
Deciphering the Genetic Basis of Root and Biomass Traits in Rapeseed (Brassica napus L.) through the Integration of GWAS and RNA-Seq under Nitrogen Stress

 ,
, 

Abstract
:1. Introduction
2. Results
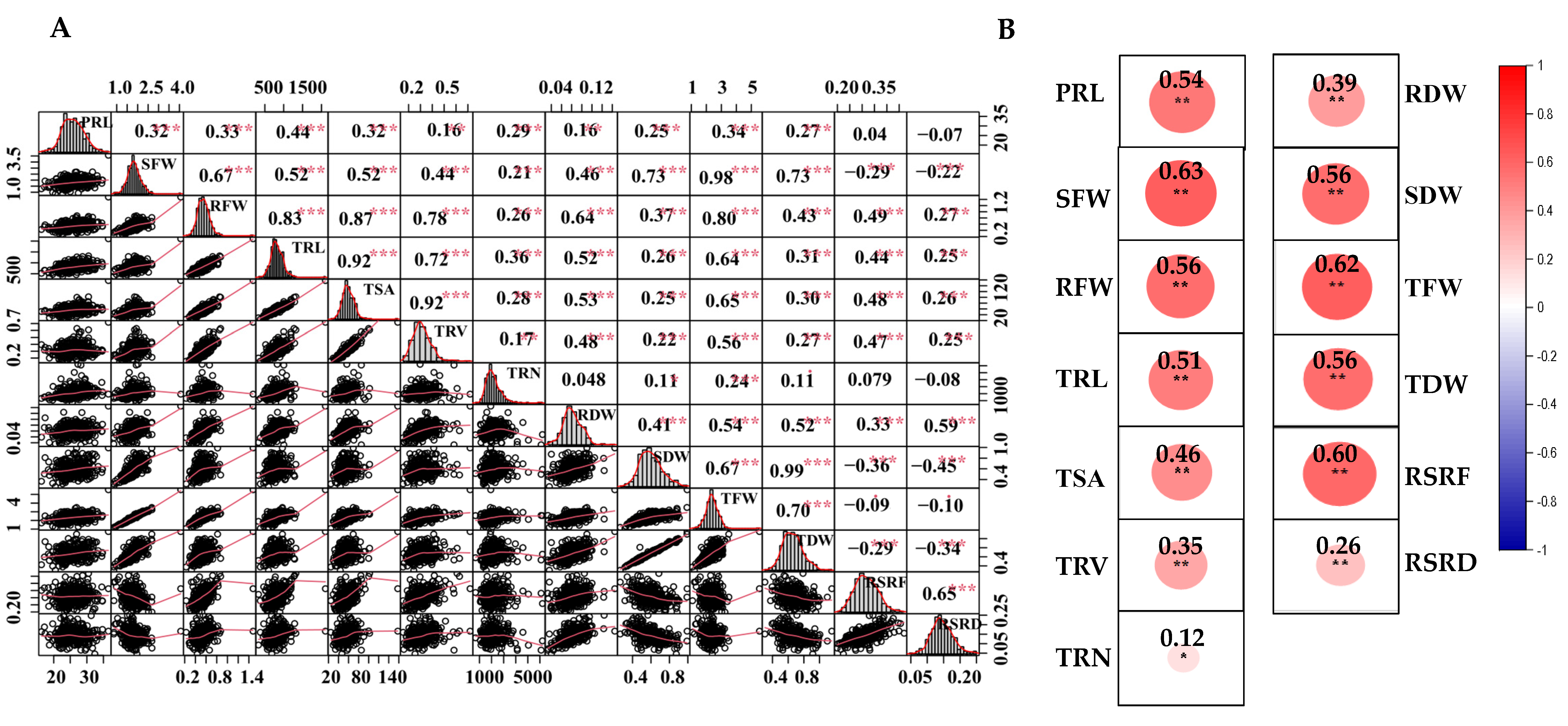
2.1. Phenotypic Analysis of Root and Shoot Biomass Traits under the LN Stress
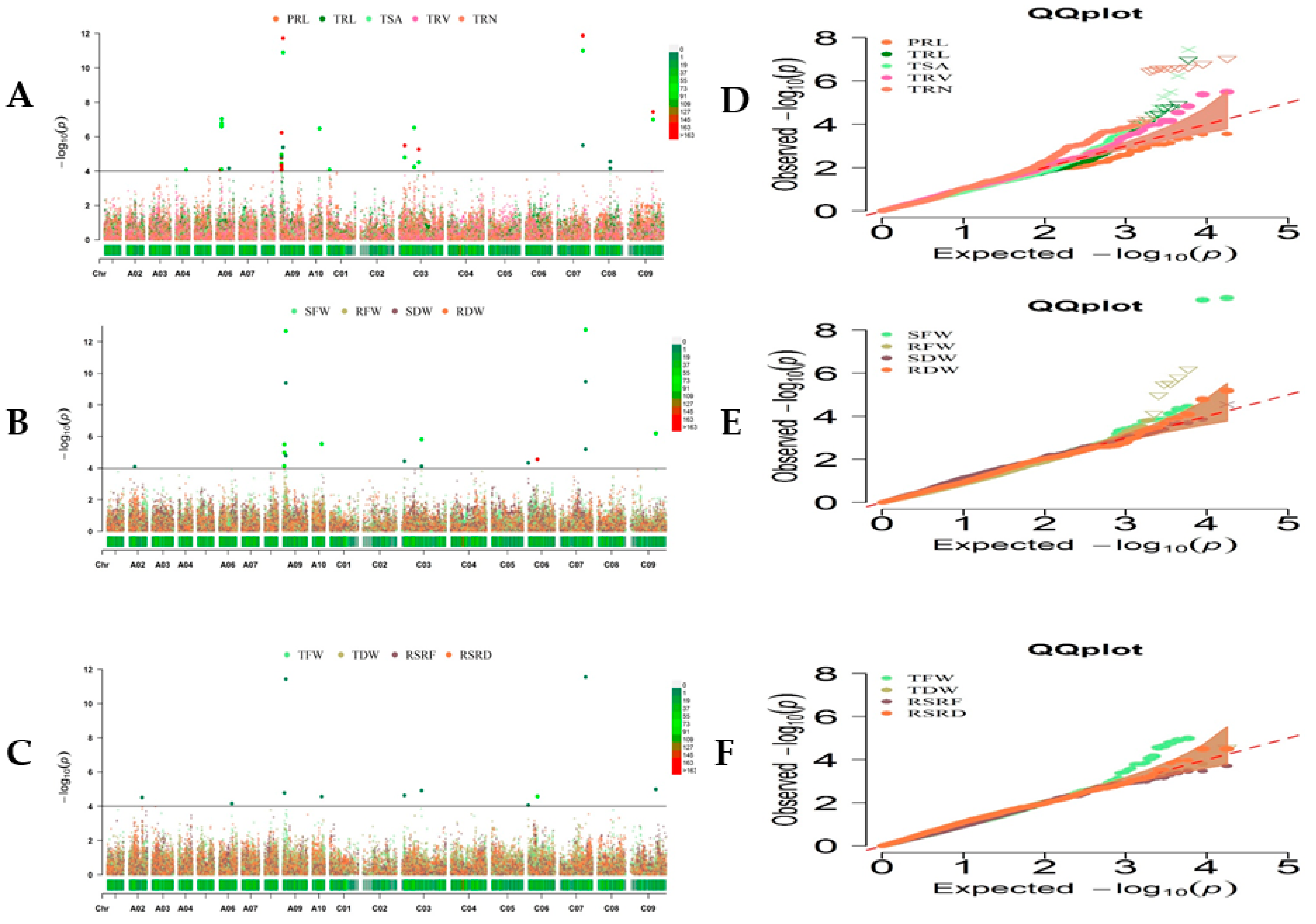
2.2. Marker–Trait Association Analysis for Root and Biomass Traits under LN Stress
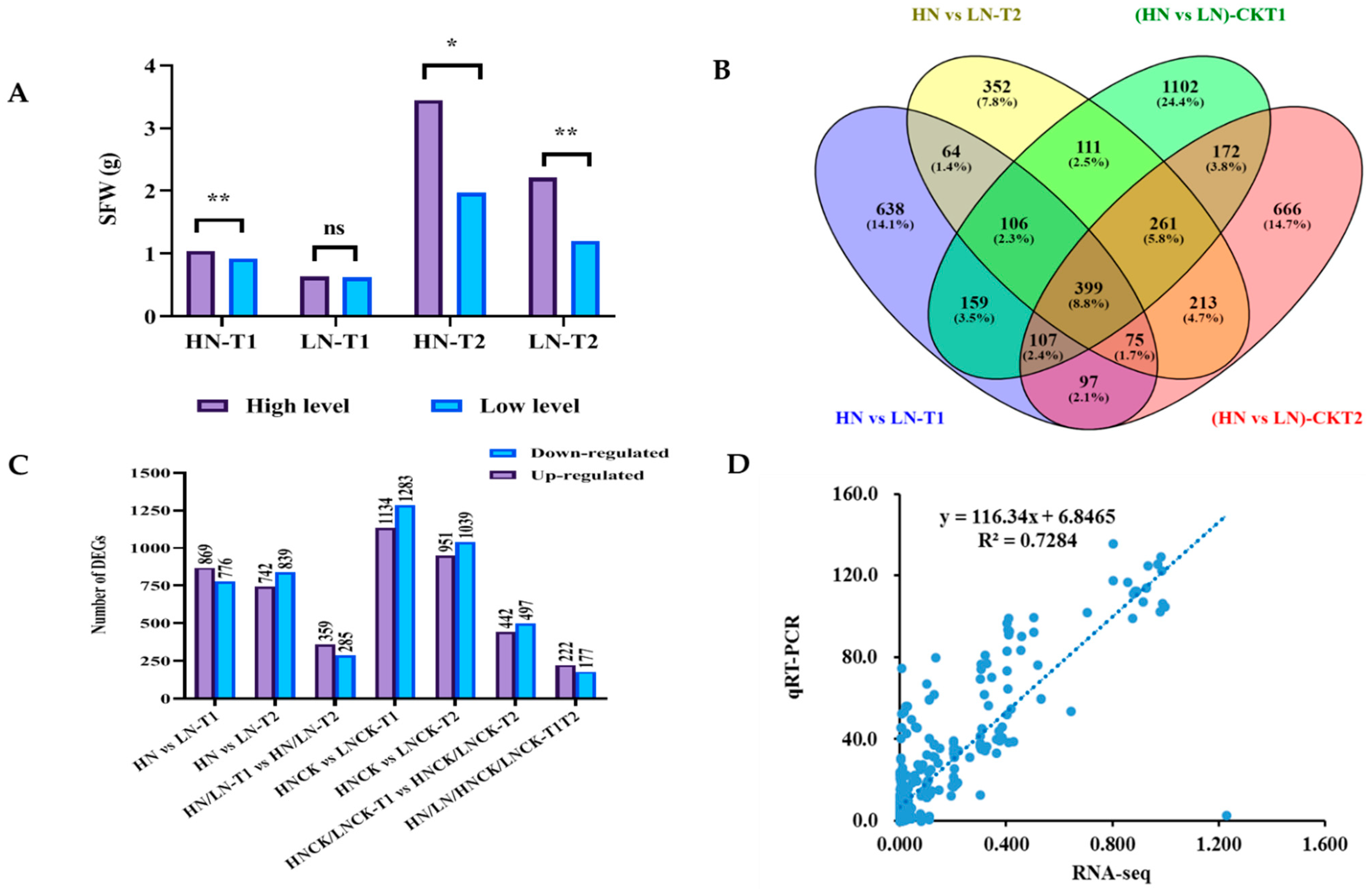
2.3. Differentially Expressed Genes (DEGs) between High and Low Nitrogen Efficient Group
2.4. Functional Classification of DEGs Involved in High and Low Nitrogen Efficiency
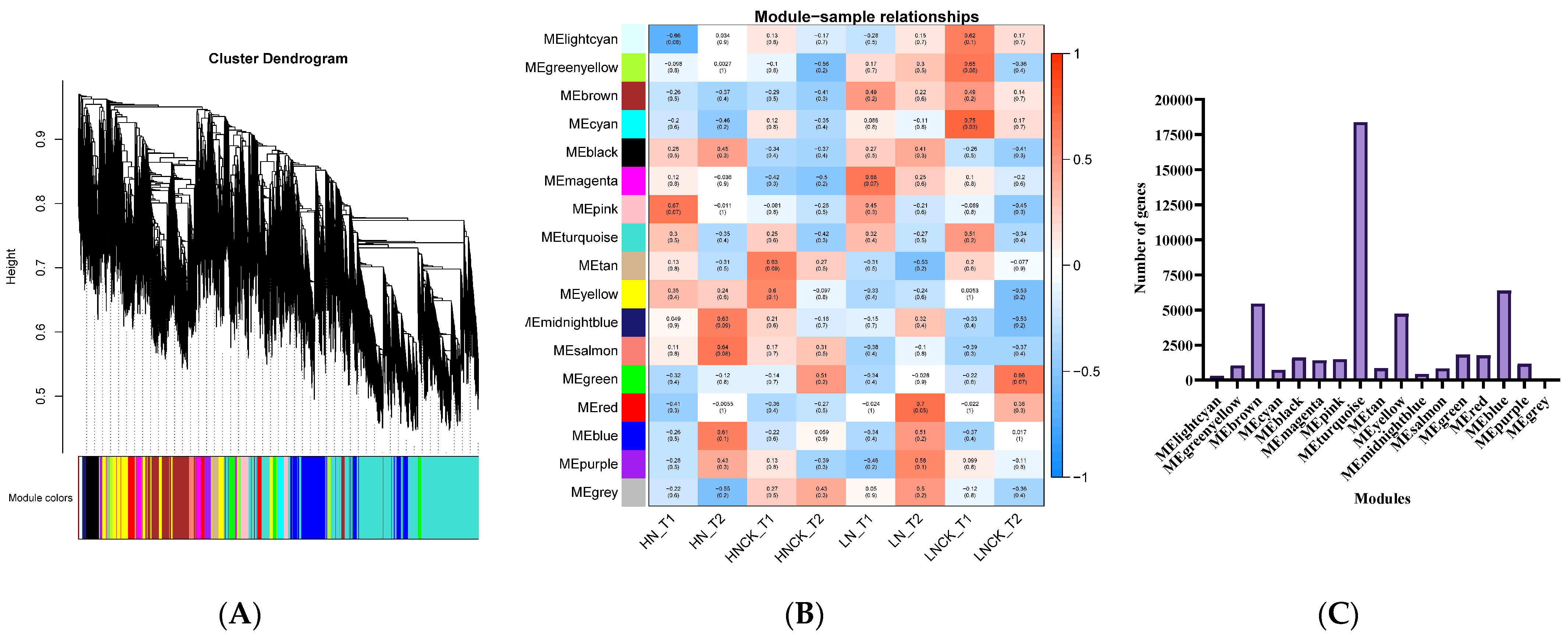
2.5. Gene Co-Expression Network Construction and Analysis (WGCNA)
2.6. Candidate Genes’ Prediction and Prioritization by Integrating GWAS, DEGs, and WGCNA
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Phenotypic Investigation
4.3. Statistical Analysis
4.4. Association Analysis
4.5. Candidate Gene Prediction
4.6. Transcriptome Sequencing and Analysis
4.7. Validation of DEGs by Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The, S.V.; Snyder, R.; Tegeder, M. Targeting Nitrogen Metabolism and Transport Processes to Improve Plant Nitrogen Use Efficiency. Front. Plant Sci. 2021, 11, 628366. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Munné-Bosch, S. Hormonal impact on photosynthesis and photoprotection in plants. Plant Physiol. 2021, 185, 1500–1522. [Google Scholar] [CrossRef] [PubMed]
- Hirel, B.; Le Gouis, J.; Ney, B.; Gallais, A. The challenge of improving nitrogen use efficiency in crop plants: Towards a more central role for genetic variability and quantitative genetics within integrated approaches. J. Exp. Bot. 2007, 58, 2369–2387. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Bali, S.K. A review of methods to improve nitrogen use efficiency in agriculture. Sustainability 2017, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.W.; Sung, Y.; Chen, B.C.; Lai, H.Y. Effects of nitrogen fertilizers on the growth and nitrate content of lettuce (Lactuca sativa L.). Int. J. Environ. Res. Public Health 2014, 11, 4427–4440. [Google Scholar] [CrossRef] [Green Version]
- Ali, J.; Jewel, Z.A.; Mahender, A.; Anandan, A.; Hernandez, J.; Li, Z. Molecular Genetics and Breeding for Nutrient Use Efficiency in Rice. Int. J. Mol. Sci. 2018, 19, 1762. [Google Scholar] [CrossRef] [Green Version]
- Lammerts van Bueren, E.T.; Struik, P.C. Diverse concepts of breeding for nitrogen use efficiency. A review. Agron. Sustain. Dev. 2017, 37, 50. [Google Scholar] [CrossRef] [Green Version]
- Raboanatahiry, N.; Li, H.; Yu, L.; Li, M. Rapeseed (Brassica napus L.): Processing, utilization, and genetic improvement. Agronomy 2021, 11, 1776. [Google Scholar] [CrossRef]
- Snowdon, R.J.; Wittkop, B.; Chen, T.W.; Stahl, A. Crop adaptation to climate change as a consequence of long-term breeding. Theor. Appl. Genet. 2021, 134, 1613–1623. [Google Scholar] [CrossRef]
- Vazquez-Carrasquer, V.; Laperche, A.; Bissuel-Bélaygue, C.; Chelle, M.; Richard-Molard, C. Nitrogen Uptake Efficiency, Mediated by Fine Root Growth, Early Determines Temporal and Genotypic Variations in Nitrogen Use Efficiency of Winter Oilseed Rape. Front. Plant Sci. 2021, 12, 641459. [Google Scholar] [CrossRef]
- Williams, S.T.; Vail, S.; Arcand, M.M. Nitrogen Use Efficiency in Parent vs. Hybrid Canola under Varying Nitrogen Availabilities. Plants 2021, 10, 2364. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, C. Genotypic diversity and plasticity of root system architecture to nitrogen availability in oilseed rape. PLoS ONE 2021, 16, e0250966. [Google Scholar] [CrossRef] [PubMed]
- Calleja-Cabrera, J.; Boter, M.; Oñate-Sánchez, L.; Pernas, M. Root Growth Adaptation to Climate Change in Crops. Front. Plant Sci. 2020, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, F.K.Y. QTL mapping for seedling traits in wheat grown under varying concentrations of N, P and K nutrients. Theor. Appl. Genet. 2012, 124, 851–865. [Google Scholar] [CrossRef]
- Ober, E.S.; Alahmad, S.; Cockram, J.; Forestan, C.; Hickey, L.T.; Kant, J.; Maccaferri, M.; Marr, E.; Milner, M.; Pinto, F.; et al. Wheat root systems as a breeding target for climate resilience. Theor. Appl. Genet. 2021, 134, 1645–1662. [Google Scholar] [CrossRef]
- Siddiqui, M.N.; León, J.; Naz, A.A.; Ballvora, A. Genetics and genomics of root system variation in adaptation to drought stress in cereal crops. J. Exp. Bot. 2021, 72, 1007–1019. [Google Scholar] [CrossRef]
- Kitomi, Y.; Hanzawa, E.; Kuya, N.; Inoue, H.; Hara, N.; Kawai, S.; Kanno, N.; Endo, M.; Sugimoto, K.; Yamazaki, T. Root angle modifications by the DRO1 homolog improve rice yields in saline paddy fields. Proc. Natl. Acad. Sci. USA 2020, 117, 21242–21250. [Google Scholar] [CrossRef]
- Dossa, K.; Zhou, R.; Li, D.; Liu, A.; Qin, L.; Mmadi, M.A.; Su, R.; Zhang, Y.; Wang, J.; Gao, Y.; et al. A novel motif in the 5′-UTR of an orphan gene ‘Big Root Biomass’ modulates root biomass in sesame. Plant Biotechnol. J. 2021, 19, 1065–1079. [Google Scholar] [CrossRef]
- Duan, X.; Jin, K.; Ding, G.; Wang, C.; Cai, H.; Wang, S.; White, P.J.; Xu, F.; Shi, L. The impact of different morphological and biochemical root traits on phosphorus acquisition and seed yield of Brassica napus. Field Crops Res. 2020, 258, 107960. [Google Scholar] [CrossRef]
- Xu, F.; Chen, S.; Yang, X.; Zhou, S.; Wang, J. Genome-Wide Association Study on Root Traits Under Different Growing Environments in Wheat (Triticum aestivum L.). Front. Genet. 2021, 12, 646712. [Google Scholar] [CrossRef]
- Xu, X.; Ye, J.; Yang, Y.; Zhang, M.; Xu, Q.; Feng, Y.; Yuan, X.; Yu, H. Genome-Wide Association Study of Rice Rooting Ability at the Seedling Stage. Rice 2020, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Mandozai, A.; Moussa, A.A.; Zhang, Q.; Qu, J.; Du, Y. Genome-Wide Association Study of Root and Shoot Related Traits in Spring Soybean (Glycine max L.) at Seedling Stages Using SLAF-Seq. Front. Plant Sci. 2021, 12, 568995. [Google Scholar] [CrossRef] [PubMed]
- Moussa, A.A.; Mandozai, A.; Jin, Y.; Qu, J.; Zhang, Q.; Zhao, H.; Anwari, G. Genome-wide association screening and verification of potential genes associated with root architectural traits in maize (Zea mays L.) at multiple seedling stages. BMC Genom. 2021, 22, 558. [Google Scholar] [CrossRef] [PubMed]
- Kiran, A.; Wakeel, A.; Snowdon, R.; Friedt, W. Genetic dissection of root architectural traits by QTL and genome-wide association mapping in rapeseed (Brassica napus L.). Plant Breed. 2019, 138, 184–192. [Google Scholar] [CrossRef]
- Ibrahim, S.; Li, K.; Ahmad, N.; Kuang, L.; Sadau, S.B.; Tian, Z.; Huang, L.; Wang, X.; Dun, X.; Wang, H. Genetic Dissection of Mature Root Characteristics by Genome-Wide Association Studies in Rapeseed (Brassica napus L.). Plants 2021, 10, 2569. [Google Scholar] [CrossRef]
- Wang, J.; Dun, X.; Shi, J.; Wang, X.; Liu, G.; Wang, H. Genetic dissection of root morphological traits related to nitrogen use efficiency in brassica napus L. Under two contrasting nitrogen conditions. Front. Plant Sci. 2017, 8, 1709. [Google Scholar] [CrossRef] [Green Version]
- De Kesel, J.; Bonneure, E.; Mangelinckx, S. The Use of PTI-Marker Genes to Identify Novel Compounds that Establish Induced Resistance in Rice. Int. J. Mol. Sci. 2020, 21, 317. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Zhang, B.; Tang, X.; Zhang, J.; Lin, J. Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways. Int. J. Mol. Sci. 2020, 21, 861. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zeng, X.; Song, Q.; Sun, Y.; Feng, Y.; Lai, Y. Identification of key genes and modules in response to Cadmium stress in different rice varieties and stem nodes by weighted gene co-expression network analysis. Sci. Rep. 2020, 10, 9525. [Google Scholar] [CrossRef]
- Li, K.; Wang, J.; Kuang, L.; Tian, Z.; Wang, X.; Dun, X.; Tu, J.; Wang, H. Genome-wide association study and transcriptome analysis reveal key genes affecting root growth dynamics in rapeseed. Biotechnol. Biofuels 2021, 14, 178. [Google Scholar] [CrossRef]
- Tracy, S.R.; Nagel, K.A.; Postma, J.A.; Fassbender, H.; Wasson, A.; Watt, M. Crop Improvement from Phenotyping Roots: Highlights Reveal Expanding Opportunities. Trends Plant Sci. 2020, 25, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, J.M.; Alvaro, F. Discovering consensus genomic regions in wheat for root-related traits by QTL meta-analysis. Sci. Rep. 2019, 9, 10537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Chen, W.; Yao, Q.; Guo, B.; Valliyod, B.; Wang, Z.; Nguyen, H.T. Genome-wide transcriptional profiling for elucidating the effects of brassinosteroids on Glycine max during early vegetative development. Sci. Rep. 2019, 9, 16085. [Google Scholar] [CrossRef]
- Sandhu, N.; Subedi, S.R.; Yadaw, R.B.; Chaudhary, B.; Prasai, H. Root Traits Enhancing Rice Grain Yield under Alternate Wetting and Drying Condition. Front. Plant Sci. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xie, W.; Yang, C.; Xu, J.; Li, J.; Wang, H.; Chen, X.; Huang, C. transporter, is required for Arabidopsis root growth under manganese deficiency. New Phytol. 2018, 217, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Fan, C.; Li, J.; Cai, G.; Yang, Q.; Wu, J.; Yi, X.; Zhang, C.; Zhou, Y. A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor. Appl. Genet. 2016, 129, 1203–1215. [Google Scholar] [CrossRef]
- Xu, L.; Hu, K.; Zhang, Z.; Guan, C.; Chen, S.; Hua, W.; Li, J.; Wen, J.; Yi, B.; Shen, J.; et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res. 2015, 23, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; Qian, W.; Snowdon, R.J. Sub-genomic selection patterns as a signature of breeding in the allopolyploid Brassica napus genome. BMC Genom. 2014, 15, 1170. [Google Scholar] [CrossRef] [Green Version]
- Anas, M.; Liao, F.; Verma, K.K.; Sarwar, M.A.; Mahmood, A.; Chen, Z.L.; Li, Q.; Zeng, X.P.; Liu, Y.; Li, Y.R. Fate of nitrogen in agriculture and environment: Agronomic, eco-physiological and molecular approaches to improve nitrogen use efficiency. Biol. Res. 2020, 53, 47. [Google Scholar] [CrossRef]
- Wu, B.; Ren, W.; Zhao, L.; Li, Q.; Sun, J.; Chen, F.; Pan, Q. Genome-Wide Association Study of Root System Architecture in Maize. Genes 2022, 13, 181. [Google Scholar] [CrossRef]
- Wang, J.; Kuang, L.; Wang, X.; Liu, G.; Dun, X.; Wang, H. Temporal genetic patterns of root growth in Brassica napus L. revealed by a low-cost, high-efficiency hydroponic system. Theor. Appl. Genet. 2019, 132, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Kupcsik, L.; Chiodi, C.; Moturu, T.R.; De Gernier, H.; Haelterman, L.; Louvieaux, J.; Tillard, P.; Sturrock, C.J.; Bennett, M.; Nacry, P.; et al. Oilseed Rape Cultivars Show Diversity of Root Morphologies with the Potential for Better Capture of Nitrogen. Nitrogen 2021, 2, 491–505. [Google Scholar] [CrossRef]
- Dun, X.; Tao, Z.; Wang, J.; Wang, X.; Liu, G.; Wang, H. Comparative transcriptome analysis of primary roots of brassica napus seedlings with extremely different primary root lengths using RNA sequencing. Front. Plant Sci. 2016, 7, 1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Peterkin, J.H. Clothing the Emperor: Dynamic Root-Shoot Allocation Trajectories in Relation to Whole-Plant Growth Rate and in Response to Temperature. Plants 2019, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Kuang, L.; Ahmad, N.; Su, B.; Huang, L.; Li, K.; Wang, H.; Wang, X.; Dun, X. Discovery of Genomic Regions and Candidate Genes Controlling Root Development Using a Recombinant Inbred Line Population in Rapeseed (Brassica napus L.). Int. J. Mol. Sci. 2022, 23, 4781. [Google Scholar] [CrossRef]
- Falik, O.; Mordoch, Y.; Ben-Natan, D.; Vanunu, M.; Goldstein, O.; Novoplansky, A. Plant responsiveness to root-root communication of stress cues. Ann. Bot. 2012, 110, 271–280. [Google Scholar] [CrossRef]
- Canales, J.; Verdejo, J.; Carrasco-Puga, G.; Castillo, F.M.; Arenas-M, A.; Calderini, D.F. Transcriptome analysis of seed weight plasticity in Brassica napus. Int. J. Mol. Sci. 2021, 22, 4449. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Zhang, Q.; Yuan, P.; Liu, L.; King, G.J.; Ding, G.; Wang, S.; Cai, H.; Wang, C.; et al. Integrating a genome-wide association study with transcriptomic data to predict candidate genes and favourable haplotypes influencing Brassica napus seed phytate. DNA Res. 2021, 28, dsab011. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Chen, X.; Xu, F.; Ding, M.; Ye, W.; Kawai, Y.; Toda, Y.; Hayashi, Y.; Suzuki, T.; et al. Plasma membrane H+-ATPase overexpression increases rice yield via simultaneous enhancement of nutrient uptake and photosynthesis. Nat. Commun. 2021, 12, 735. [Google Scholar] [CrossRef]
- Samuilov, S.; Brilhaus, D.; Rademacher, N.; Flachbart, S.; Arab, L.; Alfarraj, S.; Kuhnert, F.; Kopriva, S.; Weber, A.P.M.; Mettler-Altmann, T.; et al. The photorespiratory BOU gene mutation alters sulfur assimilation and its crosstalk with carbon and nitrogen metabolism in Arabidopsis thaliana. Front. Plant Sci. 2018, 9, 1709. [Google Scholar] [CrossRef]
- Liao, Q.; Tang, T.; Zhou, T.; Song, H.; Hua, Y. Integrated Transcriptional and Proteomic Profiling Reveals Potential Amino Acid Transporters Targeted by Nitrogen Limitation Adaptation. Int. J. Mol. Sci. 2020, 2, 2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Cai, X.T.; Wang, Y.; Xing, L.; Chen, Q.; Xiang, C. Bin HDG11 upregulates cell-wall-loosening protein genes to promote root elongation in Arabidopsis. J. Exp. Bot. 2014, 65, 4285–4295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, C.; König, J.; Altmann, F.; Strasser, R. Processing of the terminal alpha-1,2-linked mannose residues from oligomannosidic n-glycans is critical for proper root growth. Front. Plant Sci. 2018, 9, 1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellero, Y. Manipulating Amino Acid Metabolism to Improve Crop Nitrogen Use Efficiency for a Sustainable Agriculture. Front. Plant Sci. 2020, 11, 602548. [Google Scholar] [CrossRef]
- Li, H.; Cheng, X.; Zhang, L.; Hu, J.; Zhang, F.; Chen, B.; Xu, K.; Gao, G.; Li, H.; Li, L.; et al. An integration of genome-wide association study and gene co-expression network analysis identifies candidate genes of stem lodging-related traits in Brassica napus. Front. Plant Sci. 2018, 9, 796. [Google Scholar] [CrossRef]
- Lu, B.; Wen, S.; Zhu, P.; Cao, H.; Zhou, Y.; Bie, Z.; Cheng, J. Overexpression of melon tonoplast sugar transporter cmtst1 improved root growth under high sugar content. Int. J. Mol. Sci. 2020, 21, 3524. [Google Scholar] [CrossRef]
- Schofield, R.A.; Bi, Y.M.; Kant, S.; Rothstein, S.J. Over-expression of STP13, a hexose transporter, improves plant growth and nitrogen use in Arabidopsis thaliana seedlings. Plant Cell Environ. 2009, 32, 271–285. [Google Scholar] [CrossRef]
- Wang, D.; Xu, T.; Yin, Z.; Wu, W.; Geng, H.; Li, L.; Yang, M.; Cai, H.; Lian, X. Overexpression of OsMYB305 in Rice Enhances the Nitrogen Uptake Under Low-Nitrogen Condition. Front. Plant Sci. 2020, 11, 369. [Google Scholar] [CrossRef] [Green Version]
- Vandenbussche, F.; Fierro, A.C.; Wiedemann, G.; Reski, R.; Van Der Straeten, D. Evolutionary conservation of plant gibberellin signalling pathway components. BMC Plant Biol. 2007, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Rui, C.; Peng, F.; Fan, Y.; Zhang, Y.; Zhang, Z.; Xu, N.; Zhang, H.; Wang, J.; Li, S.; Yang, T.; et al. Genome-wide expression analysis of carboxylesterase (CXE) gene family implies GBCXE49 functional responding to alkaline stress in cotton. BMC Plant Biol. 2022, 22, 194. [Google Scholar] [CrossRef]
- Li, Y.; Dai, X.; Cheng, Y.; Zhao, Y. NPY genes play an essential role in root gravitropic responses in Arabidopsis. Mol. Plant 2011, 4, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Y.M.; Wang, R.L.; Zhu, T.; Rothstein, S.J. Global transcription profiling reveals differential responses to chronic nitrogen stress and putative nitrogen regulatory components in Arabidopsis. BMC Genom. 2007, 8, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moison, M.; Marmagne, A.; Dinant, S.; Soulay, F.; Azzopardi, M.; Lothier, J.; Citerne, S.; Morin, H.; Legay, N.; Chardon, F.; et al. Three cytosolic glutamine synthetase isoforms localized in different-order veins act together for N remobilization and seed filling in Arabidopsis. J. Exp. Bot. 2018, 69, 4379–4393. [Google Scholar] [CrossRef]
- Gan, Y.; Bernreiter, A.; Filleur, S.; Abram, B.; Forde, B.G. Overexpressing the ANR1 MADS-box gene in transgenic plants provides new insights into its role in the nitrate regulation of root development. Plant Cell Physiol. 2012, 53, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furutani, M.; Hirano, Y.; Nishimura, T.; Nakamura, M.; Taniguchi, M.; Suzuki, K.; Oshida, R.; Kondo, C.; Sun, S.; Kato, K.; et al. Polar recruitment of RLD by LAZY1-like protein during gravity signaling in root branch angle control. Nat. Commun. 2020, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Iyer-Pascuzzi, A.S. Shedding the last layer: Mechanisms of root cap cell release. Plants 2020, 9, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaro, M.; De Paoli, E.; Tomasi, N.; Morgante, M.; Pinton, R.; Zanin, L. Transgenerational Response to Nitrogen Deprivation in Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 5587. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Fang, X.; Xiao, C.; Ma, Z.; Huang, X.; Su, J.; Li, J.; Wang, J.; Wang, S.; Luan, S.; et al. Kinase SnRK1.1 regulates nitrate channel SLAH3 engaged in nitrate-dependent alleviation of ammonium toxicity. Plant Physiol. 2021, 186, 731–749. [Google Scholar] [CrossRef]
- Lhamo, D.; Luan, S. Potential Networks of Channels and Transporters in Arabidopsis Roots at a Single Cell Resolution. Front. Plant Sci. 2021, 12, 689545. [Google Scholar] [CrossRef]
- Hoagland, D.R. Optimum nutrient solutions for plants. Science 1920, 52, 562–564. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Gómez-Rubio, V. ggplot2—Elegant Graphics for Data Analysis (2nd Edition). J. Stat. Softw. 2017, 77, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Jian, H.; Lu, K.; Filardo, F.; Yin, N.; Liu, L.; Qu, C.; Li, W.; Du, H.; Li, J. Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol. J. 2016, 14, 1368–1380. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Huang, H.; Yi, X.; Zhang, Y.; Yang, Q.; Zhang, C.; Fan, C.; Zhou, Y. Dissection of genetic architecture for glucosinolate accumulations in leaves and seeds of Brassica napus by genome-wide association study. Plant Biotechnol. J. 2020, 18, 1472–1484. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Guo, W.; Lu, J.; Yu, H.; Qu, C.; Tang, Z. Genome-Wide Survey and Expression Profile Analysis of the Mitogen-Activated Protein Kinase (MAPK) Gene Family in Brassica rapa. PLoS ONE 2015, 10, e0132051. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Trait Description | Abbreviations | Units |
|---|---|---|---|
| Root morphological traits (MT) | Primary root length | PRL | cm |
| Total root length | TRL | cm | |
| Total root surface area | TSA | cm2 | |
| Total root volume | TRV | cm3 | |
| Total number of roots | TNR | number | |
| Biomass-related traits (BM) | Shoot fresh weight | SFW | g |
| Root fresh weight | RFW | g | |
| Shoot dry weight | SDW | g | |
| Root dry weight | RDW | g | |
| Total fresh weight | TFW | g | |
| Total dry weight | TDW | g | |
| Fresh root–shoot ratio | RSRF | RFW/SFW | |
| Dry root–shoot ratio | RSRD | RDW/SDW |
| Trait | Control | LN | Control × N Stress | N Stress Impact (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | Mean | CV (%) | h2 | Min | Max | Mean | CV (%) | h2 | |||
| PRL (cm) | 12.2 | 34.3 | 24.3 | 14.5 | 0.62 | 16.3 | 36.5 | 25.9 | 12.8 | 0.58 | ** | 6.4 |
| TRL (cm) | 434.3 | 1435.7 | 843.8 | 20.7 | 0.66 | 330.7 | 2104.9 | 912.1 | 21.6 | 0.58 | * | 7.5 |
| TSA (cm2) | 18.4 | 99.9 | 56.5 | 23.6 | 0.62 | 24.6 | 158.0 | 65.8 | 23.1 | 0.55 | * | 14.1 |
| TRV (cm3) | 0.062 | 0.604 | 0.311 | 29.9 | 0.57 | 0.154 | 0.719 | 0.319 | 25.4 | 0.53 | ns | 2.4 |
| TRN | 673.5 | 4804.5 | 1654.2 | 45.6 | 0.55 | 667 | 6213.9 | 2166.8 | 31.9 | 0.53 | ** | 23.7 |
| SFW (g) | 1.291 | 4.889 | 3.243 | 20.1 | 0.72 | 0.858 | 4.062 | 1.799 | 18.7 | 0.66 | ** | −80.3 |
| SDW (g) | 0.234 | 1.377 | 0.741 | 26.5 | - | 0.266 | 0.992 | 0.594 | 20.8 | - | ** | −24.7 |
| RFW (g) | 0.261 | 0.788 | 0.503 | 19.2 | 0.60 | 0.227 | 1.479 | 0.538 | 22.2 | 0.60 | ** | 6.4 |
| RDW (g) | 0.013 | 0.134 | 0.070 | 28.2 | - | 0.033 | 0.165 | 0.081 | 23.4 | - | ** | 14.2 |
| TFW (g) | 1.617 | 5.599 | 3.746 | 19.3 | 0.70 | 1.077 | 5.541 | 2.345 | 18.3 | 0.65 | ** | −59.7 |
| TDW (g) | 0.277 | 1.476 | 0.809 | 25.9 | - | 0.302 | 1.157 | 0.676 | 19.6 | - | ** | −19.6 |
| RSRF | 0.102 | 0.248 | 0.159 | 15.9 | 0.70 | 0.199 | 0.470 | 0.311 | 16.1 | 0.55 | ** | 48.9 |
| RSRD | 0.031 | 0.298 | 0.095 | 27.5 | - | 0.046 | 0.250 | 0.143 | 23.5 | - | ** | 33.8 |
| QTL Cluster | No. of SNPs | Peak SNP | Chr. | Position | PVE (%) | −log10P | Haplotype Block (Mb) | Traits |
|---|---|---|---|---|---|---|---|---|
| RT-A02-1 | 4 | Bn-A02-p18994312 | A02 | 17,997,697 | 7.0 | 4.5 | 17.95–18.25 | RSRD |
| RT-A06-1 | 8 | seq-new-rs31601 | A06 | 7,556,204 | 10.5 | 6.8 | 7.25–7.72 | TRN |
| RT-A09-1 | 19 | Bn-A09-p1552993 | A09 | 2,375,212 | 9.9 | 6.2 | 2.18–2.51 | TRL, TSA, RFW, RDW, TRV, TFW, SFW |
| RT-A09-2 | 9 | seq-new-rs41996 | A09 | 4,405,703 | 21.2 | 12.7 | 4.35–4.41 | TRL, TFW, RFW, TSA, SFW, TRV, RDW |
| RT-A10-1 | 4 | Bn-A10-p11396195 | A10 | 12,709,829 | 8.9 | 5.5 | 12.67–12.72 | RFW, TFW, TRL, SFW |
| RT-A10-2 | 1 | Bn-A10-p13659996 | A10 | 13,700,523 | 10.0 | 6.5 | 13.56–13.70 | TRN |
| RT-C03-1 | 2 | seq-new-rs49231 | C03 | 4,066,558 | 7.0 | 4.6 | 4.00–4.18 | TFW, SFW |
| RT-C03-2 | 3 | seq-new-rs28219 | C03 | 8,417,394 | 8.4 | 5.5 | 8.32–8.41 | TSA, TRL, TRV |
| RT-C03-3 | 2 | seq-new-rs39672 | C03 | 21,177,143 | 10.0 | 6.5 | 20.85–21.30 | TRN |
| RT-C03-4 | 11 | seq-new-rs41373 | C03 | 27,421,341 | 9.0 | 5.8 | 27.42–27.46 | TRV, TSA, TFW, TRL, RFW, SFW |
| RT-C06-1 | 2 | seq-new-rs23016 | C06 | 12,887,745 | 7.1 | 4.6 | 12.80–13.08 | TDW, SDW |
| RT-C07-1 | 7 | seq-new-rs46512 | C07 | 35,123,112 | 20.4 | 12.8 | 35.12–35.17 | RFW, TSA, TFW, TRL, SFW, TRV, RDW |
| RT-C08-1 | 4 | seq-new-rs29850 | C08 | 21,323,655 | 5.7 | 4.5 | 21.23–21.32 | TRV, TSA, TFW |
| RT-C09-1 | 5 | seq-new-rs34959 | C09 | 34,541,392 | 11.9 | 7.4 | 34.38–34.54 | TSA, TRL, RFW, TFW, SFW |
| Hub Genes in the MEsalmon Module (GWAS + WGCNA) | |||||||||||
| Darmor_ID | HN-T1 | HN-T2 | HNCK-T1 | HNCK-T2 | LN-T1 | LN-T2 | LNCK-T1 | LNCK-T2 | QTN/QTL Cluster | Distance from Lead SNP (Kb) | Description |
| BnaA09g04260D | 7.22 | 27.94 | 9.28 | 37.08 | 7.39 | 25.75 | 6.49 | 34.92 | RT-A09-1 | 273.55 | Sugar transport protein 13 |
| BnaA09g05270D | 4.92 | 1.84 | 5.39 | 1.88 | 7.99 | 2.68 | 7.09 | 3.29 | RT-A09-1 | −205.92 | Probable carboxylesterase 18 |
| Hub genes in the MEbrown module (GWAS+WGCNA) | |||||||||||
| BnaA09g08440D | 1.93 | 4.34 | 1.84 | 3.41 | 2.12 | 4.73 | 1.70 | 3.23 | RT-A09-2 | 249.99 | BTB/POZ domain-containing protein |
| BnaC07g30970D | 13.56 | 13.81 | 15.98 | 12.58 | 15.33 | 13.72 | 18.75 | 14.97 | RT-C07-1 | −118.79 | Exocyst complex component |
| Significant DEGs from GWAS + RNA-sequencing data | |||||||||||
| BnaA09g04280D | 0.72 | 0.65 | 0.55 | 0.68 | 1.69 | 1.43 | 1.10 | 2.25 | RT-A09-1 | 264.71 | TRAF-like family protein |
| BnaA09g08450D | 9.19 | 13.66 | 11.39 | 15.52 | 12.12 | 12.26 | 9.48 | 12.54 | RT-A09-2 | 241.43 | MVB pathway protein |
| BnaA10g16560D | 0.00 | 0.08 | 0.08 | 3.80 | 7.23 | 0.23 | 0.00 | 0.00 | RT-A10-1 | 147.27 | / |
| BnaA10g17620D | 5.56 | 6.81 | 6.50 | 6.40 | 4.37 | 3.03 | 4.04 | 4.40 | RT-A10-1 | −268.85 | hydrolase II (PTH2) family protein |
| BnaA10g19550D | 3.06 | 3.46 | 2.79 | 4.28 | 5.05 | 3.37 | 2.45 | 3.75 | RT-A10-2 | −150.98 | unknown protein |
| BnaA10g19700D | 46.34 | 27.83 | 38.00 | 18.89 | 16.09 | 12.86 | 18.94 | 12.44 | RT-A10-2 | −202.00 | xyloglucan hydrolase 5 (XTH5) |
| BnaC03g34740D | 1.22 | 1.04 | 1.49 | 0.92 | 1.15 | 0.96 | 1.05 | 1.08 | RT-C03-3 | 105.06 | GNS1/SUR4 membrane protein |
| BnaC03g42900D | 16.87 | 15.62 | 28.99 | 14.44 | 26.24 | 17.19 | 27.69 | 21.44 | RT-C03-4 | −217.19 | lipid-transfer protein |
| BnaC06g11180D | 1.10 | 1.63 | 1.28 | 1.72 | 0.06 | 0.11 | 1.42 | 0.10 | RT-C06-1 | −262.51 | / |
| BnaC06g11230D | 0.81 | 0.50 | 1.57 | 1.02 | 0.83 | 1.10 | 1.22 | 1.01 | RT-C06-1 | −280.51 | nicotinamidase 1 (NIC1) |
| BnaC07g30250D | 2.51 | 1.76 | 2.50 | 1.18 | 1.07 | 0.60 | 0.89 | 0.43 | RT-C07-1 | 295.55 | / |
| BnaC07g30400D | 5.52 | 3.82 | 10.18 | 8.55 | 3.83 | 5.54 | 9.20 | 10.00 | RT-C07-1 | 223.14 | SLAC1 homologue 3 (SLAH3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, N.; Su, B.; Ibrahim, S.; Kuang, L.; Tian, Z.; Wang, X.; Wang, H.; Dun, X. Deciphering the Genetic Basis of Root and Biomass Traits in Rapeseed (Brassica napus L.) through the Integration of GWAS and RNA-Seq under Nitrogen Stress. Int. J. Mol. Sci. 2022, 23, 7958. https://doi.org/10.3390/ijms23147958
Ahmad N, Su B, Ibrahim S, Kuang L, Tian Z, Wang X, Wang H, Dun X. Deciphering the Genetic Basis of Root and Biomass Traits in Rapeseed (Brassica napus L.) through the Integration of GWAS and RNA-Seq under Nitrogen Stress. International Journal of Molecular Sciences. 2022; 23(14):7958. https://doi.org/10.3390/ijms23147958
Chicago/Turabian StyleAhmad, Nazir, Bin Su, Sani Ibrahim, Lieqiong Kuang, Ze Tian, Xinfa Wang, Hanzhong Wang, and Xiaoling Dun. 2022. "Deciphering the Genetic Basis of Root and Biomass Traits in Rapeseed (Brassica napus L.) through the Integration of GWAS and RNA-Seq under Nitrogen Stress" International Journal of Molecular Sciences 23, no. 14: 7958. https://doi.org/10.3390/ijms23147958
APA StyleAhmad, N., Su, B., Ibrahim, S., Kuang, L., Tian, Z., Wang, X., Wang, H., & Dun, X. (2022). Deciphering the Genetic Basis of Root and Biomass Traits in Rapeseed (Brassica napus L.) through the Integration of GWAS and RNA-Seq under Nitrogen Stress. International Journal of Molecular Sciences, 23(14), 7958. https://doi.org/10.3390/ijms23147958