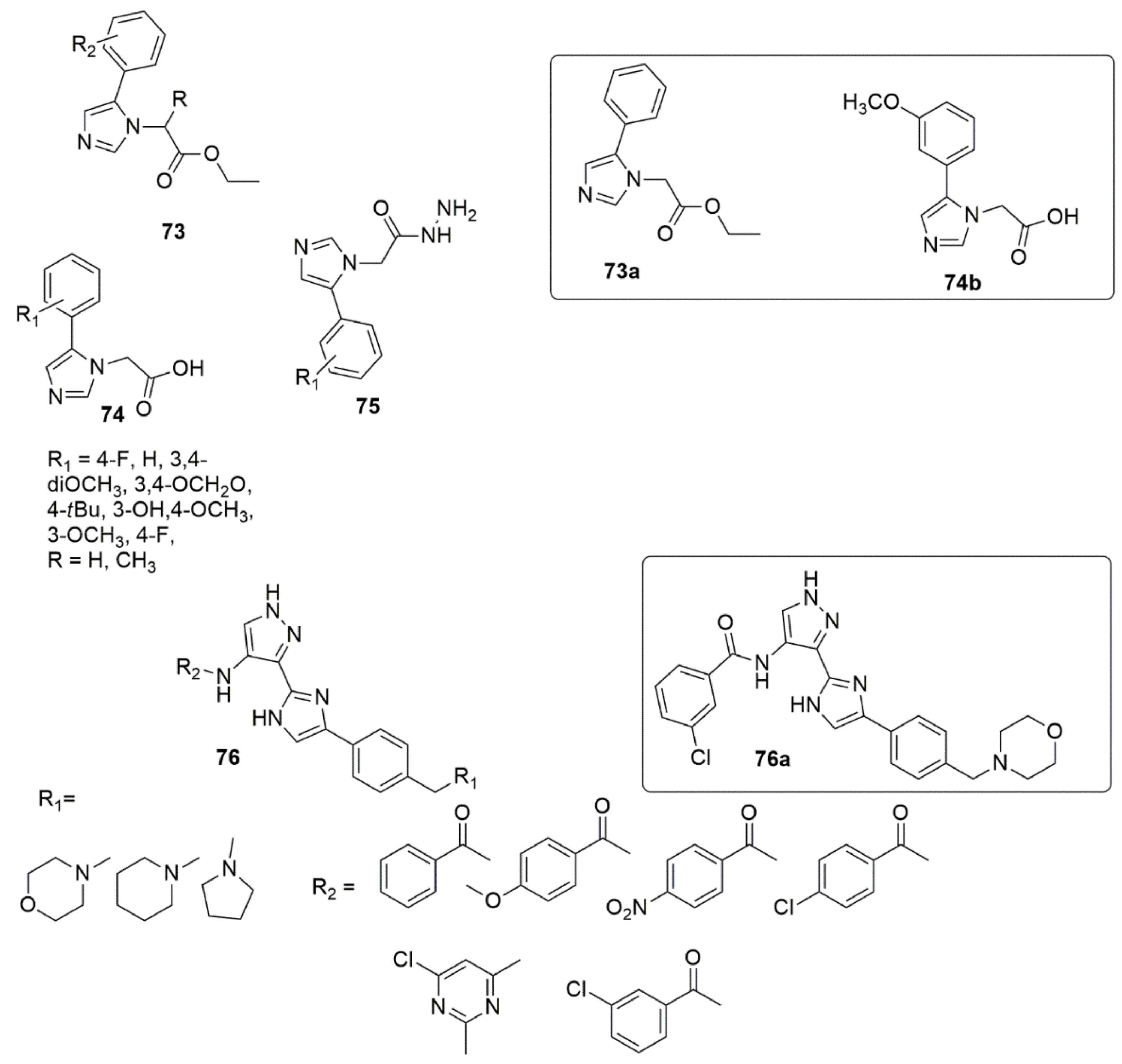
2.5. Imidazole/Benzimidazole
3. Six-Membered Heterocyclic Compounds
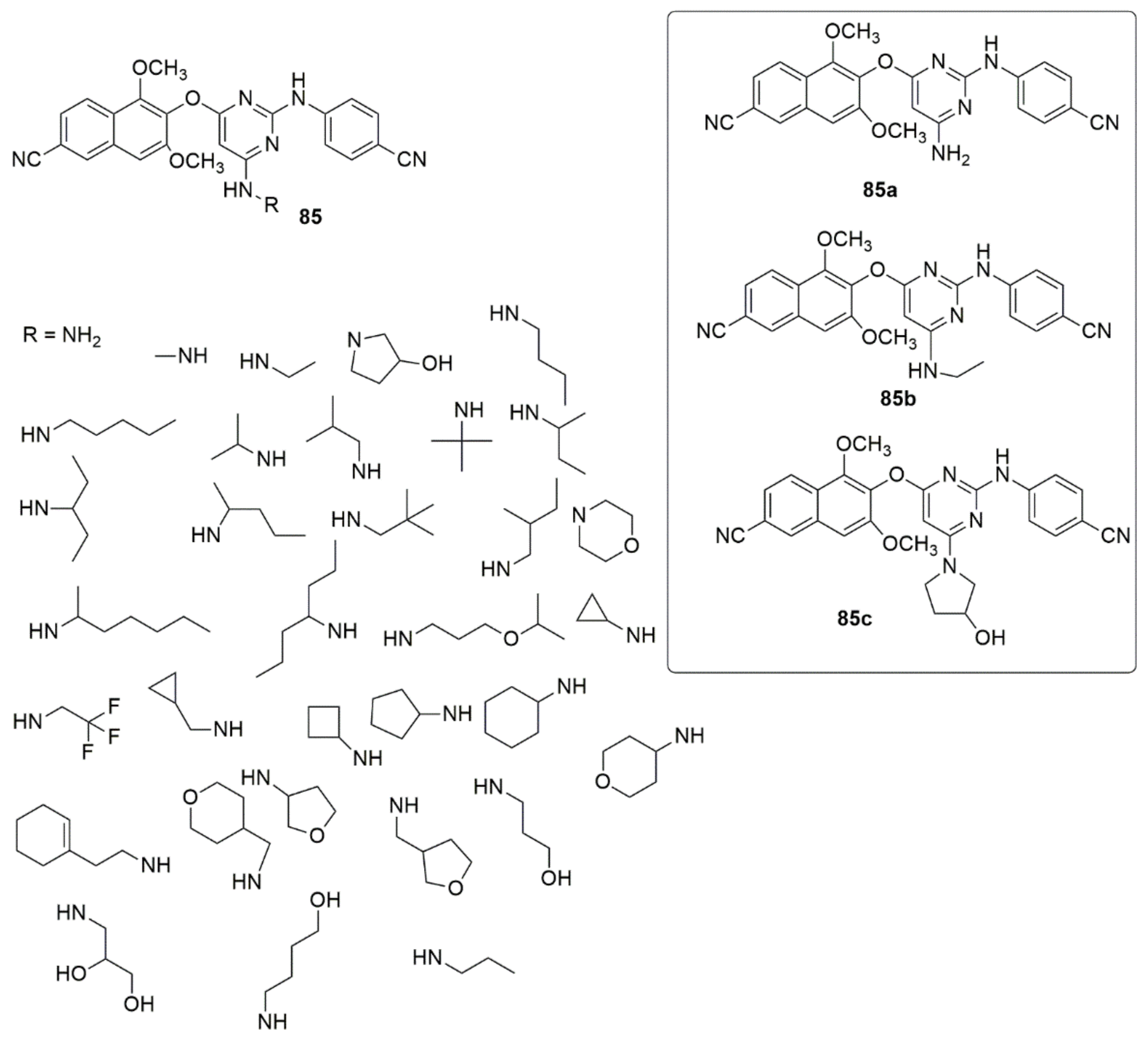
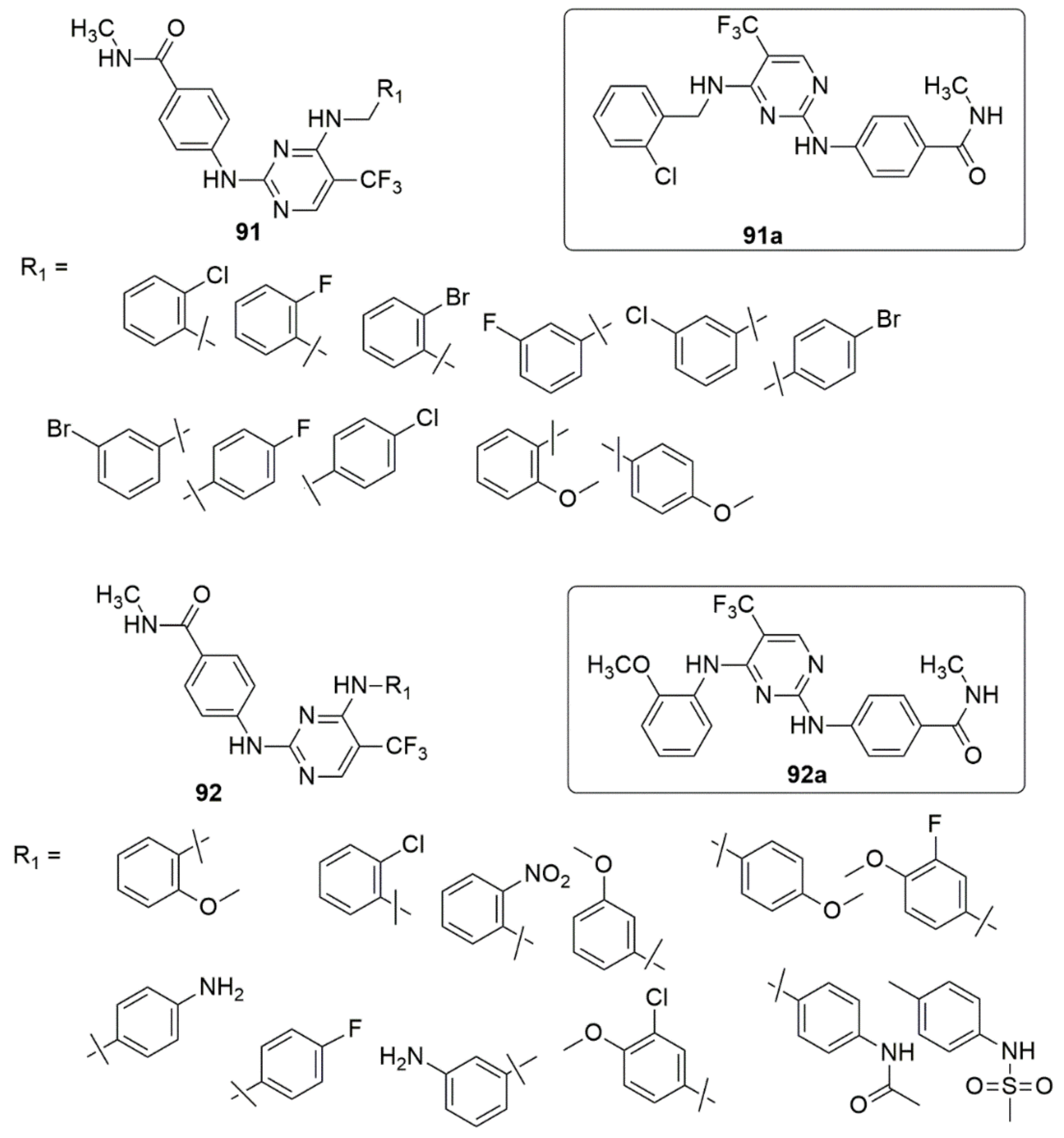
3.1. Pyrimidine
3.2. Quinoline
3.3. Quinoxaline
3.4. Purines
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, W.T.; Hwang, D.R.; Chen, C.P.; Shen, C.W.; Huang, C.L.; Chen, T.W.; Lin, C.H.; Chang, Y.L.; Chang, Y.Y.; Lo, Y.K.; et al. Synthesis and biological evaluation of N-heterocyclic indolyl glyoxylamides as orally active anticancer agents. J. Med. Chem. 2003, 46, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Sörgel, F.; Kinzig, M. Pharmacokinetics of gyrase inhibitors, Part 1: Basic chemistry and gastrointestinal disposition. Am. J. Med. 1993, 94, 44–55. [Google Scholar] [CrossRef]
- Hagen, S.E.; Domagala, J.; Gajda, C.; Lovdahl, M.; Tait, B.D.; Wise, E.; Holler, T.; Hupe, D.; Nouhan, C.; Urumov, A.; et al. 4-Hydroxy-5,6-dihydropyrones as inhibitors of HIV protease: The effect of heterocyclic substituents at C-6 on antiviral potency and pharmacokinetic parameters. J. Med. Chem. 2001, 44, 2319–2332. [Google Scholar] [CrossRef]
- Chu-Moyer, M.Y.; Ballinger, W.E.; Beebe, D.A.; Berger, R.; Coutcher, J.B.; Day, W.W.; Li, J.; Mylari, B.L.; Oates, P.J.; Weekly, R.M. Orally-effective, long-acting sorbitol dehydrogenase inhibitors: Synthesis, structure-activity relationships, and in vivo evaluations of novel heterocycle-substituted piperazino-pyrimidines. J. Med. Chem. 2002, 45, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.K.; Kaur, R.; Arora, R.; Saini, B.; Arora, S. Nitrogen-Containing Heterocycles as Anticancer Agents: An Overview. Anti-Cancer Agents Med. Chem. 2020, 20, 2150–2168. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Yao, C.; Zhong, Z.; Ge, J.; Bai, Z.; Ye, X.; Xie, T.; Xie, Y. Discovery of natural anti-inflammatory alkaloids: Potential leads for the drug discovery for the treatment of inflammation. Eur. J. Med. Chem. 2021, 213, 113165. [Google Scholar] [CrossRef]
- Dhingra, A.K.; Chopra, B.; Dua, J.S.; Prasad, D.N. Therapeutic Potential of N-heterocyclic Analogs as Anti-inflammatory Agents. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2017, 16, 136–152. [Google Scholar] [CrossRef]
- Antoci, V.; Cucu, D.; Zbancioc, G.; Moldoveanu, C.; Mangalagiu, V.; Amariucai-Mantu, D.; Aricu, A.; Mangalagiu, I.I. Bis-(imidazole/benzimidazole)-pyridine derivatives: Synthesis, structure and antimycobacterial activity. Future Med. Chem. 2020, 12, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Wakode, S.; Fayaz, F.; Khasimbi, S.; Pottoo, F.H.; Kaur, A. An Overview of Piperazine Scaffold as Promising Nucleus for Different Therapeutic Targets. Curr. Pharm. Des. 2020, 26, 4373–4385. [Google Scholar] [CrossRef]
- Khatik, G.L.; Datusalia, A.K.; Ahsan, W.; Kaur, P.; Vyas, M.; Mittal, A.; Nayak, S.K. A Retrospect Study on Thiazole Derivatives as the Potential Antidiabetic Agents in Drug Discovery and Developments. Curr. Drug Discov. Technol. 2018, 15, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.; Nath, R.; Bhatia, R.; Akhtar, M.J. Synthetic and therapeutic perspectives of nitrogen containing heterocycles as anti-convulsants. Bioorg. Med. Chem. 2020, 28, 115585. [Google Scholar] [CrossRef] [PubMed]
- Hurtevent, A.; Le Naour, M.; Leclerc, V.; Carato, P.; Melnyk, P.; Hennuyer, N.; Staels, B.; Beucher-Gaudin, M.; Caignard, D.H.; Dacquet, C.; et al. Effect of 6-Benzoyl-benzothiazol-2-one scaffold on the pharmacological profile of α-alkoxyphenylpropionic acid derived PPAR agonists. J. Enzym. Inhib. Med. Chem. 2020, 35, 524–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novel Drug Approvals for 2022. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2022 (accessed on 14 July 2022).
- Zhou, C.H.; Wang, Y. Recent researches in triazole compounds as medicinal drugs. Curr. Med. Chem. 2012, 19, 239–280. [Google Scholar] [CrossRef]
- Riu, F.; Sanna, L.; Ibba, R.; Piras, S.; Bordoni, V.; Scorciapino, M.A.; Lai, M.; Sestito, S.; Bagella, L.; Carta, A. A comprehensive assessment of a new series of 5′,6′-difluorobenzotriazole-acrylonitrile derivatives as microtubule targeting agents (MTAs). Eur. J. Med. Chem. 2021, 222, 113590. [Google Scholar] [CrossRef]
- Kasemsuk, T.; Saehlim, N.; Arsakhant, P.; Sittithumcharee, G.; Okada, S.; Saeeng, R. A novel synthetic acanthoic acid analogues and their cytotoxic activity in cholangiocarcinoma cells. Bioorg. Med. Chem. 2021, 29, 115886. [Google Scholar] [CrossRef]
- Felipe, J.L.; Cassamale, T.B.; Lourenço, L.D.; Carvalho, D.B.; das Neves, A.R.; Duarte, R.C.F.; Carvalho, M.G.; Toffoli-Kadri, M.C.; Baroni, A.C.M. Anti-inflammatory, ulcerogenic and platelet activation evaluation of novel 1,4-diaryl-1,2,3-triazole neolignan-celecoxib hybrids. Bioorg. Chem. 2022, 119, 105485. [Google Scholar] [CrossRef]
- Holanda, V.N.; da Silva, W.V.; do Nascimento, P.H.; Silva, S.R.B.; Cabral Filho, P.E.; de Oliveira Assis, S.P.; da Silva, C.A.; de Oliveira, R.N.; de Figueiredo, R.C.B.Q.; de Menezes Lima, V.L. Antileishmanial activity of 4-phenyl-1-[2-(phthalimido-2-yl) ethyl]-1H-1, 2, 3-triazole (PT4) derivative on Leishmania amazonensis and Leishmania braziliensis: In silico ADMET, in vitro activity, docking and molecular dynamic simulations. Bioorg. Chem. 2020, 105, 104437. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Singh, A.K.; Singh, A.S.; Singh, M.; Maji, P.; Yadav, S.; Rajkhowa, S.; Prakash, P.; Tiwari, V.K. Click inspired synthesis of p-tert-butyl calix [4] arene tethered benzotriazolyl dendrimers and their evaluation as anti-bacterial and anti-biofilm agents. New J. Chem. 2020, 44, 19300–19313. [Google Scholar] [CrossRef]
- El Malah, T.; Mageid, R.E.A.; Awad, H.M.; Nour, H.F. Copper (i)-catalysed azide–alkyne cycloaddition and antiproliferative activity of mono-and bis-1, 2, 3-triazole derivatives. New J. Chem. 2020, 44, 18256–18263. [Google Scholar] [CrossRef]
- Şahin, İ.; Çeşme, M.; Özgeriş, F.B.; Güngör, Ö.; Tümer, F. Design and synthesis of 1,4-disubstituted 1,2,3-triazoles: Biological evaluation, in silico molecular docking and ADME screening. J. Mol. Struct. 2022, 1247, 131344. [Google Scholar] [CrossRef]
- Zhang, G.-Y.; Zhang, Z.; Li, K.; Liu, J.; Li, B.; Jin, Z.; Liu, Y.-H.; Tang, Y.-Z. Design, synthesis and biological evaluation of novel pleuromutilin derivatives containing piperazine and 1, 2, 3-triazole linker. Bioorg. Chem. 2020, 105, 104398. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Cheng, X.; Wang, X.; An, R.; Xu, H.; Guo, M.; Li, C.; Wang, Y.; Hou, Z.; Guo, C. Design, synthesis and biological evaluation of novel carbohydrate-based sulfonamide derivatives as antitumor agents. Bioorg. Chem. 2020, 104, 104237. [Google Scholar] [CrossRef]
- Pal, T.; Bhimaneni, S.; Sharma, A.; Flora, S. Design, synthesis, biological evaluation and molecular docking study of novel pyridoxine–triazoles as anti-Alzheimer’s agents. RSC Adv. 2020, 10, 26006–26021. [Google Scholar] [CrossRef] [PubMed]
- Aneja, B.; Queen, A.; Khan, P.; Shamsi, F.; Hussain, A.; Hasan, P.; Rizvi, M.M.A.; Daniliuc, C.G.; Alajmi, M.F.; Mohsin, M. Design, synthesis & biological evaluation of ferulic acid-based small molecule inhibitors against tumor-associated carbonic anhydrase IX. Bioorg. Med. Chem. 2020, 28, 115424. [Google Scholar]
- Suryanarayana, K.; Maddila, S.; Nagaraju, K.; Jonnalagadda, S.B. Design, synthesis, docking study and biological evaluation of novel thieno[2,3-d]-pyrimidine tethered 1,2,3-triazole scaffolds. J. Mol. Struct. 2022, 1250, 131713. [Google Scholar] [CrossRef]
- Nemati, F.; Salehi, P.; Bararjanian, M.; Hadian, N.; Mohebbi, M.; Lauro, G.; Ruggiero, D.; Terracciano, S.; Bifulco, G.; Bruno, I. Discovery of noscapine derivatives as potential β-tubulin inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127489. [Google Scholar] [CrossRef]
- Sepehri, N.; Asemanipoor, N.; Mousavianfard, S.A.; Hoseini, S.; Faramarzi, M.A.; Adib, M.; Biglar, M.; Larijani, B.; Hamedifar, H.; Mohammadi-Khanaposhtani, M. New acridine-9-carboxamide linked to 1, 2, 3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: Design, synthesis, in vitro, and in silico biological evaluations. Med. Chem. Res. 2020, 29, 1836–1845. [Google Scholar] [CrossRef]
- Cherif, M.; Horchani, M.; Al-Ghamdi, Y.O.; Almalki, S.G.; Alqurashi, Y.E.; Jannet, H.B.; Romdhane, A. New pyrano-1, 2, 3-triazolopyrimidinone derivatives as anticholinesterase and antibacterial agents: Design, microwave-assisted synthesis and molecular docking study. J. Mol. Struct. 2020, 1220, 128685. [Google Scholar] [CrossRef]
- Sun, P.; Zhu, Y.; Han, Y.; Hu, K.; Huang, S.; Wang, M.; Wu, H.; Tang, G. Radiosynthesis and biological evaluation of an fluorine-18 labeled galactose derivative [18F] FPGal for imaging the hepatic asialoglycoprotein receptor. Bioorg. Med. Chem. Lett. 2020, 30, 127187. [Google Scholar] [CrossRef]
- Shi, S.; Wang, H.; Wang, J.; Wang, Y.; Xue, X.; Hou, Z.; Yao, G.-D.; Huang, X.-X.; Zhao, H.; Liu, Q. Semi-synthesis and biological evaluation of flavone hybrids as multifunctional agents for the potential treatment of Alzheimer’s disease. Bioorg. Chem. 2020, 100, 103917. [Google Scholar] [CrossRef] [PubMed]
- Tangadanchu, V.K.R.; Jiang, H.; Yu, Y.; Graham, T.J.; Liu, H.; Rogers, B.E.; Gropler, R.; Perlmutter, J.; Tu, Z. Structure-activity relationship studies and bioactivity evaluation of 1, 2, 3-triazole containing analogues as a selective sphingosine kinase-2 inhibitors. Eur. J. Med. Chem. 2020, 206, 112713. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, W.; Zhang, Y.; Fu, X.; Ping, K.; Zhao, J.; Lei, Y.; Mou, Y.; Wang, S. Synthesis and biological evaluation of celastrol derivatives as potential anti-glioma agents by activating RIP1/RIP3/MLKL pathway to induce necroptosis. Eur. J. Med. Chem. 2022, 229, 114070. [Google Scholar] [CrossRef] [PubMed]
- Chaidam, S.; Saehlim, N.; Athipornchai, A.; Sirion, U.; Saeeng, R. Synthesis and biological evaluation of 1, 6-bis-triazole-2, 3, 4-tri-O-benzyl-α-d-glucopyranosides as a novel α-glucosidase inhibitor in the treatment of Type 2 diabetes. Bioorg. Med. Chem. Lett. 2021, 50, 128331. [Google Scholar] [CrossRef] [PubMed]
- Payne, M.; Bottomley, A.L.; Och, A.; Hiscocks, H.G.; Asmara, A.P.; Harry, E.J.; Ung, A.T. Synthesis and biological evaluation of tetrahydroisoquinoline-derived antibacterial compounds. Bioorg. Med. Chem. 2022, 57, 116648. [Google Scholar] [CrossRef]
- Le-Nhat-Thuy, G.; Thi, N.N.; Pham-The, H.; Thi, T.A.D.; Thi, H.N.; Thi, T.H.N.; Hoang, S.N.; Van Nguyen, T. Synthesis and biological evaluation of novel quinazoline-triazole hybrid compounds with potential use in Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2020, 30, 127404. [Google Scholar] [CrossRef]
- Begam, R.; Shajahan, A.; Shefin, B.; Murugan, V. Synthesis of novel naphthalimide tethered 1,2,3-triazoles: In vitro biological evaluation and docking study of anti-inflammatory inhibitors. J. Mol. Struct. 2022, 1254, 132364. [Google Scholar] [CrossRef]
- Hosseini, S.; Pourmousavi, S.A.; Mahdavi, M.; Taslimi, P. Synthesis, and in vitro biological evaluations of novel naphthoquinone conjugated to aryl triazole acetamide derivatives as potential anti-Alzheimer agents. J. Mol. Struct. 2022, 1255, 132229. [Google Scholar] [CrossRef]
- Gurrapu, N.; Kumar, E.P.; Kolluri, P.K.; Putta, S.; Sivan, S.K.; Subhashini, N. Synthesis, biological evaluation and molecular docking studies of novel 1, 2, 3-triazole tethered chalcone hybrids as potential anticancer agents. J. Mol. Struct. 2020, 1217, 128356. [Google Scholar] [CrossRef]
- Abdel-Hafez, G.A.; Mohamed, A.-M.I.; Youssef, A.F.; Simons, C.; Aboraia, A.S. Synthesis, computational study and biological evaluation of 9-acridinyl and 1-coumarinyl-1, 2, 3-triazole-4-yl derivatives as topoisomerase II inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 502–513. [Google Scholar] [CrossRef]
- Vo, D.V.; Hong, K.H.; Lee, J.; Park, H. Synthesis, in vitro evaluation, and computational simulations studies of 1,2,3-triazole analogues as DPP-4 inhibitors. Bioorg. Med. Chem. 2021, 29, 115861. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.H.; Sojo, F.; Arvelo, F.; Calderón, C.; Morales, A.; López, S.E. Anticancer potential of new 3-nitroaryl-6-(N-methyl) piperazin-1, 2, 4-triazolo [3, 4-a] phthalazines targeting voltage-gated K+ channel: Copper-catalyzed one-pot synthesis from 4-chloro-1-phthalazinyl-arylhydrazones. Bioorg. Chem. 2020, 101, 104031. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-E.; Li, S.-M.; Tseng, C.-C.; Chung, C.-Y.; Zeng, Y.-H.; Lin, C.C.; Fuh, M.-T.; Yang, L.-C.; Yang, Y.-C.; Wong, F.-F. Chlorotrimethylsilane promoted one-flask heterocyclic synthesis of 1, 2, 4-triazoles from nitrilimines: Modeling studies and bioactivity evaluation of LH-21 and Rimonabant analogues. Bioorg. Chem. 2020, 104, 104299. [Google Scholar] [CrossRef] [PubMed]
- Cebeci, Y.U.; Ceylan, Ş.; Karaoğlu, Ş.A. Conventional and microwave irradiated synthesis, biological activity evaluation of highly substituted indole-triazole hybrids. J. Mol. Struct. 2022, 1250, 131799. [Google Scholar] [CrossRef]
- Wang, N.-Y.; Xu, Y.; Xiao, K.-J.; Zuo, W.-Q.; Zhu, Y.-X.; Hu, R.; Wang, W.-L.; Shi, Y.-J.; Yu, L.-T.; Liu, Z.-H. Design, synthesis, and biological evaluation of 4, 5-dihydro-[1, 2, 4] triazolo [4, 3-f] pteridine derivatives as novel dual-PLK1/BRD4 inhibitors. Eur. J. Med. Chem. 2020, 191, 112152. [Google Scholar] [CrossRef]
- Wu, C.J.; Wu, J.Q.; Hu, Y.; Pu, S.; Lin, Y.; Zeng, Z.; Hu, J.; Chen, W.H. Design, synthesis and biological evaluation of indole-based [1,2,4]triazolo[4,3-a] pyridine derivatives as novel microtubule polymerization inhibitors. Eur. J. Med. Chem. 2021, 223, 113629. [Google Scholar] [CrossRef]
- Luo, R.; Wang, Z.; Luo, D.; Qin, Y.; Zhao, C.; Yang, D.; Lu, T.; Zhou, Z.; Huang, Z. Design, synthesis, and biological evaluation of novel triazoloquinazolinone derivatives as SHP2 protein inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 2170–2182. [Google Scholar] [CrossRef]
- Jain, A.; Piplani, P. Design, synthesis and biological evaluation of triazole-oxadiazole conjugates for the management of cognitive dysfunction. Bioorg. Chem. 2020, 103, 104151. [Google Scholar] [CrossRef]
- Li, S.-M.; Tsai, S.-E.; Chiang, C.-Y.; Chung, C.-Y.; Chuang, T.-J.; Tseng, C.-C.; Jiang, W.-P.; Huang, G.-J.; Lin, C.-Y.; Yang, Y.-C. New methyl 5-(halomethyl)-1-aryl-1H-1, 2, 4-triazole-3-carboxylates as selective COX-2 inhibitors and anti-inflammatory agents: Design, synthesis, biological evaluation, and docking study. Bioorg. Chem. 2020, 104, 104333. [Google Scholar] [CrossRef]
- Yang, F.; Jian, X.-E.; Diao, P.-C.; Huo, X.-S.; You, W.-W.; Zhao, P.-L. Synthesis, and biological evaluation of 3, 6-diaryl-[1, 2, 4] triazolo [4, 3-a] pyridine analogues as new potent tubulin polymerization inhibitors. Eur. J. Med. Chem. 2020, 204, 112625. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; Alqahtani, A.M.; Omar, H.A.; Bukhari, S.N.A.; Gouda, A.M. Synthesis, biological evaluation and kinase profiling of novel S-benzo [4, 5] thiazolo [2, 3-c][1, 2, 4] triazole derivatives as cytotoxic agents with apoptosis-inducing activity. J. Mol. Struct. 2020, 1219, 128567. [Google Scholar] [CrossRef]
- Ma, W.; Chen, P.; Huo, X.; Ma, Y.; Li, Y.; Diao, P.; Yang, F.; Zheng, S.; Hu, M.; You, W. Development of triazolothiadiazine derivatives as highly potent tubulin polymerization inhibitors: Structure-activity relationship, in vitro and in vivo study. Eur. J. Med. Chem. 2020, 208, 112847. [Google Scholar] [CrossRef] [PubMed]
- Wittenberger, S.J. Recent developments in tetrazole chemistry. A review. Org. Prep. Proced. Int. 1994, 26, 499–531. [Google Scholar] [CrossRef]
- Myznikov, L.; Hrabalek, A.; Koldobskii, G. Drugs in the tetrazole series. Chem. Heterocycl. Compd. 2007, 43, 1–9. [Google Scholar] [CrossRef]
- Zhao, H.; Qu, Z.-R.; Ye, H.-Y.; Xiong, R.-G. In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties. Chem. Soc. Rev. 2008, 37, 84–100. [Google Scholar] [CrossRef]
- Yoneyama, H.; Usami, Y.; Komeda, S.; Harusawa, S. Efficient transformation of inactive nitriles into 5-substituted 1H-tetrazoles using microwave irradiation and their applications. Synthesis 2013, 45, 1051–1059. [Google Scholar] [CrossRef]
- Herr, R.J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: Medicinal chemistry and synthetic methods. Bioorg. Med. Chem. 2002, 10, 3379–3393. [Google Scholar] [CrossRef]
- Wei, C.X.; Bian, M.; Gong, G.H. Tetrazolium compounds: Synthesis and applications in medicine. Molecules 2015, 20, 5528–5553. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, Y.; Liu, Z.; Wang, Z.; Liu, Z.; Man, S.; Zhang, Y.; Bao, K.; Wu, Y.; Guan, Q.; et al. Design, synthesis and biological evaluation of 1-Aryl-5-(4-arylpiperazine-1-carbonyl)-1H-tetrazols as novel microtubule destabilizers. J. Enzym. Inhib. Med. Chem. 2021, 36, 549–560. [Google Scholar] [CrossRef]
- Ulgheri, F.; Spanu, P.; Deligia, F.; Loriga, G.; Fuggetta, M.P.; de Haan, I.; Chandgudge, A.; Groves, M.; Domling, A. Design, synthesis and biological evaluation of 1,5-disubstituted α-amino tetrazole derivatives as non-covalent inflammasome-caspase-1 complex inhibitors with potential application against immune and inflammatory disorders. Eur. J. Med. Chem. 2022, 229, 114002. [Google Scholar] [CrossRef]
- Shekouhy, M.; Karimian, S.; Moaddeli, A.; Faghih, Z.; Delshad, Y.; Khalafi-Nezhad, A. The synthesis and biological evaluation of nucleobases/tetrazole hybrid compounds: A new class of phosphodiesterase type 3 (PDE3) inhibitors. Bioorg. Med. Chem. 2020, 28, 115540. [Google Scholar] [CrossRef] [PubMed]
- Rashidipour, A.; Alizadeh, R.; Sadeghi Mohammadi, S.; Tohidlou, M.; Amani, V.; Seyfi, S. Synthesis, crystal structures and biological activity of palladium(II) complexes with 1-methyl-1H-1,2,3,4-tetrazole-5-thiol and substituted 2,2′-bipyridines. J. Coord. Chem. 2020, 73, 3249–3266. [Google Scholar] [CrossRef]
- Pooi, B.; Lee, J.; Choi, K.; Hirao, H.; Hong, S.H. Tandem insertion-cyclization reaction of isocyanides in the synthesis of 1,4-diaryl-1H-imidazoles: Presence of N-arylformimidate intermediate. J. Org. Chem. 2014, 79, 9231–9245. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. The role of imidazole and benzimidazole heterocycles in Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112692. [Google Scholar] [CrossRef] [PubMed]
- Daraji, D.G.; Rajani, D.P.; Rajani, S.D.; Pithawala, E.A.; Jayanthi, S.; Patel, H.D. Structure based design, synthesis, and biological evaluation of imidazole derivatives targeting dihydropteroate synthase enzyme. Bioorg. Med. Chem. Lett. 2021, 36, 127819. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, Z.; Zhang, Z.; Yan, D.; Zhang, W. Synthesis and biological evaluation of naphthoquinone phenacylimidazolium derivatives. Bioorg. Med. Chem. Lett. 2021, 41, 127977. [Google Scholar] [CrossRef]
- Al-Hamashi, A.A.; Koranne, R.; Dlamini, S.; Alqahtani, A.; Karaj, E.; Rashid, M.S.; Knoff, J.R.; Dunworth, M.; Pflum, M.K.H.; Casero, R.A.; et al. A new class of cytotoxic agents targets tubulin and disrupts microtubule dynamics. Bioorg. Chem. 2021, 116, 105297. [Google Scholar] [CrossRef]
- Wu, Z.; Xia, M.B.; Bertsetseg, D.; Wang, Y.H.; Bao, X.L.; Zhu, W.B.; Tao, X.; Chen, P.R.; Tang, H.S.; Yan, Y.J.; et al. Design, synthesis and biological evaluation of novel fluoro-substituted benzimidazole derivatives with anti-hypertension activities. Bioorg. Chem. 2020, 101, 104042. [Google Scholar] [CrossRef]
- Dhameliya, T.M.; Patel, K.I.; Tiwari, R.; Vagolu, S.K.; Panda, D.; Sriram, D.; Chakraborti, A.K. Design, synthesis, and biological evaluation of benzo[d]imidazole-2-carboxamides as new anti-TB agents. Bioorg. Chem. 2021, 107, 104538. [Google Scholar] [CrossRef]
- Askin, S.; Tahtaci, H.; Türkeş, C.; Demir, Y.; Ece, A.; Akalın Çiftçi, G.; Beydemir, Ş. Design, synthesis, characterization, in vitro and in silico evaluation of novel imidazo[2,1-b][1,3,4]thiadiazoles as highly potent acetylcholinesterase and non-classical carbonic anhydrase inhibitors. Bioorg. Chem 2021, 113, 105009. [Google Scholar] [CrossRef]
- Liu, J.; Liu, F.; Li, Z.; Li, C.; Wu, S.; Shen, J.; Wang, H.; Du, S.; Wei, H.; Hou, Y.; et al. Novel 4-phenoxypyridine derivatives bearing imidazole-4-carboxamide and 1,2,4-triazole-3-carboxamide moieties: Design, synthesis and biological evaluation as potent antitumor agents. Bioorg. Chem. 2022, 120, 105629. [Google Scholar] [CrossRef] [PubMed]
- Gadekar, P.K.; Urunkar, G.; Roychowdhury, A.; Sharma, R.; Bose, J.; Khanna, S.; Damre, A.; Sarveswari, S. Design, synthesis and biological evaluation of 2,3-dihydroimidazo[2,1-b]thiazoles as dual EGFR and IGF1R inhibitors. Bioorg. Chem. 2021, 115, 105151. [Google Scholar] [CrossRef] [PubMed]
- Rashamuse, T.J.; Harrison, A.T.; Mosebi, S.; van Vuuren, S.; Coyanis, E.M.; Bode, M.L. Design, synthesis and biological evaluation of imidazole and oxazole fragments as HIV-1 integrase-LEDGF/p75 disruptors and inhibitors of microbial pathogens. Bioorg. Med. Chem. 2020, 28, 115210. [Google Scholar] [CrossRef] [PubMed]
- Rashamuse, T.J.; Njengele, Z.; Coyanis, E.M.; Sayed, Y.; Mosebi, S.; Bode, M.L. Design, synthesis and biological evaluation of novel 2-(5-aryl-1H-imidazol-1-yl) derivatives as potential inhibitors of the HIV-1 Vpu and host BST-2 protein interaction. Eur. J. Med. Chem. 2020, 190, 112111. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.G.; Wang, J.A.; Meng, L.; Pei, X.; Zhang, L.; An, L.; Li, C.L.; Miao, Y.L. Design, synthesis, biological activity evaluation of 3-(4-phenyl-1H-imidazol-2-yl)-1H-pyrazole derivatives as potent JAK 2/3 and aurora A/B kinases multi-targeted inhibitors. Eur. J. Med. Chem 2021, 209, 112934. [Google Scholar] [CrossRef]
- Reddy, G.L.; Sarma, R.; Liu, S.; Huang, W.; Lei, J.; Fu, J.; Hu, W. Design, synthesis and biological evaluation of novel scaffold benzo[4,5]imidazo [1,2-a]pyrazin-1-amine: Towards adenosine A2A receptor (A2A AR) antagonist. Eur. J. Med. Chem. 2021, 210, 113040. [Google Scholar] [CrossRef]
- Ali, E.M.H.; El-Telbany, R.F.A.; Abdel-Maksoud, M.S.; Ammar, U.M.; Mersal, K.I.; Zaraei, S.O.; El-Gamal, M.I.; Choi, S.I.; Lee, K.T.; Kim, H.K.; et al. Design, synthesis, biological evaluation, and docking studies of novel (imidazol-5-yl)pyrimidine-based derivatives as dual BRAFV600E/p38α inhibitors. Eur. J. Med. Chem. 2021, 215, 113277. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, B.; Zhang, Y.; Dai, X.; Duan, Y.; Mao, Q.; Gao, J.; Yang, Y.; Bao, Z.; Fu, X.; et al. Design, synthesis and biological evaluation of novel FXIa inhibitors with 2-phenyl-1H-imidazole-5-carboxamide moiety as P1 fragment. Eur. J. Med. Chem. 2021, 220, 113437. [Google Scholar] [CrossRef]
- Sekioka, R.; Honda, S.; Akashiba, H.; Yarimizu, J.; Mitani, Y.; Yamasaki, S. Optimization and biological evaluation of imidazopyridine derivatives as a novel scaffold for γ-secretase modulators with oral efficacy against cognitive deficits in Alzheimer’s disease model mice. Bioorg. Med. Chem. 2020, 28, 115455. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, P.; Zhao, L.; Zhang, B.; Xu, C.; Zhang, H.; Zhou, J. Design, synthesis and biological evaluation of imidazolopyridone derivatives as novel BRD4 inhibitors. Bioorg. Med. Chem. 2021, 29, 115857. [Google Scholar] [CrossRef]
- Bu, H.; Yuan, X.; Wu, H.; Zhou, J.; Zhang, H. Design, synthesis and biological evaluation of imidazopyridazine derivatives containing isoquinoline group as potent MNK1/2 inhibitors. Bioorg. Med. Chem. 2021, 40, 116186. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Piao, H.R.; Pannecouque, C.; De Clercq, E.; Zhuang, C.; Chen, F.E. Design of the naphthyl-diarylpyrimidines as potent non-nucleoside reverse transcriptase inhibitors (NNRTIs) via structure-based extension into the entrance channel. Eur. J. Med. Chem. 2021, 226, 113868. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Z.; Gao, C.; Dai, H.; Si, X.; Zhang, Y.; Meng, Y.; Zheng, J.; Ke, Y.; Liu, H.; et al. Design, synthesis and antitumor activity evaluation of trifluoromethyl-substituted pyrimidine derivatives. Bioorg. Med. Chem. Lett. 2021, 51, 128268. [Google Scholar] [CrossRef]
- Senapathi, J.; Bommakanti, A.; Kusuma, V.; Vangara, S.; Kondapi, A.K. Design, Synthesis, and Antiviral activity of 1,2,3,4-Tetrahydropyrimidine derivatives acting as novel entry inhibitors to target at “Phe43 cavity” of HIV-1 gp120. Bioorg. Med. Chem. 2021, 52, 116526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, P.; Duan, Y.; Xiong, H.; Li, H.; Zeng, Y.; Liang, G.; Tang, Q.; Wu, D. Design, synthesis and biological evaluation of 7H-pyrrolo[2,3-d]pyrimidine derivatives containing 1,8-naphthyridine-4-one fragment. Eur. J. Med. Chem. 2021, 215, 113273. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, R.H.; Zhang, H.; Wang, Y.C.; Yang, D.; Zhao, Y.L.; Yan, G.Y.; Xu, G.B.; Guan, H.Y.; Zhou, Y.H.; et al. Design, synthesis, and biological evaluation of 2,4-diamino pyrimidine derivatives as potent FAK inhibitors with anti-cancer and anti-angiogenesis activities. Eur. J. Med. Chem. 2021, 222, 113573. [Google Scholar] [CrossRef]
- Wei, W.; Feng, Z.; Liu, Z.; Li, X.; He, H.; Ran, K.; Shi, Y.; Zhu, Y.; Ye, T.; Gao, C.; et al. Design, synthesis and biological evaluation of 7-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-2,3-dihydro-1H-inden-1-one derivatives as potent FAK inhibitors for the treatment of ovarian cancer. Eur. J. Med. Chem. 2022, 228, 113978. [Google Scholar] [CrossRef]
- Li, Y.; Chang, Y.; Fu, J.; Ding, R.; Zhang, L.; Liang, T.; Liu, Y.; Liu, Y.; Hu, J. Design, synthesis and biological evaluation of aminopyrimidine derivatives bearing a 4,5,6,7-tetrahydrothieno [3,2-c]pyridine as potent EGFR inhibitors. Eur. J. Med. Chem. 2021, 226, 113845. [Google Scholar] [CrossRef]
- Wu, B.; Yang, S.; Deng, T.; Wang, C.; Jin, Y.; Yu, J.; Xu, Y.; Chen, L.; Li, Y.; Ma, X. Design, synthesis, and biological evaluation of cyano-substituted 2,4-diarylaminopyrimidines as potent JAK3 inhibitors for the treatment of B-cell lymphoma. Bioorg. Chem. 2021, 116, 105330. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Wang, C.; Tang, Z.; Meng, Q.; Sun, H.; Qi, Y.; Ma, X.; Li, L.; Li, Y.; et al. Design, synthesis, and biological evaluation of hydroxamic acid-substituted 2,4-diaryl aminopyrimidines as potent EGFRT790M/L858R inhibitors for the treatment of NSCLC. Bioorg. Chem. 2021, 114, 105045. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, Y.; Yang, Y.; Mao, Q.; Lin, F.; Gao, J.; Dai, X.; Zhang, P.; Li, Q.; Li, J.; et al. Design, synthesis, and biological evaluation of N-(3-cyano-1H-indol-5/6-yl)-6-oxo-1,6-dihydropyrimidine-4-carboxamides and 5-(6-oxo-1,6-dihydropyrimidin-2-yl)-1H-indole-3-carbonitriles as novel xanthine oxidase inhibitors. Eur. J. Med. Chem. 2022, 227, 113928. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, S.A.; Taher, E.S.; Lan, P.; Asaad, G.F.; Gomaa, H.A.M.; El-Koussi, N.A.; Youssif, B.G.M. Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1,3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 2021, 111, 104890. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, N.B.; Bansod, S.; Mara, A.; Garise, R.; Srinivas, N.; Godugu, C.; Yaddanapudi, V.M. Design, synthesis, and biological evaluation of N-(4-substituted)-3-phenylisoxazolo[5,4–d]pyrimidin-4-amine derivatives as apoptosis-inducing cytotoxic agents. Bioorg. Med. Chem. Lett. 2021, 49, 128294. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; An, B.; Song, X.; Zhang, Q.; Chen, C.; Wei, S.; Fan, R.; Li, X.; Zou, Y. Design, synthesis and biological evaluation of novel 2,4-diaryl pyrimidine derivatives as selective EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2021, 212, 113019. [Google Scholar] [CrossRef]
- Sherbiny, F.F.; Bayoumi, A.H.; El-Morsy, A.M.; Sobhy, M.; Hagras, M. Design, Synthesis, biological Evaluation, and molecular docking studies of novel Pyrazolo[3,4-d]Pyrimidine derivative scaffolds as potent EGFR inhibitors and cell apoptosis inducers. Bioorg. Chem. 2021, 116. [Google Scholar] [CrossRef]
- Zhai, S.; Zhang, H.; Chen, R.; Wu, J.; Ai, D.; Tao, S.; Cai, Y.; Zhang, J.Q.; Wang, L. Design, synthesis and biological evaluation of novel hybrids targeting mTOR and HDACs for potential treatment of hepatocellular carcinoma. Eur. J. Med. Chem. 2021, 225, 113824. [Google Scholar] [CrossRef]
- Manzoor, S.; Prajapati, S.K.; Majumdar, S.; Raza, K.; Gabr, M.T.; Kumar, S.; Pal, K.; Rashid, H.; Kumar, S.; Krishnamurthy, S.; et al. Discovery of new phenyl sulfonyl-pyrimidine carboxylate derivatives as the potential multi-target drugs with effective anti-Alzheimer’s action: Design, synthesis, crystal structure and in-vitro biological evaluation. Eur. J. Med. Chem. 2021, 215, 113224. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Jain, P.K.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef] [Green Version]
- El-Shershaby, M.H.; El-Gamal, K.M.; Bayoumi, A.H.; El-Adl, K.; Ahmed, H.E.A.; Abulkhair, H.S. Synthesis, antimicrobial evaluation, DNA gyrase inhibition, and in silico pharmacokinetic studies of novel quinoline derivatives. Arch. Pharm. 2021, 354, e2000277. [Google Scholar] [CrossRef]
- Gaikwad, N.B.; Bansode, S.; Biradar, S.; Ban, M.; Srinivas, N.; Godugu, C.; Yaddanapudi, V.M. New 3-(1H-benzo[d]imidazol-2-yl)quinolin-2(1H)-one-based triazole derivatives: Design, synthesis, and biological evaluation as antiproliferative and apoptosis-inducing agents. Arch. Pharm. 2021, 354, e2100074. [Google Scholar] [CrossRef]
- Karnik, K.S.; Sarkate, A.P.; Tiwari, S.V.; Azad, R.; Burra, P.; Wakte, P.S. Computational and Synthetic approach with Biological Evaluation of Substituted Quinoline derivatives as small molecule L858R/T790M/C797S triple mutant EGFR inhibitors targeting resistance in Non-Small Cell Lung Cancer (NSCLC). Bioorg. Chem. 2021, 107, 104612. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Xu, B.; Ping, L.; Lv, X. Structural optimization towards promising β-methyl-4-acrylamido quinoline derivatives as PI3K/mTOR dual inhibitors for anti-cancer therapy: The in vitro and in vivo biological evaluation. Eur. J. Med. Chem. 2021, 214, 113249. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wu, X.; Yan, S.; Liu, T.; Yin, X. Synthesis and in vitro evaluation of novel spiroketopyrazoles as acetyl-CoA carboxylase inhibitors and potential antitumor agents. Eur. J. Med. Chem. 2021, 212, 113036. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.D.; Amor, P.; Bader, S.; Buzon, L.M.; Coffey, S.B.; Corbett, J.W.; Dirico, K.J.; Doran, S.D.; Elliott, R.L.; Esler, W.; et al. Maximizing lipophilic efficiency: The use of Free-Wilson analysis in the design of inhibitors of acetyl-CoA carboxylase. J. Med. Chem. 2012, 55, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Mirzazadeh, R.; Asgari, M.S.; Barzegari, E.; Pedrood, K.; Mohammadi-Khanaposhtani, M.; Sherafati, M.; Larijani, B.; Rastegar, H.; Rahmani, H.; Mahdavi, M.; et al. New quinoxalin-1,3,4-oxadiazole derivatives: Synthesis, characterization, in vitro biological evaluations, and molecular modeling studies. Arch. Pharm. 2021, 354, 202000471. [Google Scholar] [CrossRef]
- Kumar Jain, A.; Gupta, A.; Karthikeyan, C.; Trivedi, P.; Dutt Konar, A. Unravelling the Selectivity of 6,7-Dimethyl Quinoxaline Analogs for Kinase Inhibition: An Insight towards the Development of Alzheimer’s Therapeutics. Chem. Biodivers. 2021, 18, 202100364. [Google Scholar] [CrossRef]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.M.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2(1H)-one-based anticancer agents targeting VEGFR-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Mahdy, H.A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Dahab, M.A.; Eissa, I.H. New bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem. 2021, 112, 104949. [Google Scholar] [CrossRef]
- Poulsen, A.; Nagaraj, H.; Lee, A.; Blanchard, S.; Soh, C.K.; Chen, D.; Wang, H.; Hart, S.; Goh, K.C.; Dymock, B.; et al. Structure and ligand-based design of mTOR and PI3-kinase inhibitors leading to the clinical candidates VS-5584 (SB2343) and SB2602. J. Chem. Inf. Model. 2014, 54, 3238–3250. [Google Scholar] [CrossRef]
- Chen, D.; Soh, C.K.; Goh, W.H.; Wang, Z.; Wang, H. Synthesis and biological evaluation of 6-phenylpurine linked hydroxamates as novel histone deacetylase inhibitors. Bioorg. Chem. 2020, 98, 103724. [Google Scholar] [CrossRef]
- Kim, Y.S.; Cheon, M.G.; Boggu, P.R.; Koh, S.Y.; Park, G.M.; Kim, G.; Park, S.H.; Park, S.L.; Lee, C.W.; Kim, J.W.; et al. Synthesis and biological evaluation of novel purinyl quinazolinone derivatives as PI3Kδ-specific inhibitors for the treatment of hematologic malignancies. Bioorg. Med. Chem. 2021, 45, 116312. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.T.; He, W.B.; Mai, X.; Feng, L.H.; Li, N.; Liao, Y.J.; Zhu, C.S.; Li, J.; Chen, T.; Liu, S.H.; et al. Synthesis and biological evaluation of aminobenzamides containing purine moiety as class I histone deacetylases inhibitors. Bioorg. Med. Chem. 2022, 56, 116599. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebenezer, O.; Jordaan, M.A.; Carena, G.; Bono, T.; Shapi, M.; Tuszynski, J.A. An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds. Int. J. Mol. Sci. 2022, 23, 8117. https://doi.org/10.3390/ijms23158117
Ebenezer O, Jordaan MA, Carena G, Bono T, Shapi M, Tuszynski JA. An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds. International Journal of Molecular Sciences. 2022; 23(15):8117. https://doi.org/10.3390/ijms23158117
Chicago/Turabian StyleEbenezer, Oluwakemi, Maryam Amra. Jordaan, Gea Carena, Tommaso Bono, Michael Shapi, and Jack A. Tuszynski. 2022. "An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds" International Journal of Molecular Sciences 23, no. 15: 8117. https://doi.org/10.3390/ijms23158117
APA StyleEbenezer, O., Jordaan, M. A., Carena, G., Bono, T., Shapi, M., & Tuszynski, J. A. (2022). An Overview of the Biological Evaluation of Selected Nitrogen-Containing Heterocycle Medicinal Chemistry Compounds. International Journal of Molecular Sciences, 23(15), 8117. https://doi.org/10.3390/ijms23158117