
Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases

 , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction: Alzheimer’s Disease and Sphingolipids
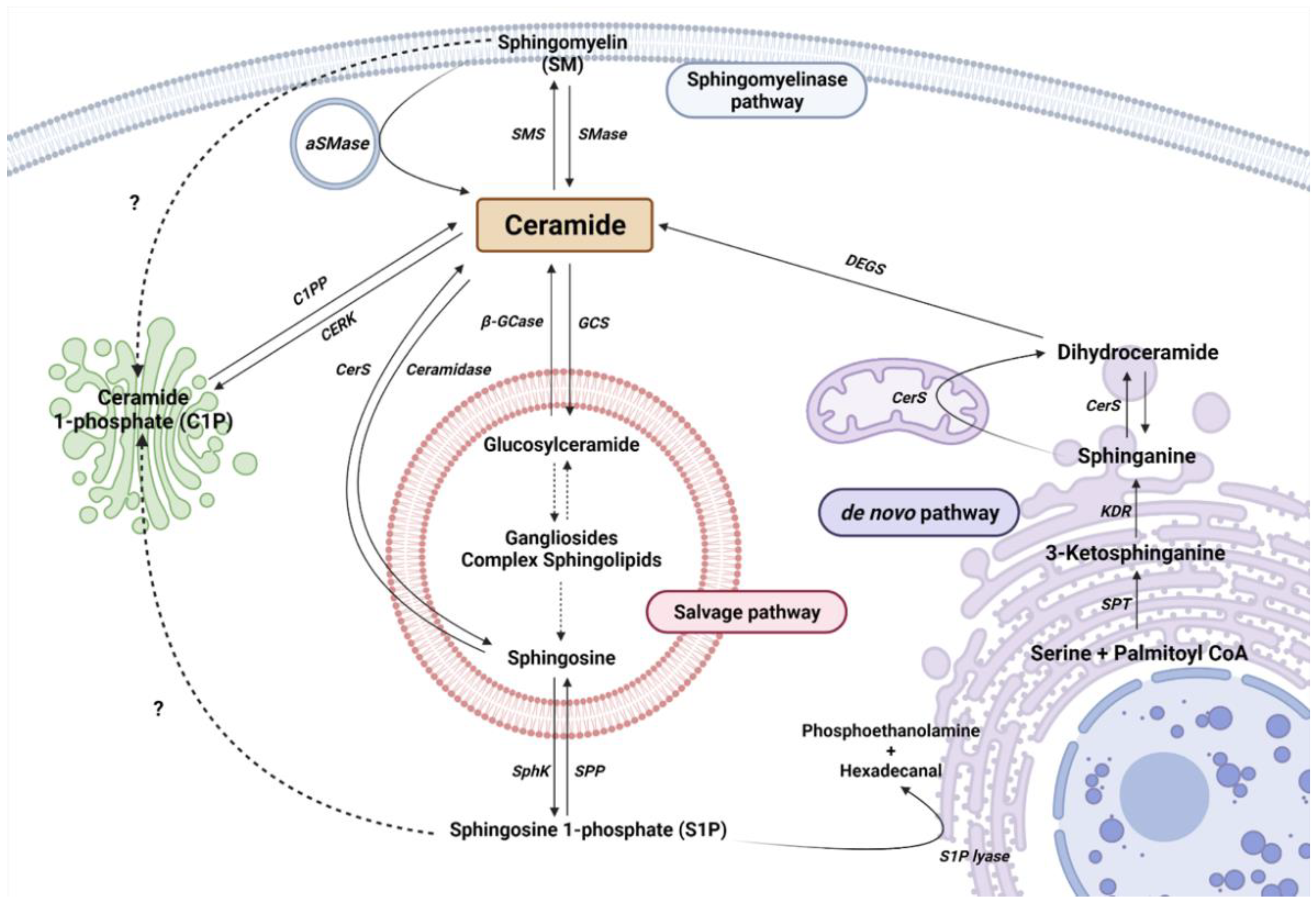
2. Sphingolipid Metabolism
2.1. The De Novo Pathway
2.2. The Sphingomyelinase (SMase Pathway)
2.3. The Salvage Pathway
2.4. Ceramide Kinase/Ceramide 1-Phosphate Phosphatase (CerK/CPP) and Sphingosine Kinase/Sphingosine 1-Phosphate Phosphatase (SphK/SPP) Axis
3. Neurodegeneration and Sphingolipid Metabolism
3.1. Parkinson’s Disease
3.2. Dementia with Lewy Bodies
3.3. Multiple Sclerosis
3.4. Amyotrophic Lateral Sclerosis
3.5. Huntington’s Disease
3.6. Lysosomal Storage Diseases

{kind=link}
{kind=link}
| Disease | Sphingolipid Species | Levels | Source | Implication in the Disease | Ref. |
|---|---|---|---|---|---|
| Parkinson’s disease | C16-Cer | ↑ | Plasma | Higher levels are associated with worse cognition | [109] |
| C18-Cer | ↑ | ||||
| C20-Cer | ↑ | ||||
| C22-Cer | ↑ | ||||
| ↓ | Anterior cingulate cortex | [108] | |||
| C23-Cer | ↓ | ||||
| C24:1-Cer | ↓ | ||||
| ↑ | Plasma | Higher levels are associated with worse cognition | [109] | ||
| CerS1 | ↑ | Anterior cingulate cortex | [108] | ||
| CerS4 | ↑ | ||||
| Dementia with Lewy bodies | C16-Cer | ↑ | Plasma | [112] | |
| C18:1-Cer | ↑ | ||||
| C20-Cer | ↑ | ||||
| C24:1-Cer | ↑ | ||||
| Multiple sclerosis | C16-Cer | ↑ | CSF | [113] | |
| C18-Cer | ↑ | ||||
| C24-Cer | ↑ | ||||
| Monohexosyl C16-Cer | ↑ | ||||
| aSMase activity | ↑ | [114] | |||
| Amyotrophic Lateral Sclerosis | C16-Cer | ↑ | Spinal cord lumbar region | Accumulation before the onset of the symptoms | [118] |
| C18-Cer | ↑ | Spinal cord cervical region | [119] | ||
| C24-Cer | ↑ | Spinal cord lumbar region | [118] | ||
| ↑ | Spinal cord cervical region | [119] | |||
| C24:1-Cer | ↑ | ||||
| C16-SM | ↑ | Spinal cord lumbar region | [118] | ||
| GM3 | ↑ | Spinal cord cervical region | [119] | ||
| Huntington’s disease | S1P lyase | ↑ | Striatum and cortex | [121] | |
| SphK1 | ↓ | Striatum | |||
| Niemann-Pick’s disease type A and B | SM | ↑ | Systemic organs and brain | Disease caused by a mutation in SMPD1 gene generating a deficiency of aSMase and the accumulation of SM | [126] |
| Gaucher’s disease | GlcCer | ↑ | Macrophage lysosomes | Disease caused by a mutation in GBA gene generating a deficiency of β-GCase and the accumulation of GlcCer | [127] |
| Faber’s disease | Cer | ↑ | Systemic organs and brain | Disease caused by a mutation in ASAH1 gene generating a decreasing of ASAH1 activity and the accumulation of Cer | [130] |
| C26-Cer | ↑ | Blood | Proposed as a diagnostic biomarker | [131] | |
| Krabbe’s disease | galactocerebrosides | ↑ | Disease caused by a mutation in GALC gene generating a deficiency of GALC and the accumulation of galactosphingolipids (including galactocerebrosides) | [132] |
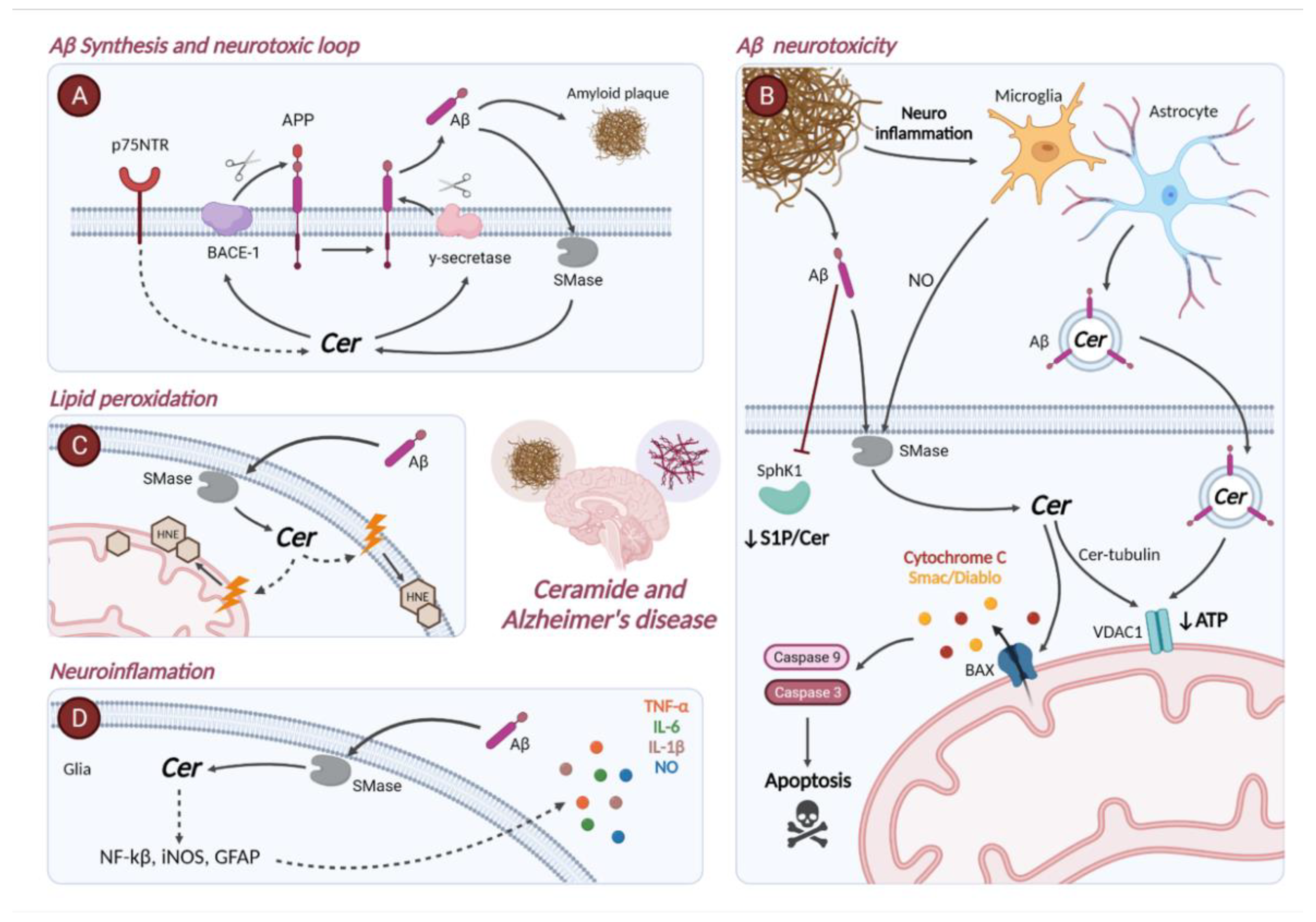
4. Ceramide Metabolism in Alzheimer’s Disease
4.1. Gene Expression of Enzymes Involved in Sphingolipid Metabolism in Alzheimer’s Disease
4.2. Role of Ceramide in the Pathophysiology of Alzheimer’s Disease
5. Sphingolipidomics in Alzheimer’s Disease
5.1. Brain Tissue
5.2. Cerebrospinal Fluid (CSF)
5.3. Blood
| Sphingolipid Species | Levels | Source | AD Stage/Condition | Notes | Ref. |
|---|---|---|---|---|---|
| Total Cer | ↑ * | Hippocampus | Cognitively normal elderly | Increased trend with age | [170] |
| ↑ | Cognitively normal elderly men | Significantly correlated with age in males | |||
| ↑ | Brain samples | Not specified | [171] | ||
| ↑ | Prefrontal cortex | [172] | |||
| ↑ | Temporal, frontal, and parietal white matter | Very early (CDR = 0.5), mild (CDR = 1), moderate (CDR = 2), and severe AD (CDR = 3) | Peak concentration at very early AD in temporal with matter | [173] | |
| ↑ | Frontal cortex | Not specified | Detection of Cer-immunoreactive astroglia in amyloid plaques in layer 2 and 3 of the frontal cortex | [175] | |
| ↑ | Occipital cortex | Braak stages 4 to 6 | Increased expression of Cer in reactive astrocytes and microglia associated with cerebral amyloid angiopathy | [177] | |
| ↑ | CSF | Not specified | Higher concentration in moderate AD compared to mild and severe stages | [175] | |
| ↓ | CSF nanoparticle fraction | Probable AD | [184] | ||
| ↑ | CSF supernatant fraction | ||||
| ↑ | Serum | Not specified | Correlation with total tau levels in CSF and brain atrophy | [194] | |
| ↑ | Elderly women without dementia (MMSE score ≥ 24) | Predicted cognitive impairment in asymptomatic individuals | [196] | ||
| ↓ | Association with cross-sectional impairment of delayed recall memory | ||||
| Short-chain Cer | ↑ | Not specified | Compared to healthy controls and iNPH patients | [186] | |
| Long-chain Cer | ↑ | ||||
| C16-Cer | ↑ | Frontal cortex | Braak stages 1 to 6 | [27] | |
| ↑ | Hippocampus | Cognitively normal elderly | Associated with age | [170] | |
| ↑ | Plasma | Mild and moderate (MMSE ≥ 20) AD | [189] | ||
| ↑ | AD (Braak stage ≥ 4 and no LB) and high-likelihood DLB (Braak stage ≤ 4) | Compared to cognitively normal controls | [112] | ||
| ↑ | Elderly men | Increased risk of AD | [192] | ||
| ↑ | Serum | Elderly women without dementia (MMSE score ≥24) | Prediction of impaired immediate recall and psychomotor speed | [196] | |
| C18-Cer | ↑ | Frontal cortex | Braak stages 1 to 6 | [27] | |
| ↑ | Middle frontal gyrus | Mild (MMSE 23–29), moderate (MMSE 11–20) and severe (MMSE 0–10) AD patients | Analysis of cell membranes. Highest values corresponded to greatest severity of AD | [147] | |
| ↑ | CSF | MCI (MMSE 24–30) | Significantly associated with AD (Aβ42 and total tau) and inflammatory (S100B) markers | [183] | |
| C18:1-Cer | ↑ | Plasma | AD (Braak stage ≥ 4 and no LB) and high-likelihood DLB (Braak stage ≤ 4) | Compared to cognitively normal controls | [112] |
| C18:1/16:0-Cer | ↑ | Serum | Elderly women without dementia (MMSE score ≥ 24) | Significantly associated with a 7 to 10-fold increase in the risk of AD | [191] |
| C18:1/18:0-Cer | ↑ | Prefrontal cortex | Not specified | [172] | |
| ↑ | Superior temporal gyrus | Braak stage 6 | Detection of Cer-enriches amyloid plaques | [176] | |
| C18:1/20:0-Cer | ↑ | ||||
| C18:1/24:0-Cer | ↑ | Serum | Elderly women without dementia (MMSE score ≥ 24) | Significantly associated with a 7 to 10-fold increase in the risk of AD | [191] |
| C20-Cer | ↑ | Frontal cortex | Braak stages 1 to 6 | [27] | |
| ↑ | Serum | MMSE score ≥ 24 | Prediction of impaired immediate recall and psychomotor speed | [196] | |
| ↑ | Plasma | AD (Braak stage ≥ 4 and no LB) and high-likelihood DLB (Braak stage ≤ 4) | Compared to cognitively normal controls | [112] | |
| C21-Cer | ↑ | Mild and moderate (MMSE ≥ 20) AD | [189] | ||
| C22-Cer | ↑ | Hippocampus | Cognitively normal elderly | Associated with age | [170] |
| ↓ | Plasma | MCI (CDR = 0.5) | Compared with AD (CDR = 1) patients and controls | [190] | |
| ↑ | Higher level predicted hippocampal volume loss and cognitive impairment | ||||
| C24-Cer | ↑ | Frontal cortex | Braak stages 1 to 6 | [27] | |
| ↑ | Middle frontal gyrus | Mild (MMSE 23–29), moderate (MMSE 11–20) and severe (MMSE 0–10) AD patients | Analysis of cell membranes. Highest values corresponded to greatest severity of AD | [147] | |
| ↑ * | CSF | Not specified | Compared to iNPH patients | [186] | |
| ↓ | Plasma | MCI (CDR = 0.5) | Compared with AD (CDR = 1) patients and controls | [190] | |
| ↑ | Higher level predicted hippocampal volume loss and cognitive impairment | ||||
| C24:1-Cer | ↑ | Temporal white matter | Very early AD | [173] | |
| ↑ | Plasma | AD (Braak stage ≥ 4 and no LB) and high-likelihood DLB (Braak stage ≤ 4) | Compared to cognitively normal controls | [112] | |
| C18:1-HexCer | ↑ | ||||
| C24:1-HexCer | ↑ | ||||
| GalCer | ↑ | Prefrontal cortex | Not specified | [172] | |
| C24-GalCer | ↑ * | Middle frontal gyrus | Mild (MMSE 23–29), moderate (MMSE 11–20) and severe (MMSE 0–10) AD patients | [147] | |
| ↑ | CSF | Not specified | Compared to iNPH patients | [186] | |
| GlcCer | ↑ | Serum | Not specified | Correlation between Aβ1–42 levels in CSF | [194] |
| LacCer | ↑ | Elderly women without dementia (MMSE score ≥24) | Significantly associated with a 7 to 10-fold increase in the risk of AD | [191] | |
| Total SM | ↑ * | Hippocampus | Cognitively normal elderly | Increased trend with age. Significantly correlated with age in males | [170] |
| ↑ * | Cognitively normal elderly men | Significantly correlated with age in males | |||
| ↓ | Brain samples | Not specified | [171] | ||
| ↓ | Middle frontal gyrus | Mild (MMSE 23–29), moderate (MMSE 11–20) and severe (MMSE 0–10) AD patients | [147] | ||
| ↑ | CSF | Prodromal AD (MMSE 24–29) | Compared to cognitively normal controls | [185] | |
| ↓ | CSF nanoparticle and supernatant fraction | Probable AD | [184] | ||
| ↑ | Plasma | Elderly men | Increased risk of AD | ||
| ↑ | Elderly women | Association with lower risk of AD. Greater association among APOEε4 carriers | [192] | ||
| ↑ | Serum | Elderly women without dementia MMSE score ≥ 24 | Predicted cognitive impairment in asymptomatic individuals | [196] | |
| ↓ | Association with cross-sectional impairment of delayed recall memory | ||||
| Monounsaturated SM | ↑ | Not specified | Correlation with total tau levels in CSF and brain atrophy. | [194] | |
| Short-chain SM | ↑ | Compared to healthy controls and iNPH patients | [186] | ||
| C18:0-SM | ↑ | Plasma | Elderly men | Increased risk of AD | [192] |
| C18:1-SM | ↑ | ||||
| C18:0/18:0-SM | ↑ | Entorhinal cortex | Not specified | [172] | |
| C18:1/14:0-SM | ↓ | CSF | Mild AD (MMSE 21–23) | Compared to cognitively normal controls | [185] |
| C18:1/16:0-SM | ↓ | ||||
| C18:1/16:1-SM | ↑ | Entorhinal cortex | Not specified | [172] | |
| C18:1/18:0-SM | ↑ | ||||
| C18:0/20:0-SM | ↓ | ||||
| C18:1/20:0-SM | ↓ | ||||
| C18:1/22:SM | ↑ | ||||
| C18:0/26:1-SM | ↑ | ||||
| C20:1-SM | ↑ | Plasma | Elderly men | Increased risk of AD | [192] |
| C22:1-SM | ↓ | Plasma | Mild and moderate (MMSE ≥ 20) AD | [189] | |
| ↑ | Elderly men | Increased risk of AD | [192] | ||
| C24:1-SM | ↓ | CSF | Not specified | Compared to healthy controls and iNPH patients | [186] |
| ↓ | Plasma | Mild and moderate (MMSE ≥ 20) AD | [189] | ||
| SM/Cer | ↑ | Plasma | Not specified | Correlation with slow progression of cognitive decline | [195] |
| dhSM/dhCer | ↑ | ||||
| Total Sph | ↑ * | Hippocampus | Cognitively normal elderly | Increased trend with age. Significantly correlated with age in male | [170] |
| ↑ | Brain samples | Not specified | |||
| S1P | ↓ | Negative correlation between Aβ, hyperphosphorylated tau, and S1P levels | |||
| ↓ | CSF | Compared to iNPH patients | [186] | ||
| ↓ | Mild AD | [188] | |||
| ↑ | MCI | Increased progressively concentration from healthy controls to MCI patients | |||
| S1P/Sph | ↑ | Hippocampus | Cognitively normal elderly women | Inversely correlated with age | [170] |
| Spha-1P | ↑ | Serum | MCI | Prediction of the conversion of MCI to AD | [193] |
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Action Plan on the Public Health Response to Dementia 2017–2025; World Health Organization: Geneva, Switzerland, 2017; Volume 27. [Google Scholar]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Javaid, S.F.; Giebel, C.; Khan, M.A.; Hashim, M.J. Epidemiology of Alzheimer’s Disease and Other Dementias: Rising Global Burden and Forecasted Trends. F1000Research 2021, 10, 425. [Google Scholar] [CrossRef]
- Nichols, E.; Abd-Allah, F.; Abdoli, A.; Abosetugn, A.E.; Abrha, W.A.; Abualhasan, A.; Abu-Gharbieh, E.; Akinyemi, R.O.; Alahdab, F.; Alanezi, F.M.; et al. Global Mortality from Dementia: Application of a New Method and Results from the Global Burden of Disease Study 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12200. [Google Scholar] [CrossRef]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [CrossRef]
- Custodia, A.; Ouro, A.; Romaus-Sanjurjo, D.; Pías-Peleteiro, J.M.; de Vries, H.E.; Castillo, J.; Sobrino, T. Endothelial Progenitor Cells and Vascular Alterations in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 13, 946. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of Oxidative Stress in Alzheimer’s Disease. Biomed. Rep. 2016, 4, 519. [Google Scholar] [CrossRef] [Green Version]
- Crivelli, S.M.; Giovagnoni, C.; Visseren, L.; Scheithauer, A.L.; de Wit, N.; den Hoedt, S.; Losen, M.; Mulder, M.T.; Walter, J.; de Vries, H.E.; et al. Sphingolipids in Alzheimer’s Disease, How Can We Target Them? Adv. Drug Deliv. Rev. 2020, 159, 214–231. [Google Scholar] [CrossRef]
- Giri, M.; Shah, A.; Upreti, B.; Rai, J.C. Unraveling the Genes Implicated in Alzheimer’s Disease (Review). Biomed. Rep. 2017, 7, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer Disease: Pathobiology and Targeting Strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Wang, C.; Gratuze, M.; Hoyle, R.; Bien-Ly, N.; Silverman, A.P.; Sullivan, P.M.; et al. APOE Immunotherapy Reduces Cerebral Amyloid Angiopathy and Amyloid Plaques While Improving Cerebrovascular Function. Sci. Transl. Med. 2021, 13, eabd7522. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol. Dis. 2014, 72PA, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Thalamuthu, A.; Mather, K.A.; Crawford, J.; Ulanova, M.; Wong, M.W.K.; Pickford, R.; Sachdev, P.S.; Braidy, N. Plasma Lipidome Is Dysregulated in Alzheimer’s Disease and Is Associated with Disease Risk Genes. Transl. Psychiatry 2021, 11, 344. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Role of Bioactive Sphingolipids in Physiology and Pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [CrossRef]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Ouro, A.; Arana, L.; Gangoiti, P.; Gomez-Muoz, A.; Gomez-Muñoz, A. Role of Ceramide 1-Phosphate in the Regulation of Cell Survival and Inflammation. In Biochemistry; InTech Open: London, UK, 2012; Volume 4. [Google Scholar] [CrossRef] [Green Version]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gómez-Muñoz, A.; Gomez-Munoz, A.; Gómez-Muñoz, A. Ceramide and Ceramide 1-Phosphate in Health and Disease. Lipids Health Dis. 2010, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabriás, G.; Abad, J.L.; Delgado, A.; et al. Control of Metabolism and Signaling of Simple Bioactive Sphingolipids: Implications in Disease. Prog. Lipid Res. 2010, 49, 316–334. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Myelination and Myelin Stability and Their Involvement in Childhood and Adult Demyelinating Disorders. J. Neurochem. 2021, 156, 403–414. [Google Scholar] [CrossRef]
- Ouro, A.; Correa-Paz, C.; Maqueda, E.; Custodia, A.; Aramburu-Nuñez, M.; Romaus-Sanjurjo, D.; Posado-Fernandez, A.; Candamo-Lourido, M.; Alonso-Alonso, M.L.; Hervella, P.; et al. Involvement of Ceramide Metabolism in Cerebral Ischemia. Front. Mol. Biosci. 2022, 9, 309. [Google Scholar] [CrossRef]
- Gomez-Larrauri, A.; das Adhikari, U.; Aramburu-Nuñez, M.; Custodia, A.; Ouro, A. Ceramide Metabolism Enzymes—Therapeutic Targets against Cancer. Medicina 2021, 57, 729. [Google Scholar] [CrossRef] [PubMed]
- Giovagnoni, C.; Ali, M.; Eijssen, L.M.T.; Maes, R.; Choe, K.; Mulder, M.; Kleinjans, J.; del Sol, A.; Glaab, E.; Mastroeni, D.; et al. Altered Sphingolipid Function in Alzheimer’s Disease; a Gene Regulatory Network Approach. Neurobiol. Aging 2021, 102, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Li, C.; Haroutunian, V. Gene Expression Alterations in the Sphingolipid Metabolism Pathways during Progression of Dementia and Alzheimer’s Disease: A Shift Toward Ceramide Accumulation at the Earliest Recognizable Stages of Alzheimer’s Disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Akyol, S.; Ugur, Z.; Yilmaz, A.; Ustun, I.; Gorti, S.K.K.; Oh, K.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Maddens, M.E.; et al. Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(Phospho)Lipid, and Sphingolipid Metabolism. Cells 2021, 10, 2591. [Google Scholar] [CrossRef] [PubMed]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased Ceramide in Brains with Alzheimer’s and Other Neurodegenerative Diseases. J. Alzheimer’s Dis. 2012, 29, 537–547. [Google Scholar] [CrossRef] [Green Version]
- de Wit, K.M.; Mol, K.; Rodríguez-Lorenzo, S.; de Vries, H.E.; Kooij, G. The Role of Sphingolipids and Specialized Pro-Resolving Mediators in Alzheimer’s Disease. Front. Immunol. 2021, 11, 620348. [Google Scholar] [CrossRef]
- Mielke, M.M.; Lyketsos, C.G. Alterations of the Sphingolipid Pathway in Alzheimer’s Disease: New Biomarkers and Treatment Targets? Neuromolecular Med. 2010, 12, 331. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.; Khan, S. Plasma Lipids as Biomarkers for Alzheimer’s Disease: A Systematic Review. Cureus 2020, 12, e12008. [Google Scholar] [CrossRef]
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 437. [Google Scholar] [CrossRef]
- Wattenberg, B.W. Kicking off Sphingolipid Biosynthesis: Structures of the Serine Palmitoyltransferase Complex. Nat. Struct. Mol. Biol. 2021, 28, 229–231. [Google Scholar] [CrossRef]
- Davis, D.L.; Gable, K.; Suemitsu, J.; Dunn, T.M.; Wattenberg, B.W. The ORMDL/Orm-Serine Palmitoyltransferase (SPT) Complex Is Directly Regulated by Ceramide: Reconstitution of SPT Regulation in Isolated Membranes. J. Biol. Chem. 2019, 294, 5146–5156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.L.; Mestre, B.; Shin, S.-H.H.; Futerman, A.H. Ceramide Synthases: Reflections on the Impact of Dr. Lina M. Obeid. Cell. Signal. 2021, 82, 109958. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal Structure of Mammalian Acid Sphingomyelinase. Nat. Commun. 2016, 7, 12196. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Rhein, C.; Müller, C.P.; Mühle, C. Secretory Sphingomyelinase in Health and Disease. Biol. Chem. 2015, 396, 707–736. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.J.; Snook, C.F.; Tani, M.; Matmati, N.; Marchesini, N.; Hannun, Y.A. The Extended Family of Neutral Sphingomyelinases. Biochemistry 2006, 45, 11247–11256. [Google Scholar] [CrossRef]
- Goi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and Membrane Activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, S.; Borrelli, A.; Ceccarini, M.R.; Nakashidze, I.; Codini, M.; Belov, O.; Ivanov, A.; Krasavin, E.; Ferri, I.; Conte, C.; et al. Acid and Neutral Sphingomyelinase Behavior in Radiation-Induced Liver Pyroptosis and in the Protective/Preventive Role of RMnSOD. Int. J. Mol. Sci. 2020, 21, 3281. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, Y. The Sphingomyelin Synthase Family: Proteins, Diseases, and Inhibitors. Biol. Chem. 2017, 398, 1319–1325. [Google Scholar] [CrossRef]
- Yabu, T.; Shiba, H.; Shibasaki, Y.; Nakanishi, T.; Imamura, S.; Touhata, K.; Yamashita, M. Stress-Induced Ceramide Generation and Apoptosis via the Phosphorylation and Activation of NSMase1 by JNK Signaling. Cell Death Differ. 2014, 22, 258–273. [Google Scholar] [CrossRef] [Green Version]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher Disease: Mutation and Polymorphism Spectrum in the Glucocerebrosidase Gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Velayati, A.; Yu, W.H.; Sidransky, E. The Role of Glucocerebrosidase Mutations in Parkinson Disease and Lewy Body Disorders. Curr. Neurol. Neurosci. Rep. 2010, 10, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Custodia, A.; Aramburu-Núñez, M.; Correa-Paz, C.; Posado-Fernández, A.; Gómez-Larrauri, A.; Castillo, J.; Gómez-Muñoz, A.; Sobrino, T.; Ouro, A. Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets. Biomolecules 2021, 11, 945. [Google Scholar] [CrossRef]
- Coant, N.; Hannun, Y.A. Neutral Ceramidase: Advances in Mechanisms, Cell Regulation, and Roles in Cancer. Adv. Biol. Regul. 2019, 71, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Romiti, E.; Meacci, E.; Tani, M.; Nuti, F.; Farnararo, M.; Ito, M.; Bruni, P. Neutral/Alkaline and Acid Ceramidase Activities Are Actively Released by Murine Endothelial Cells. Biochem. Biophys. Res. Commun. 2000, 275, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Choi, J.W. Sphingosine 1-Phosphate Receptors in Cerebral Ischemia. NeuroMol. Med. 2021, 23, 211–223. [Google Scholar] [CrossRef]
- Calise, S.; Blescia, S.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Bruni, P. Sphingosine 1-Phosphate Stimulates Proliferation and Migration of Satellite Cells: Role of S1P Receptors. Biochim. Biophys. Acta 2012, 1823, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial Sphingosine 1-Phosphate Receptors Promote Vascular Normalization and Antitumor Therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3157–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Wang, W.; McGiffert, C.; Huang, M.C.; Graler, M.H.; Gräler, M.H. Sphingosine 1-Phosphate and Its G Protein-Coupled Receptors Constitute a Multifunctional Immunoregulatory System. J. Cell Biochem. 2004, 92, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Saba, J.D. Fifty Years of Lyase and a Moment of Truth: Sphingosine Phosphate Lyase from Discovery to Disease. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef] [Green Version]
- Bornancin, F. Ceramide Kinase: The First Decade. Cell. Signal. 2011, 23, 999–1008. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordoñez, M. Control of Inflammatory Responses by Ceramide, Sphingosine 1-Phosphate and Ceramide 1-Phosphate. Prog. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Movsesyan, V.A.; Lea IV, P.M.; Faden, A.I. Ceramide-Induced Neuronal Apoptosis Is Associated with Dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and Induction of the Mitochondrial-Dependent Intrinsic Caspase Pathway. Mol. Cell Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and Activated Bax Act Synergistically to Permeabilize the Mitochondrial Outer Membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Movsesyan, V.A.; Knoblach, S.M.; Faden, A.I. Ceramide Induces Neuronal Apoptosis through Mitogen-Activated Protein Kinases and Causes Release of Multiple Mitochondrial Proteins. Mol. Cell. Neurosci. 2005, 29, 355–371. [Google Scholar] [CrossRef]
- Falluel-Morel, A.; Aubert, N.; Vaudry, D.; Basille, M.; Fontaine, M.; Fournier, A.; Vaudry, H.; Gonzalez, B.J. Opposite Regulation of the Mitochondrial Apoptotic Pathway by C2-Ceramide and PACAP through a MAP-Kinase-Dependent Mechanism in Cerebellar Granule Cells. J. Neurochem. 2004, 91, 1231–1243. [Google Scholar] [CrossRef]
- Movsesyan, V.A.; Yakovlev, A.G.; Dabaghyan, E.A.; Stoica, B.A.; Faden, A.I. Ceramide Induces Neuronal Apoptosis through the Caspase-9/Caspase-3 Pathway. Biochem. Biophys. Res. Commun. 2002, 299, 201–207. [Google Scholar] [CrossRef]
- Lai, M.; Amato, R.; La Rocca, V.; Bilgin, M.; Freer, G.; Spezia, P.; Quaranta, P.; Piomelli, D.; Pistello, M. Acid Ceramidase Controls Apoptosis and Increases Autophagy in Human Melanoma Cells Treated with Doxorubicin. Sci. Rep. 2021, 11, 11221. [Google Scholar] [CrossRef]
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of Ceramide Synthase 1 Increases C18-Ceramide and Leads to Lethal Autophagy in Human Glioma. Oncotarget 2017, 8, 104022–104036. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Bieberich, E. Sphingolipids in Neurodegeneration (with Focus on Ceramide and S1P). Adv. Biol. Regul. 2018, 70, 51. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, H.J.; Chun, Y.J.; Kim, M.Y. Ceramide Produces Apoptosis through Induction of P27(Kip1) by Protein Phosphatase 2A-Dependent Akt Dephosphorylation in PC-3 Prostate Cancer Cells. J. Toxicol. Environ. Health A 2010, 73, 1465–1476. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508. [Google Scholar] [CrossRef]
- de Wit, N.M.; den Hoedt, S.; Martinez-Martinez, P.; Rozemuller, A.J.; Mulder, M.T.; de Vries, H.E. Astrocytic Ceramide as Possible Indicator of Neuroinflammation. J. Neuroinflamm. 2019, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Togo, T.; Katsuse, O.; Iseki, E. Nitric Oxide Pathways in Alzheimer’s Disease and Other Neurodegenerative Dementias. Neurol. Res. 2004, 26, 563–566. [Google Scholar] [CrossRef]
- Pahan, K.; Sheikh, F.G.; Khan, M.; Namboodiri, A.M.S.; Singh, I. Sphingomyelinase and Ceramide Stimulate the Expression of Inducible Nitric-Oxide Synthase in Rat Primary Astrocytes. J. Biol. Chem. 1998, 273, 2591–2600. [Google Scholar] [CrossRef] [Green Version]
- Scheiblich, H.; Schlütter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 Inflammasome in Microglia: The Role of Ceramide. J. Neurochem. 2017, 143, 534–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Yu, Y.; Zhang, X.; Zhang, C.; Zhao, Y.; Liu, B.; Zhang, L.; Wang, L.; Chen, R.; Gao, X.; et al. Sphingomyelin Synthase 2 Inhibition Ameliorates Cerebral Ischemic Reperfusion Injury through Reducing the Recruitment of Toll-Like Receptor 4 to Lipid Rafts. J. Am. Heart Assoc. 2019, 8, e012885. [Google Scholar] [CrossRef]
- Martinez, T.N.; Chen, X.; Bandyopadhyay, S.; Merrill, A.H.; Tansey, M.G. Ceramide Sphingolipid Signaling Mediates Tumor Necrosis Factor (TNF)-Dependent Toxicity via Caspase Signaling in Dopaminergic Neurons. Mol. Neurodegener. 2012, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in Neurodegenerative Diseases: Pathogenesis and Therapy. Brain Pathol. 2018, 28, 3. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.P. Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 10, 433. [Google Scholar] [CrossRef]
- Schubert, K.M.; Scheid, M.P.; Duronio, V. Ceramide Inhibits Protein Kinase B/Akt by Promoting Dephosphorylation of Serine 473. J. Biol. Chem. 2000, 275, 13330–13335. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Kitatani, K.; Kondo, T.; Hashimoto-Nishimura, M.; Asano, S.; Hayashi, A.; Mitsutake, S.; Igarashi, Y.; Umehara, H.; Takeya, H.; et al. Regulation of Autophagy and Its Associated Cell Death by “Sphingolipid Rheostat”: Reciprocal Role of Ceramide and Sphingosine 1-Phosphate in the Mammalian Target of Rapamycin Pathway. J. Biol. Chem. 2012, 287, 39898–39910. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Ogretmen, B. Autophagy Paradox and Ceramide. Biochim. Biophys. Acta 2014, 1841, 783–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Costa, M.; Spezia, P.G.; Quaranta, P.; Lombardo, G.; Piomelli, D.; Pistello, M. Ablation of Acid Ceramidase Impairs Autophagy and Mitochondria Activity in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 3247. [Google Scholar] [CrossRef]
- Gulbins, A.; Schumacher, F.; Becker, K.A.; Wilker, B.; Soddemann, M.; Boldrin, F.; Müller, C.P.; Edwards, M.J.; Goodman, M.; Caldwell, C.C.; et al. Antidepressants Act by Inducing Autophagy Controlled by Sphingomyelin–Ceramide. Mol. Psychiatry 2018, 23, 2324–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Panneer Selvam, S.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide Targets Autophagosomes to Mitochondria and Induces Lethal Mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 3075–3088. [Google Scholar] [CrossRef] [PubMed]
- Law, B.A.; Liao, X.; Moore, K.S.; Southard, A.; Roddy, P.; Ji, R.; Szulc, Z.; Bielawska, A.; Schulze, P.C.; Cowart, L.A. Lipotoxic Very-Long-Chain Ceramides Cause Mitochondrial Dysfunction, Oxidative Stress, and Cell Death in Cardiomyocytes. FASEB J. 2018, 32, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Czubowicz, K.; Strosznajder, R. Ceramide in the Molecular Mechanisms of Neuronal Cell Death. The Role of Sphingosine-1-Phosphate. Mol. Neurobiol. 2014, 50, 26. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.V.T.S.; Nithipatikom, K.; Harder, D.R. Ceramide Elevates 12-Hydroxyeicosatetraenoic Acid Levels and Upregulates 12-Lipoxygenase in Rat Primary Hippocampal Cell Cultures Containing Predominantly Astrocytes. Neurochem. Int. 2008, 53, 220–229. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, Mitochondrial Dysfunction, and Alzheimer’s Disease. Pharm. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef]
- Kong, J.N.; Zhu, Z.; Itokazu, Y.; Wang, G.; Dinkins, M.B.; Zhong, L.; Lin, H.P.; Elsherbini, A.; Leanhart, S.; Jiang, X.; et al. Novel Function of Ceramide for Regulation of Mitochondrial ATP Release in Astrocytes. J. Lipid Res. 2018, 59, 488–506. [Google Scholar] [CrossRef] [Green Version]
- Bieberich, E.; MacKinnon, S.; Silva, J.; Yu, R.K. Regulation of Apoptosis during Neuronal Differentiation by Ceramide and B-Series Complex Gangliosides. J. Biol. Chem. 2001, 276, 44396–44404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, K.; Wang, G.; Silva, J.; Condie, B.G.; Bieberich, E. Ceramide Regulates Atypical PKCzeta/Lambda-Mediated Cell Polarity in Primitive Ectoderm Cells. A Novel Function of Sphingolipids in Morphogenesis. J. Biol. Chem. 2007, 282, 3379–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Krishnamurthy, K.; Chiang, Y.W.; Dasgupta, S.; Bieberich, E. Regulation of Neural Progenitor Cell Motility by Ceramide and Potential Implications for Mouse Brain Development. J. Neurochem. 2008, 106, 718–733. [Google Scholar] [CrossRef] [Green Version]
- Hea, Q.; Wang, G.; Wakade, S.; Dasgupta, S.; Dinkins, M.; Konga, J.N.; Spassieva, S.D.; Bieberich, E. Primary Cilia in Stem Cells and Neural Progenitors Are Regulated by Neutral Sphingomyelinase 2 and Ceramide. Mol. Biol. Cell 2014, 25, 1715–1729. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.N.; Hardin, K.; Dinkins, M.; Wang, G.; He, Q.; Mujadzic, T.; Zhu, G.; Bielawski, J.; Spassieva, S.; Bieberich, E. Regulation of Chlamydomonas Flagella and Ependymal Cell Motile Cilia by Ceramide-Mediated Translocation of GSK3. Mol. Biol. Cell 2015, 26, 4451–4465. [Google Scholar] [CrossRef]
- Rutherford, C.; Childs, S.; Ohotski, J.; McGlynn, L.; Riddick, M.; MacFarlane, S.; Tasker, D.; Pyne, S.; Pyne, N.J.; Edwards, J.; et al. Regulation of Cell Survival by Sphingosine-1-Phosphate Receptor S1P1 via Reciprocal ERK-Dependent Suppression of Bim and PI-3-Kinase/Protein Kinase C-Mediated Upregulation of Mcl-1. Cell Death Dis. 2013, 4, e927. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine Kinase, Sphingosine-1-Phosphate, and Apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar] [CrossRef]
- Edsall, L.C.; Pirianov, G.G.; Spiegel, S. Involvement of Sphingosine 1-Phosphate in Nerve Growth Factor-Mediated Neuronal Survival and Differentiation. J. Neurosci. 1997, 17, 6952–6960. [Google Scholar] [CrossRef]
- Moore, A.N.; Kampfl, A.W.; Zhao, X.; Hayes, R.L.; Dash, P.K. Sphingosine-1-Phosphate Induces Apoptosis of Cultured Hippocampal Neurons That Requires Protein Phosphatases and Activator Protein-1 Complexes. Neuroscience 1999, 94, 405–415. [Google Scholar] [CrossRef]
- Kanno, T.; Nishizaki, T.; Proia, R.L.; Kajimoto, T.; Jahangeer, S.; Okada, T.; Nakamura, S. Regulation of Synaptic Strength by Sphingosine 1-Phosphate in the Hippocampus. Neuroscience 2010, 171, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darios, F.; Wasser, C.; Shakirzyanova, A.; Giniatullin, A.; Goodman, K.; Munoz-Bravo, J.L.; Raingo, J.; Jorgačevski, J.; Kreft, M.; Zorec, R.; et al. Sphingosine Facilitates SNARE Complex Assembly and Activates Synaptic Vesicle Exocytosis. Neuron 2009, 62, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Hölbling, B.V.; Schumak, B.; Hübner, M.P.; Gräler, M.H.; et al. Neural Sphingosine 1-Phosphate Accumulation Activates Microglia and Links Impaired Autophagy and Inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Caraveo, A.; Sayd, A.; Maus, S.R.; Caso, J.R.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C. Lipopolysaccharide Enters the Rat Brain by a Lipoprotein-Mediated Transport Mechanism in Physiological Conditions. Sci. Rep. 2017, 7, 13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-Induced Blood-Brain Barrier Disruption: Roles of Cyclooxygenase, Oxidative Stress, Neuroinflammation, and Elements of the Neurovascular Unit. J. Neuroinflammation 2015, 12, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.; Huo, Y.; Kwang, W.X.T.; Pushparaj, P.N.; Kumar, S.D.; Ling, E.A.; Dheen, S.T. Sphingosine Kinase 1 Regulates the Expression of Proinflammatory Cytokines and Nitric Oxide in Activated Microglia. Neuroscience 2010, 166, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ting, N.-C.; Kuo, M.-L. LPS Elevates Ceramide Levels through Regulating ORMDL3 Expression in Macrophages and Acute Respiratory Distress Syndrome Model. J. Immunol. 2020, 204, 144. [Google Scholar]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-Phosphate Receptor 3 and RhoA Signaling Mediate Inflammatory Gene Expression in Astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, S.D.; Nicole, O.; Peavy, R.D.; Montoya, L.M.; Lee, C.J.; Murphy, T.J.; Traynelis, S.F.; Hepler, J.R. Common Signaling Pathways Link Activation of Murine PAR-1, LPA, and S1P Receptors to Proliferation of Astrocytes. Mol. Pharm. 2003, 64, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, S.A.; O’Sullivan, C.; Healy, L.M.; Dev, K.K.; Sheridan, G.K. Sphingosine 1-Phosphate Receptors Regulate TLR4-Induced CXCL5 Release from Astrocytes and Microglia. J. Neurochem. 2018, 144, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Moruno-Manchon, J.F.; Uzor, N.E.; Ambati, C.R.; Shetty, V.; Putluri, N.; Jagannath, C.; McCullough, L.D.; Tsvetkov, A.S. Sphingosine Kinase 1-Associated Autophagy Differs between Neurons and Astrocytes. Cell Death Dis. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, S.; Piazzesi, A.; Abd El Fatah, M.; Raucamp, M.; van Echten-Deckert, G. Neurodegeneration Caused by S1P-Lyase Deficiency Involves Calcium-Dependent Tau Pathology and Abnormal Histone Acetylation. Cells 2020, 9, 2189. [Google Scholar] [CrossRef] [PubMed]
- Karaca, I.; Tamboli, I.Y.; Glebov, K.; Richter, J.; Fell, L.H.; Grimm, M.O.; Haupenthal, V.J.; Hartmann, T.; Gräler, M.H.; Van Echten-Deckert, G.; et al. Deficiency of Sphingosine-1-Phosphate Lyase Impairs Lysosomal Metabolism of the Amyloid Precursor Protein. J. Biol. Chem. 2014, 289, 16761–16772. [Google Scholar] [CrossRef] [Green Version]
- Motyl, J.A.; Strosznajder, J.B.; Wencel, A.; Strosznajder, R.P. Recent Insights into the Interplay of Alpha-Synuclein and Sphingolipid Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6277. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered Ceramide Acyl Chain Length and Ceramide Synthase Gene Expression in Parkinson’s Disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism Is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 44. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Savica, R.; Murray, M.E.; Persson, X.M.; Kantarci, K.; Parisi, J.E.; Dickson, D.W.; Petersen, R.C.; Ferman, T.J.; Boeve, B.F.; Mielke, M.M. Plasma Sphingolipid Changes with Autopsy-Confirmed Lewy Body or Alzheimer’s Pathology. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Vidaurre, O.G.; Haines, J.D.; Katz Sand, I.; Adula, K.P.; Huynh, J.L.; Mcgraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal Fluid Ceramides from Patients with Multiple Sclerosis Impair Neuronal Bioenergetics. Brain 2014, 137, 2271–2286. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; Cicalini, I.; Lanuti, P.; Ercolino, E.; di Ioia, M.; Zucchelli, M.; Zappacosta, R.; Miscia, S.; Marchisio, M.; Sacchetta, P.; et al. Enhanced Release of Acid Sphingomyelinase-Enriched Exosomes Generates a Lipidomics Signature in CSF of Multiple Sclerosis Patients. Sci. Rep. 2018, 8, 3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, J.; Brunkhorst, R.; Foerch, C.; Blum, L.; Henke, M.; Gabriel, L.; Ulshöfer, T.; Ferreirós, N.; Parnham, M.J.; Geisslinger, G.; et al. The Relevance of Ceramides and Their Synthesizing Enzymes for Multiple Sclerosis. Clin. Sci. 2018, 132, 1963–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberle, M.; Ebel, P.; Mayer, C.A.; Barthelmes, J.; Tafferner, N.; Ferreiros, N.; Ulshöfer, T.; Henke, M.; Foerch, C.; De Bazo, A.M.; et al. Exacerbation of Experimental Autoimmune Encephalomyelitis in Ceramide Synthase 6 Knockout Mice Is Associated with Enhanced Activation/Migration of Neutrophils. Immunol. Cell Biol. 2015, 93, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Barthelmes, J.; de Bazo, A.M.; Pewzner-Jung, Y.; Schmitz, K.; Mayer, C.A.; Foerch, C.; Eberle, M.; Tafferner, N.; Ferreirós, N.; Henke, M.; et al. Lack of Ceramide Synthase 2 Suppresses the Development of Experimental Autoimmune Encephalomyelitis by Impairing the Migratory Capacity of Neutrophils. Brain Behav. Immun. 2015, 46, 280–292. [Google Scholar] [CrossRef]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence That Accumulation of Ceramides and Cholesterol Esters Mediates Oxidative Stress-Induced Death of Motor Neurons in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.L.; Sidman, R.L.; et al. Glycosphingolipids Are Modulators of Disease Pathogenesis in Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef] [Green Version]
- Gutner, U.A.; Shupik, M.A.; Maloshitskaya, O.A.; Sokolov, S.A.; Rezvykh, A.P.; Funikov, S.Y.; Lebedev, A.T.; Ustyugov, A.A.; Alessenko, A.V. Changes in the Metabolism of Sphingoid Bases in the Brain and Spinal Cord of Transgenic FUS(1-359) Mice, a Model of Amyotrophic Lateral Sclerosis. Biochemistry 2019, 84, 1166–1176. [Google Scholar] [CrossRef]
- di Pardo, A.; Amico, E.; Basit, A.; Armirotti, A.; Joshi, P.; Neely, D.M.; Vuono, R.; Castaldo, S.; Digilio, A.F.; Scalabrì, F.; et al. Defective Sphingosine-1-Phosphate Metabolism Is a Druggable Target in Huntington’s Disease. Sci. Rep. 2017, 7, 5280. [Google Scholar] [CrossRef]
- Di Pardo, A.; Basit, A.; Armirotti, A.; Amico, E.; Castaldo, S.; Pepe, G.; Marracino, F.; Buttari, F.; Digilio, A.F.; Maglione, V. De Novo Synthesis of Sphingolipids Is Defective in Experimental Models of Huntington’s Disease. Front. Neurosci. 2017, 11, 698. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Favellato, M.; Castrataro, R.; Fucile, S.; Squitieri, F.; Maglione, V. FTY720 (Fingolimod) Is a Neuroprotective and Disease-Modifying Agent in Cellular and Mouse Models of Huntington Disease. Hum. Mol. Genet. 2014, 23, 2251–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, F.M. Sphingolipid Lysosomal Storage Disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M. Lysosomal Diseases and Neuropsychiatry: Opportunities to Rebalance the Mind. Front. Mol. Biosci. 2020, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick Diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Baris, H.N.; Cohen, I.J.; Mistry, P.K. Gaucher Disease: The Metabolic Defect, Pathophysiology, Phenotypes and Natural History. Pediatric Endocrinol. Rev. 2014, 12, 72–81. [Google Scholar]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine Accumulation in Tissues from Patients with Gaucher Disease: Correlation with Phenotype and Genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Indellicato, R.; Trinchera, M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 3304. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.P.S.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid Ceramidase Deficiency: Farber Disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Cozma, C.; Iuraşcu, M.I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.K.; Lukas, J.; Rolfs, A. C26-Ceramide as Highly Sensitive Biomarker for the Diagnosis of Farber Disease. Sci. Rep. 2017, 7, 6149. [Google Scholar] [CrossRef] [Green Version]
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic 2016, 17, 908–922. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Ellis, B.C.; Saunders, A.J.; Kovacs, D.M. Ceramide Stabilizes β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 and Promotes Amyloid β-Peptide Biogenesis. J. Biol. Chem. 2003, 278, 19777–19783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasugi, N.; Sasaki, T.; Shinohara, M.; Iwatsubo, T.; Tomita, T. Synthetic Ceramide Analogues Increase Amyloid-β 42 Production by Modulating γ-Secretase Activity. Biochem. Biophys. Res. Commun. 2015, 457, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Weindruch, R.; Della Valle, G.; Puglielli, L. A TrkA-to-P75NTR Molecular Switch Activates Amyloid Beta-Peptide Generation during Aging. Biochem. J. 2005, 391, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; et al. BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci. 2011, 31, 6850–6857. [Google Scholar] [CrossRef] [Green Version]
- Dinkins, M.B.; Enasko, J.; Hernandez, C.; Wang, G.; Kong, J.; Helwa, I.; Liu, Y.; Terry, A.V.; Bieberich, E. Neutral Sphingomyelinase-2 Deficiency Ameliorates Alzheimer’s Disease Pathology and Improves Cognition in the 5XFAD Mouse. J. Neurosci. 2016, 36, 8653–8667. [Google Scholar] [CrossRef] [Green Version]
- Jana, A.; Pahan, K. Fibrillar Amyloid-Beta Peptides Kill Human Primary Neurons via NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase. Implications for Alzheimer’s Disease. J. Biol. Chem. 2004, 279, 51451–51459. [Google Scholar] [CrossRef] [Green Version]
- Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble Oligomers of Amyloid-β Peptide Induce Neuronal Apoptosis by Activating a CPLA2-Dependent Sphingomyelinase-Ceramide Pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Jana, A.; Pahan, K. Fibrillar Amyloid-β-Activated Human Astroglia Kill Primary Human Neurons via Neutral Sphingomyelinase: Implications for Alzheimer’s Disease. J. Neurosci. 2010, 30, 12676–12689. [Google Scholar] [CrossRef]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, M.; Bieberich, E. Astrocytes Secrete Exosomes Enriched with Proapoptotic Ceramide and Prostate Apoptosis Response 4 (PAR-4): Potential Mechanism of Apoptosis Induction in Alzheimer Disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Brouchet, A.; Pchejetski, D.; Brizuela, L.; Garcia, V.; Altié, M.F.; Maddelein, M.L.; Delisle, M.B.; Cuvillier, O. Critical Role for Sphingosine Kinase-1 in Regulating Survival of Neuroblastoma Cells Exposed to Amyloid-Beta Peptide. Mol. Pharm. 2007, 72, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-T.; Xu, J.; Lee, J.-M.; Ku, G.; Han, X.; Yang, D.I.; Chen, S.; Hsu, C.Y. Amyloid-β Peptide Induces Oligodendrocyte Death by Activating the Neutral Sphingomyelinase-Ceramide Pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Crivelli, S.M.; Luo, Q.; Stevens, J.A.A.; Giovagnoni, C.; van Kruining, D.; Bode, G.; den Hoedt, S.; Hobo, B.; Scheithauer, A.-L.; Walter, J.; et al. CERTL Reduces C16 Ceramide, Amyloid-β Levels, and Inflammation in a Model of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Ferré-González, L.; Peña-Bautista, C.; Baquero, M.; Cháfer-Pericás, C. Assessment of Lipid Peroxidation in Alzheimer’s Disease Differential Diagnosis and Prognosis. Antioxidants 2022, 11, 551. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of Oxidative Stress-Induced Abnormalities in Ceramide and Cholesterol Metabolism in Brain Aging and Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Alessenko, A.V.; Bugrova, A.E.; Dudnik, L.B. Connection of Lipid Peroxide Oxidation with the Sphingomyelin Pathway in the Development of Alzheimer’s Disease. Biochem. Soc. Trans. 2004, 32, 144–146. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Zeng, C.; Lee, J.T.; Chen, H.; Chen, S.; Hsu, C.Y.; Xu, J. Amyloid-β Peptide Enhances Tumor Necrosis Factor-α-Induced INOS through Neutral Sphingomyelinase/Ceramide Pathway in Oligodendrocytes. J. Neurochem. 2005, 94, 703–712. [Google Scholar] [CrossRef]
- De Vita, T.; Albani, C.; Realini, N.; Migliore, M.; Basit, A.; Ottonello, G.; Cavalli, A. Inhibition of Serine Palmitoyltransferase by a Small Organic Molecule Promotes Neuronal Survival after Astrocyte Amyloid Beta 1-42 Injury. ACS Chem. Neurosci. 2019, 10, 1627–1635. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 Contributes to Microglial Activation and M1 Polarization Following Cerebral Ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-Phosphate Receptor Subtype 3 (S1P3) Contributes to Brain Injury after Transient Focal Cerebral Ischemia via Modulating Microglial Activation and Their M1 Polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A Key Player in Microglial Biology and Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Ji, J.; Sun, Y.; Huang, X.; Cai, Z.; Yang, J.; Guo, W.; Guo, R.; Cheng, H.; Sun, X. Sphingosine-1-Phosphate, a Novel TREM2 Ligand, Promotes Microglial Phagocytosis to Protect against Ischemic Brain Injury. Acta Pharm. Sin. B 2021, 12, 1885–1898. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Jiang, X.; Zhu, Z.; Qin, H.; Dinkins, M.B.; Kong, J.N.; Leanhart, S.; Wang, R.; Elsherbini, A.; Bieberich, E.; et al. Lipid Transporter Spns2 Promotes Microglia Pro-Inflammatory Activation in Response to Amyloid-Beta Peptide. Glia 2019, 67, 498. [Google Scholar] [CrossRef]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted Role of Matrix Metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, M.; Rivera, I.G.; Presa, N.; Gomez-Muñoz, A. Implication of Matrix Metalloproteinases 2 and 9 in Ceramide 1-Phosphate-Stimulated Macrophage Migration. Cell. Signal. 2016, 28, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Xuan, L.; Han, F.; Gong, L.; Lv, Y.; Wan, Z.; Liu, H.; Ren, L.; Yang, S.; Zhang, W.; Li, T.; et al. Ceramide Induces MMP-9 Expression through JAK2/STAT3 Pathway in Airway Epithelium. Lipids Health Dis. 2020, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Yamada, M.; Sasabe, J.; Terashita, K.; Shimoda, M.; Matsuoka, M.; Aiso, S. Amyloid-β Causes Memory Impairment by Disturbing the JAK2/STAT3 Axis in Hippocampal Neurons. Mol. Psychiatry 2008, 14, 206–222. [Google Scholar] [CrossRef] [Green Version]
- Brelstaff, J.; Tolkovsky, A.M.; Ghetti, B.; Goedert, M.; Spillantini, M.G. Living Neurons with Tau Filaments Aberrantly Expose Phosphatidylserine and Are Phagocytosed by Microglia. Cell Rep. 2018, 24, 1939–1948.e4. [Google Scholar] [CrossRef] [Green Version]
- Pampuscenko, K.; Morkuniene, R.; Sneideris, T.; Smirnovas, V.; Budvytyte, R.; Valincius, G.; Brown, G.C.; Borutaite, V. Extracellular Tau Induces Microglial Phagocytosis of Living Neurons in Cell Cultures. J. Neurochem. 2020, 154, 316–329. [Google Scholar] [CrossRef]
- Randez-Gil, F.; Bojunga, L.; Estruch, F.; Winderickx, J.; Del Poeta, M.; Prieto, J.A. Sphingolipids and Inositol Phosphates Regulate the Tau Protein Phosphorylation Status in Humanized Yeast. Front. Cell Dev. Biol. 2020, 8, 1329. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.M.; Ghonaim, G.A.; Gharib, S.M.; Chopra, H.; Farag, A.K.; Hassanin, M.H.; Nagah, A.; Emad-Eldin, M.; Hashem, N.E.; Yahya, G.; et al. Exosomes in Alzheimer’s Disease: From Being Pathological Players to Potential Diagnostics and Therapeutics. Int. J. Mol. Sci. 2021, 22, 794. [Google Scholar] [CrossRef] [PubMed]
- Soares Martins, T.; Trindade, D.; Vaz, M.; Campelo, I.; Almeida, M.; Trigo, G.; da Cruz e Silva, O.A.B.; Henriques, A.G. Diagnostic and Therapeutic Potential of Exosomes in Alzheimer’s Disease. J. Neurochem. 2021, 156, 162–181. [Google Scholar] [CrossRef] [PubMed]
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and Regulation of Neutral Sphingomyelinase-2 in Cellular and Pathological Processes. Adv. Biol. Regul. 2015, 57, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuyama, K.; Takahashi, K.; Usuki, S.; Mikami, D.; Sun, H.; Hanamatsu, H.; Furukawa, J.; Mukai, K.; Igarashi, Y. Plant Sphingolipids Promote Extracellular Vesicle Release and Alleviate Amyloid-β Pathologies in a Mouse Model of Alzheimer’s Disease. Sci. Rep. 2019, 9, 16827. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Mikami, D.; Sun, H.; Tsumita, T.; Takahashi, K.; Mukai, K.; Yuyama, K.; Igarashi, Y. Blood-Brain Barrier Permeability Analysis of Plant Ceramides. PLoS ONE 2020, 15, e0241640. [Google Scholar] [CrossRef]
- Elsherbini, A.; Kirov, A.S.; Dinkins, M.B.; Wang, G.; Qin, H.; Zhu, Z.; Tripathi, P.; Crivelli, S.M.; Bieberich, E. Association of A with Ceramide-Enriched Astrosomes Mediates A Neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 60. [Google Scholar] [CrossRef]
- Couttas, T.A.; Kain, N.; Tran, C.; Chatterton, Z.; Kwok, J.B.; Don, A.S. Age-Dependent Changes to Sphingolipid Balance in the Human Hippocampus Are Gender-Specific and May Sensitize to Neurodegeneration. J. Alzheimer’s Dis. 2018, 63, 503–514. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of Sphingolipid Metabolism in Alzheimer’s Disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative Lipidomic Analysis of Mouse and Human Brain with Alzheimer Disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Holtzman, D.M.; McKeel, D.W.; Kelley, J.; Morris, J.C. Substantial Sulfatide Deficiency and Ceramide Elevation in Very Early Alzheimer’s Disease: Potential Role in Disease Pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef]
- Crivello, N.A.; Rosenberg, I.H.; Dallal, G.E.; Bielinski, D.; Joseph, J.A. Age-Related Changes in Neutral Sphingomyelin-Specific Phospholipase C Activity in Striatum, Hippocampus, and Frontal Cortex: Implication for Sensitivity to Stress and Inflammation. Neurochem. Int. 2005, 47, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Satoi, H.; Tomimoto, H.; Ohtani, R.; Kitano, T.; Kondo, T.; Watanabe, M.; Oka, N.; Akiguchi, I.; Furuya, S.; Hirabayashi, Y.; et al. Astroglial Expression of Ceramide in Alzheimer’s Disease Brains: A Role during Neuronal Apoptosis. Neuroscience 2005, 130, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchal, M.; Gaudin, M.; Lazar, A.N.; Salvati, E.; Rivals, I.; Ayciriex, S.; Dauphinot, L.; Dargère, D.; Auzeil, N.; Masserini, M.; et al. Ceramides and Sphingomyelinases in Senile Plaques. Neurobiol. Dis. 2014, 65, 193–201. [Google Scholar] [CrossRef] [PubMed]
- De Wit, N.M.; Snkhchyan, H.; Den Hoedt, S.; Wattimena, D.; De Vos, R.; Mulder, M.T.; Walter, J.; Martinez-Martinez, P.; Hoozemans, J.J.; Rozemuller, A.J.; et al. Altered Sphingolipid Balance in Capillary Cerebral Amyloid Angiopathy. J. Alzheimer’s Dis. 2017, 60, 795–807. [Google Scholar] [CrossRef]
- Ceccom, J.; Loukh, N.; Lauwers-Cances, V.; Touriol, C.; Nicaise, Y.; Gentil, C.; Uro-Coste, E.; Pitson, S.; Maurage, C.A.; Duyckaerts, C.; et al. Reduced Sphingosine Kinase-1 and Enhanced Sphingosine 1-Phosphate Lyase Expression Demonstrate Deregulated Sphingosine 1-Phosphate Signaling in Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 12. [Google Scholar] [CrossRef]
- Nema, R.; Kumar, A. Sphingosine-1-Phosphate Catabolizing Enzymes Predict Better Prognosis in Triple-Negative Breast Cancer Patients and Correlates With Tumor-Infiltrating Immune Cells. Front. Mol. Biosci. 2021, 8, 697922. [Google Scholar] [CrossRef]
- Diaz Escarcega, R.; McCullough, L.D.; Tsvetkov, A.S. The Functional Role of Sphingosine Kinase 2. Front. Mol. Biosci. 2021, 8, 428. [Google Scholar] [CrossRef]
- Dominguez, G.; Maddelein, M.L.; Pucelle, M.; Nicaise, Y.; Maurage, C.A.; Duyckaerts, C.; Cuvillier, O.; Delisle, M.B. Neuronal Sphingosine Kinase 2 Subcellular Localization Is Altered in Alzheimer’s Disease Brain. Acta Neuropathol. Commun. 2018, 6, 25. [Google Scholar] [CrossRef]
- Weigert, A.; Cremer, S.; Schmidt, M.V.; Von Knethen, A.; Angioni, C.; Geisslinger, G.; Brüne, B. Cleavage of Sphingosine Kinase 2 by Caspase-1 Provokes Its Release from Apoptotic Cells. Blood 2010, 115, 3531–3540. [Google Scholar] [CrossRef] [Green Version]
- Teitsdottir, U.D.; Halldorsson, S.; Rolfsson, O.; Lund, S.H.; Jonsdottir, M.K.; Snaedal, J.; Petersen, P.H. Cerebrospinal Fluid C18 Ceramide Associates with Markers of Alzheimer’s Disease and Inflammation at the Pre- and Early Stages of Dementia. J. Alzheimer’s Dis. 2021, 81, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Ormseth, C.; Chiang, J.; Cipolla, M.; Arakaki, X.; Harrington, M.G. Sphingolipid Metabolism Correlates with Cerebrospinal Fluid Beta Amyloid Levels in Alzheimer’s Disease. PLoS ONE 2015, 10, e0125597. [Google Scholar] [CrossRef]
- Kosicek, M.; Zetterberg, H.; Andreasen, N.; Peter-Katalinic, J.; Hecimovic, S. Elevated Cerebrospinal Fluid Sphingomyelin Levels in Prodromal Alzheimer’s Disease. Neurosci. Lett. 2012, 516, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Torretta, E.; Arosio, B.; Barbacini, P.; Casati, M.; Capitanio, D.; Mancuso, R.; Mari, D.; Cesari, M.; Clerici, M.; Gelfi, C. Particular CSF Sphingolipid Patterns Identify INPH and AD Patients. Sci. Rep. 2018, 8, 13639. [Google Scholar] [CrossRef]
- Morrow, A.; Panyard, D.J.; Deming, Y.K.; Jonaitis, E.; Dong, R.; Vasiljevic, E.; Betthauser, T.J.; Kollmorgen, G.; Suridjan, I.; Bayfield, A.; et al. CSF Sphingomyelins in Alzheimer’s Disease, Neurodegeneration, and Neuroinflammation. medRxiv 2022. [Google Scholar] [CrossRef]
- Ibáñez, C.; Simó, C.; Barupal, D.K.; Fiehn, O.; Kivipelto, M.; Cedazo-Mínguez, A.; Cifuentes, A. A New Metabolomic Workflow for Early Detection of Alzheimer’s Disease. J. Chromatogr. A 2013, 1302, 65–71. [Google Scholar] [CrossRef]
- Han, X.; Rozen, S.; Boyle, S.H.; Hellegers, C.; Cheng, H.; Burke, J.R.; Welsh-Bohmer, K.A.; Doraiswamy, P.M.; Kaddurah-Daouk, R. Metabolomics in Early Alzheimer’s Disease: Identification of Altered Plasma Sphingolipidome Using Shotgun Lipidomics. PLoS ONE 2011, 6, e21643. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Ratnam Bandaru, V.V.; Schech, S.; Carrick, R.; Carlson, M.C.; Mori, S.; Miller, M.I.; Ceritoglu, C.; Brown, T.; et al. Plasma Ceramides Are Altered in Mild Cognitive Impairment and Predict Cognitive Decline and Hippocampal Volume Loss. Alzheimer’s Dement. 2010, 6, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B.; et al. Serum Ceramides Increase the Risk of Alzheimer Disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Haughey, N.J.; Han, D.; An, Y.; Bandaru, V.V.R.; Lyketsos, C.G.; Ferrucci, L.; Resnick, S.M. The Association between Plasma Ceramides and Sphingomyelins and Risk of Alzheimer’s Disease Differs by Sex and APOE in the Baltimore Longitudinal Study of Aging. J. Alzheimer’s Dis. 2017, 60, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Liu, H.; Zhang, T.; Jiang, Y.; Xing, H.; Zhang, A.H. Discovery of Serum Metabolites for Diagnosis of Progression of Mild Cognitive Impairment to Alzheimer’s Disease Using an Optimized Metabolomics Method. RSC Adv. 2016, 6, 3586–3591. [Google Scholar] [CrossRef]
- Barupal, D.K.; Baillie, R.; Fan, S.; Saykin, A.J.; Meikle, P.J.; Arnold, M.; Nho, K.; Fiehn, O.; Kaddurah-Daouk, R. Sets of Coregulated Serum Lipids Are Associated with Alzheimer’s Disease Pathophysiology. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 619. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Weinberg, D.D.; Darby, E.; Zaidi, N.; Pavlik, V.; Doody, R.S.; Lyketsos, C.G. Plasma Sphingomyelins Are Associated with Cognitive Progression in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Rabins, P.V.; Lyketsos, C.G.; Carlson, M.C. Serum Sphingomyelins and Ceramides Are Early Predictors of Memory Impairment. Neurobiol. Aging 2010, 31, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Custodia, A.; Romaus-Sanjurjo, D.; Aramburu-Núñez, M.; Álvarez-Rafael, D.; Vázquez-Vázquez, L.; Camino-Castiñeiras, J.; Leira, Y.; Pías-Peleteiro, J.M.; Aldrey, J.M.; Sobrino, T.; et al. Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 8082. https://doi.org/10.3390/ijms23158082
Custodia A, Romaus-Sanjurjo D, Aramburu-Núñez M, Álvarez-Rafael D, Vázquez-Vázquez L, Camino-Castiñeiras J, Leira Y, Pías-Peleteiro JM, Aldrey JM, Sobrino T, et al. Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(15):8082. https://doi.org/10.3390/ijms23158082
Chicago/Turabian StyleCustodia, Antía, Daniel Romaus-Sanjurjo, Marta Aramburu-Núñez, Diego Álvarez-Rafael, Laura Vázquez-Vázquez, Javier Camino-Castiñeiras, Yago Leira, Juan Manuel Pías-Peleteiro, José Manuel Aldrey, Tomás Sobrino, and et al. 2022. "Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 15: 8082. https://doi.org/10.3390/ijms23158082
APA StyleCustodia, A., Romaus-Sanjurjo, D., Aramburu-Núñez, M., Álvarez-Rafael, D., Vázquez-Vázquez, L., Camino-Castiñeiras, J., Leira, Y., Pías-Peleteiro, J. M., Aldrey, J. M., Sobrino, T., & Ouro, A. (2022). Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases. International Journal of Molecular Sciences, 23(15), 8082. https://doi.org/10.3390/ijms23158082