
Influence of Association on Binding of Disaccharides to YKL-39 and hHyal-1 Enzymes

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
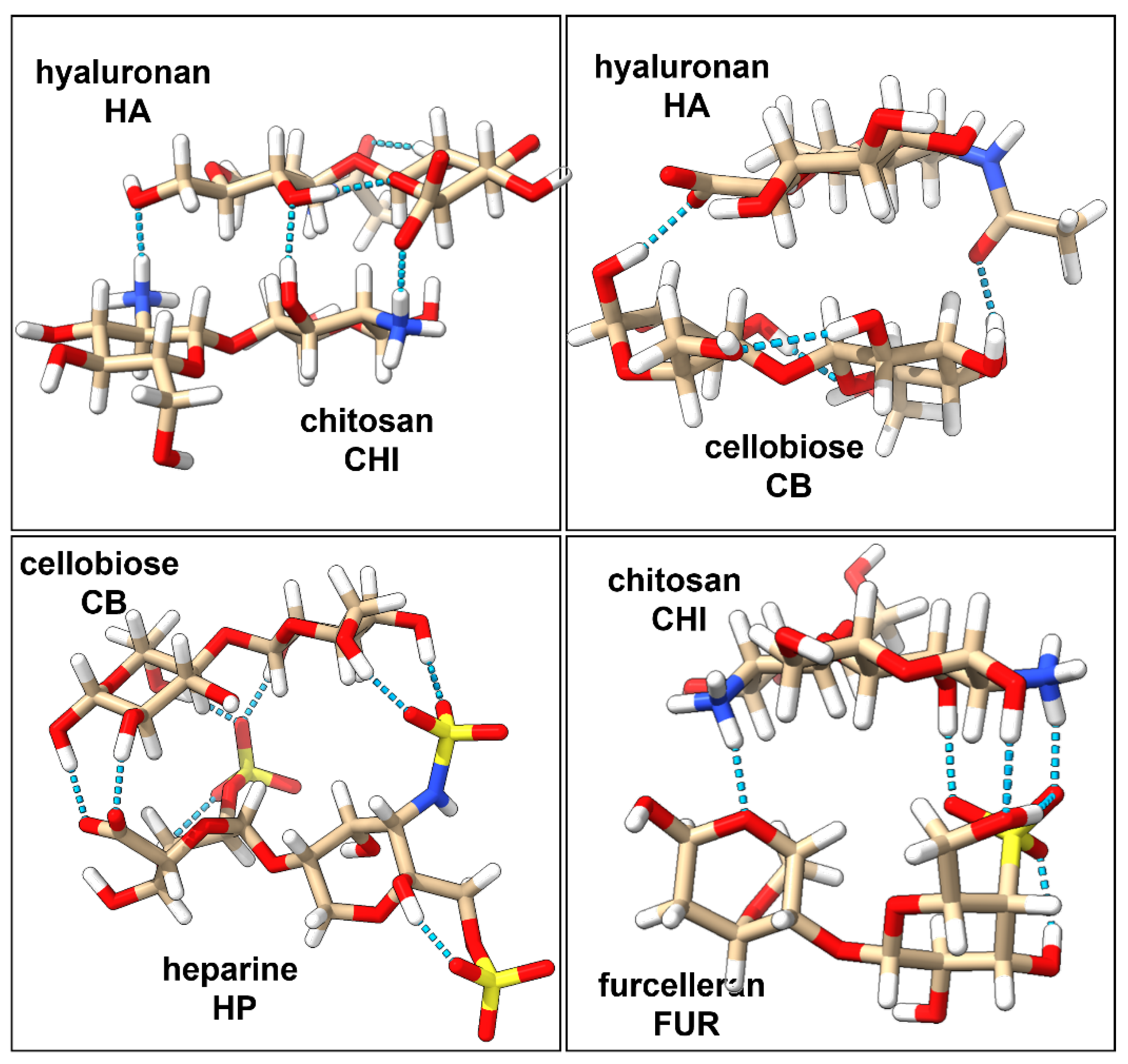
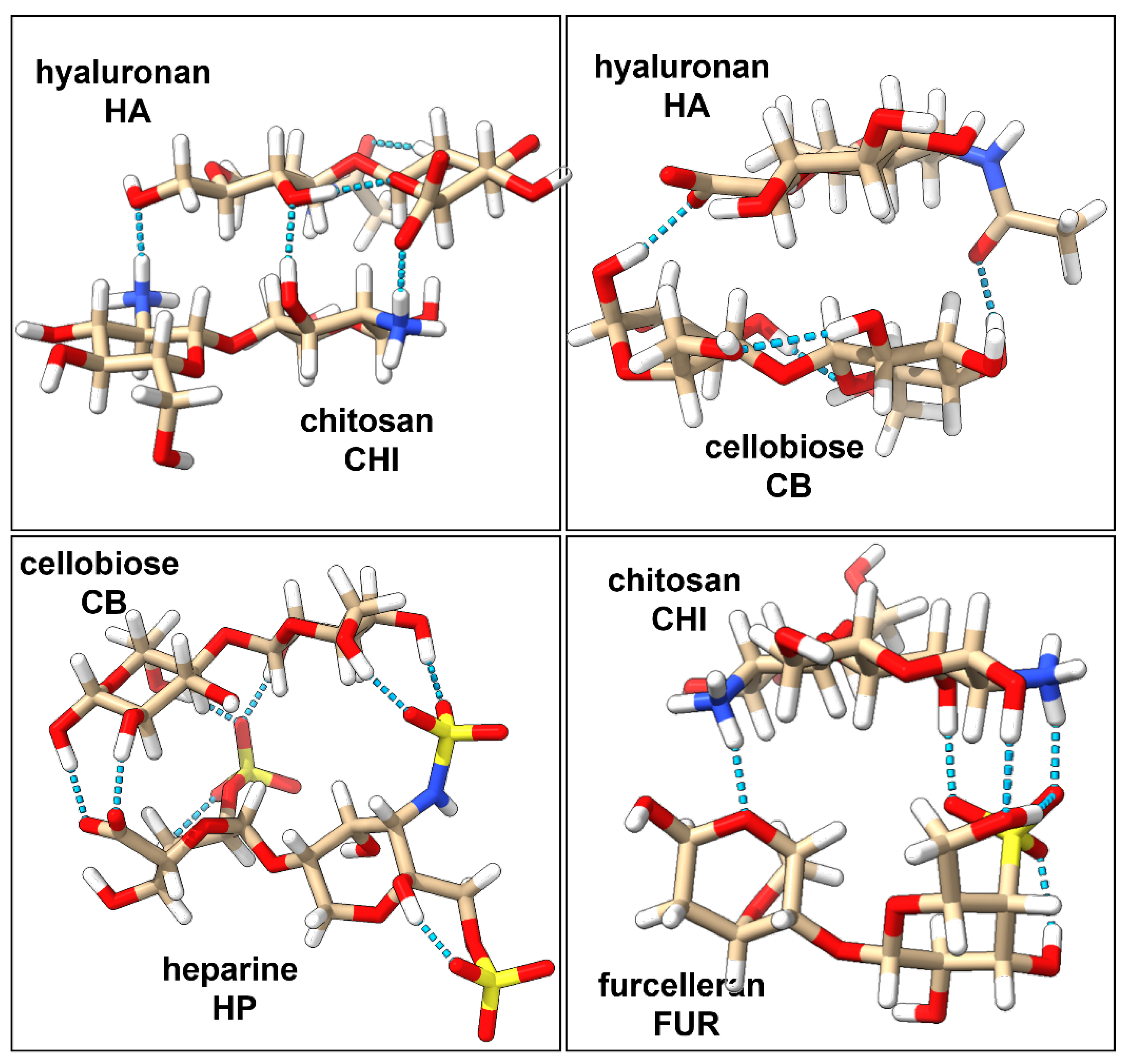
2.1. Characterization of Interactions in Polyelectrolyte Complexes
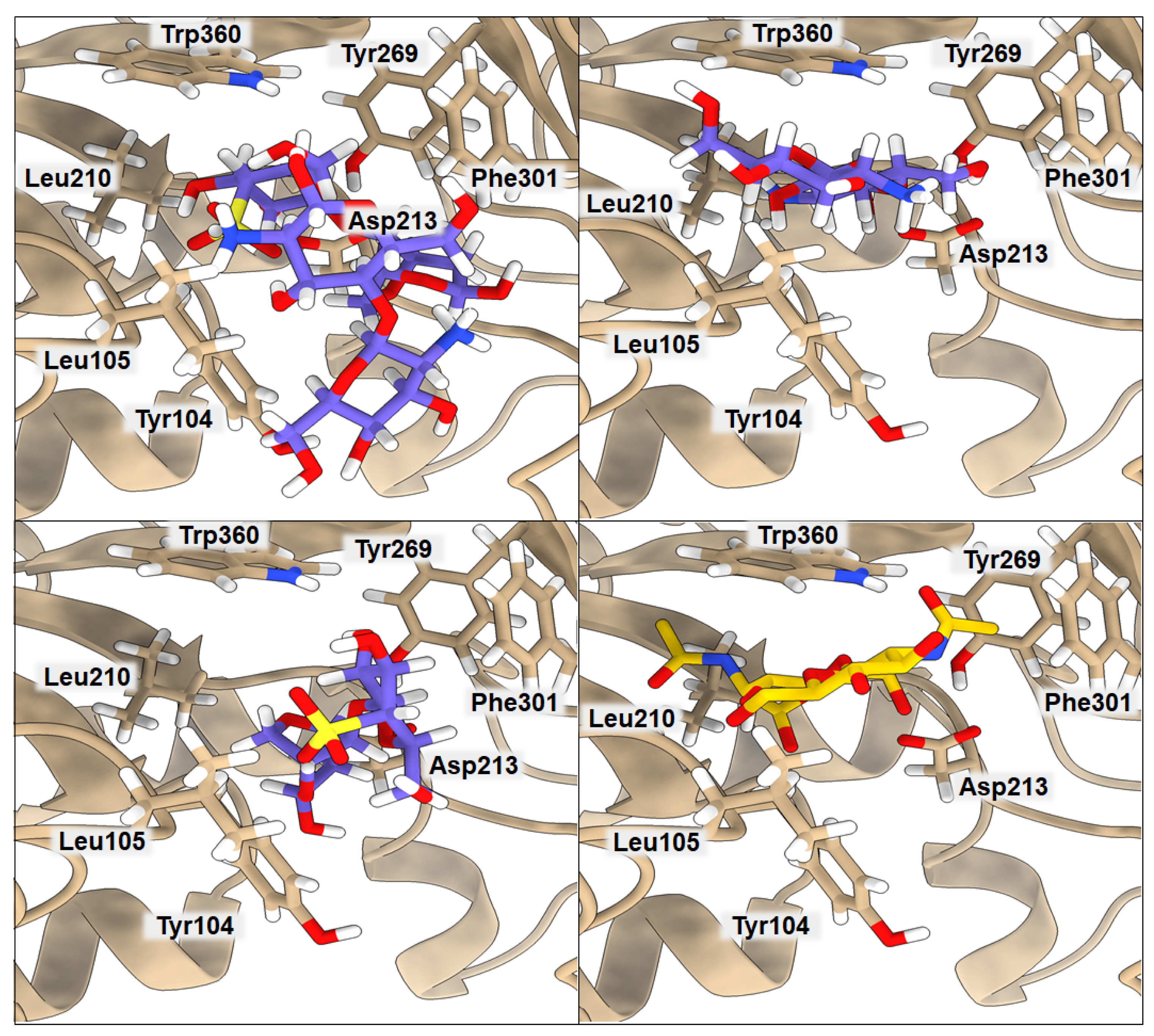
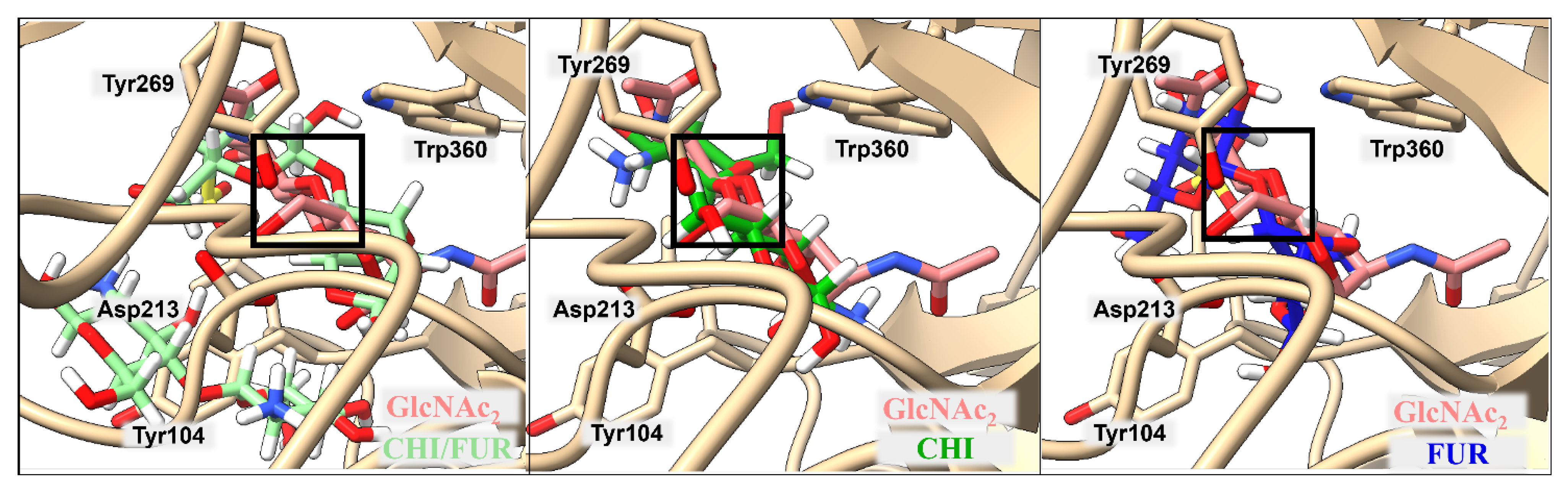
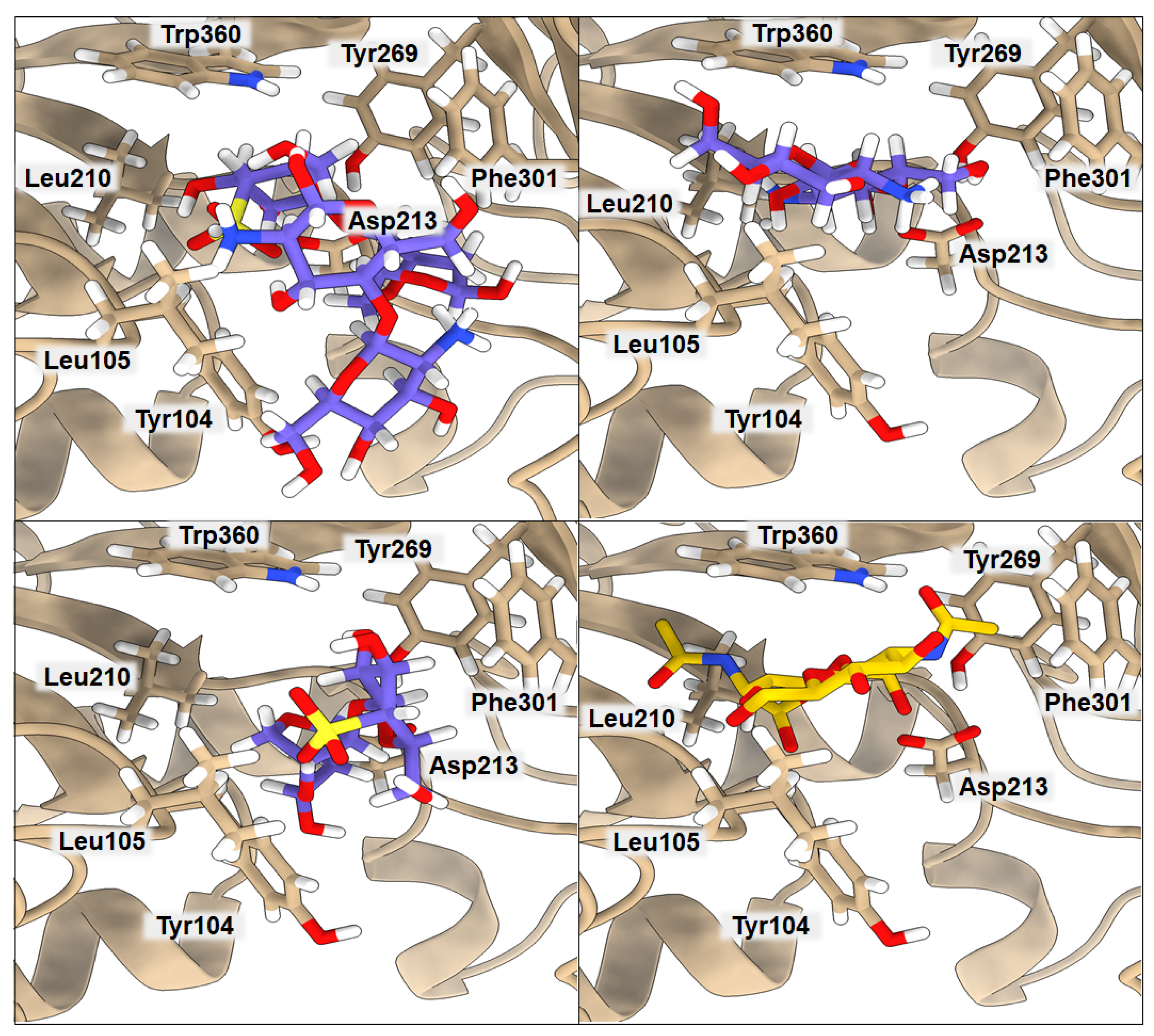
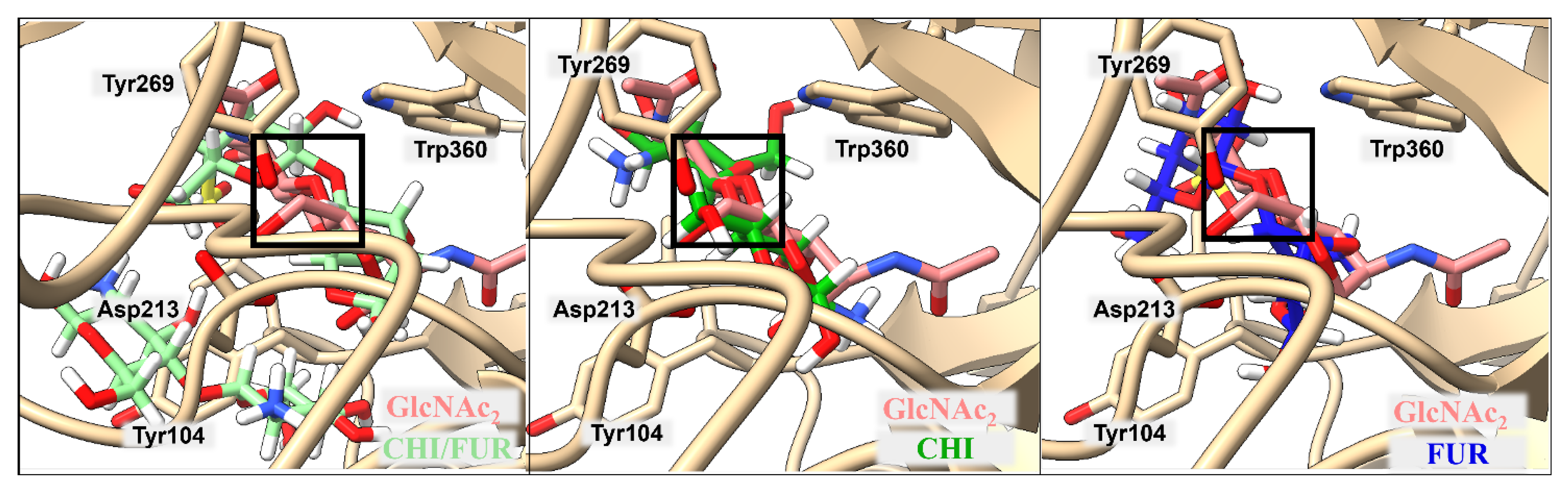
2.2. Binding of Isolated Ligands and CHI/FUR Complex to YKL-39
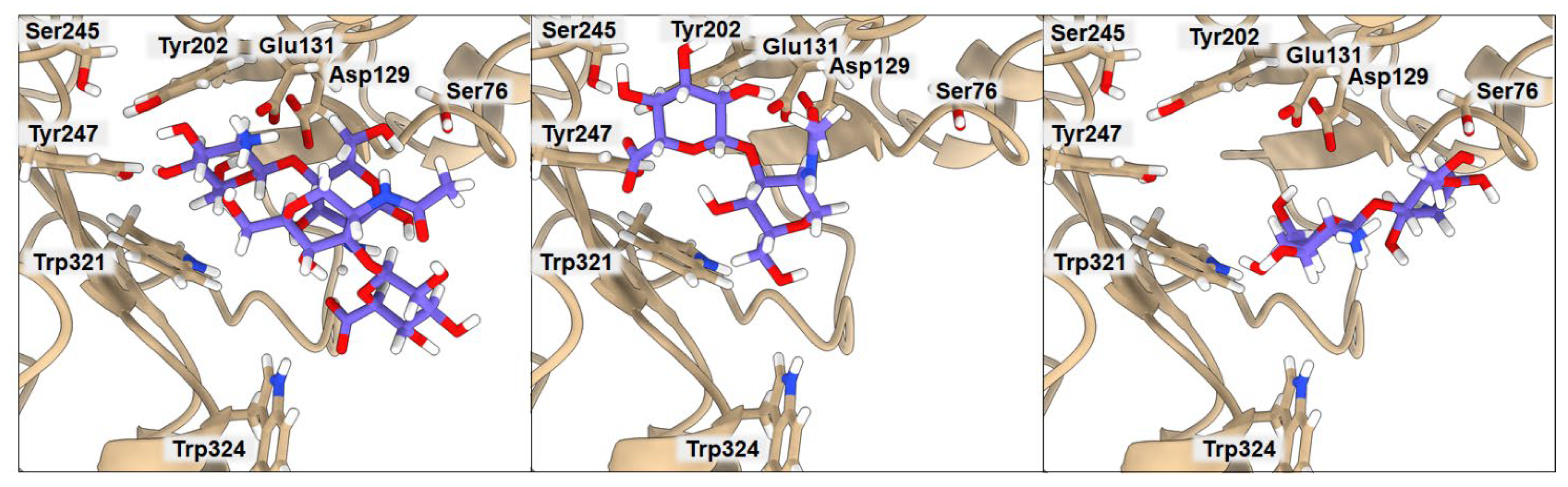
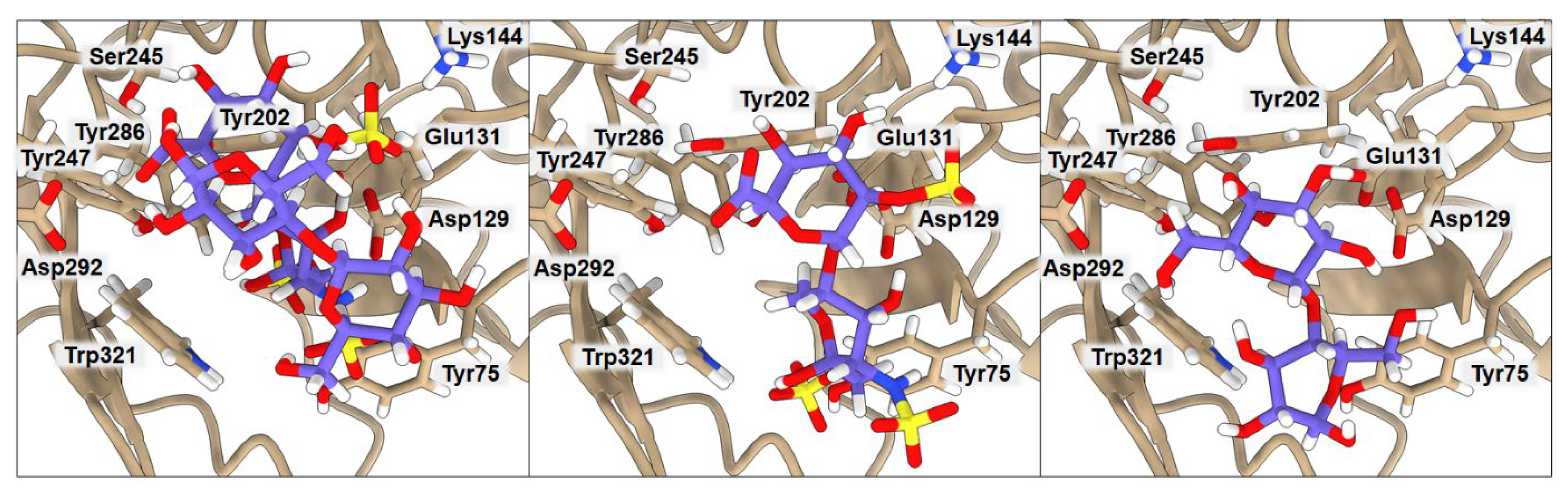
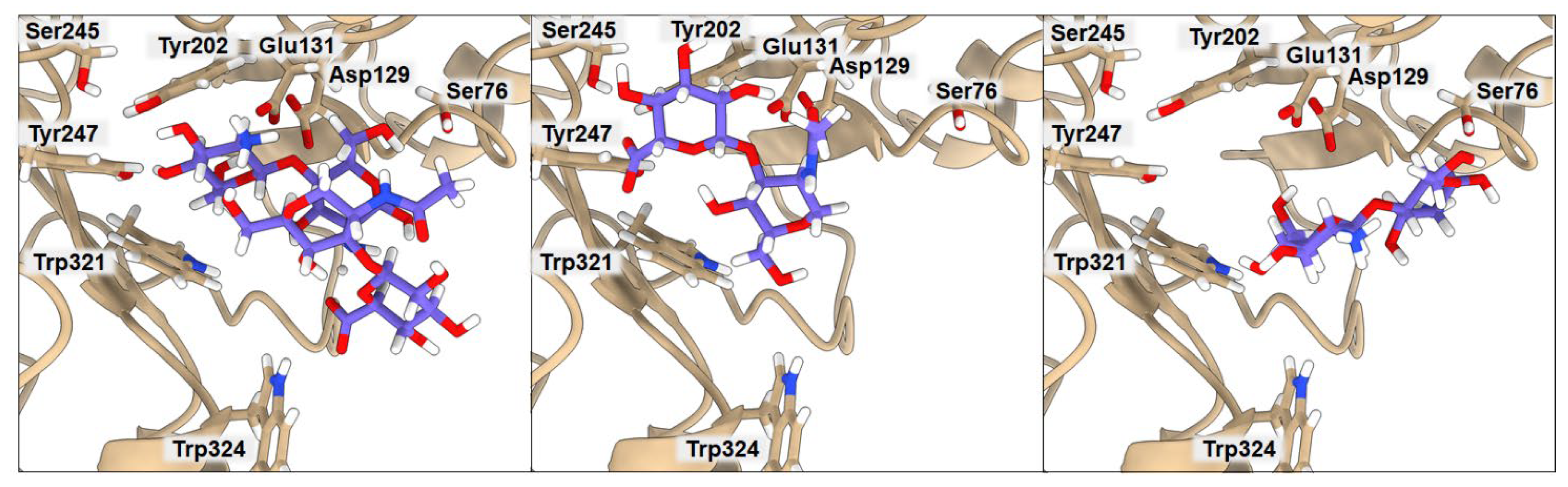
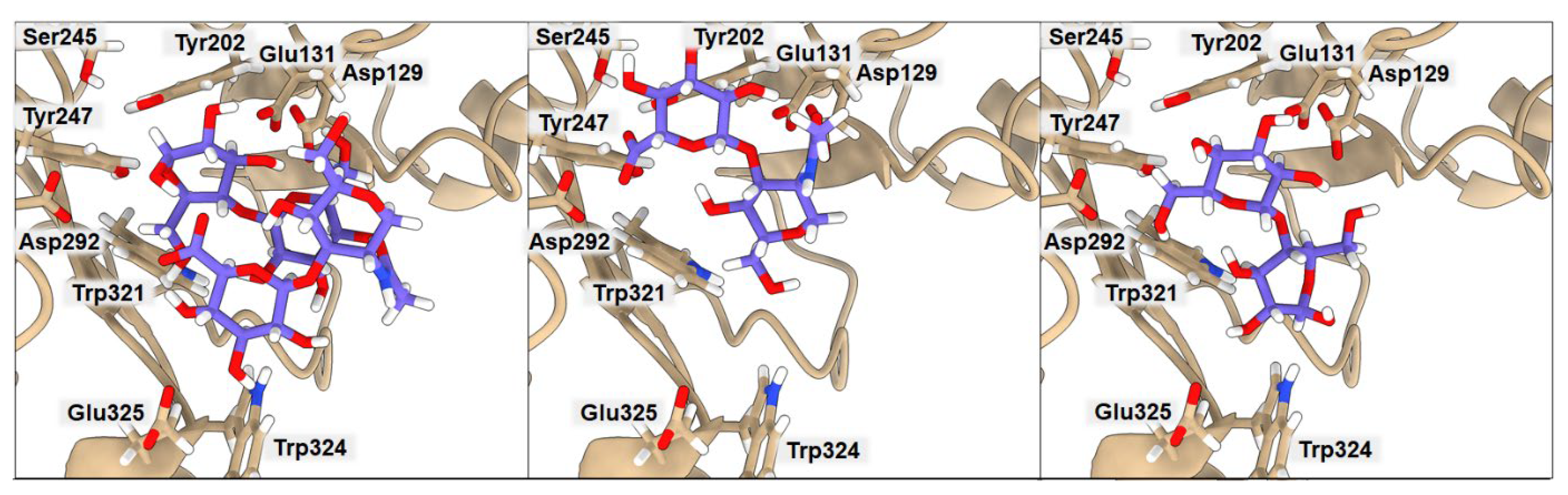
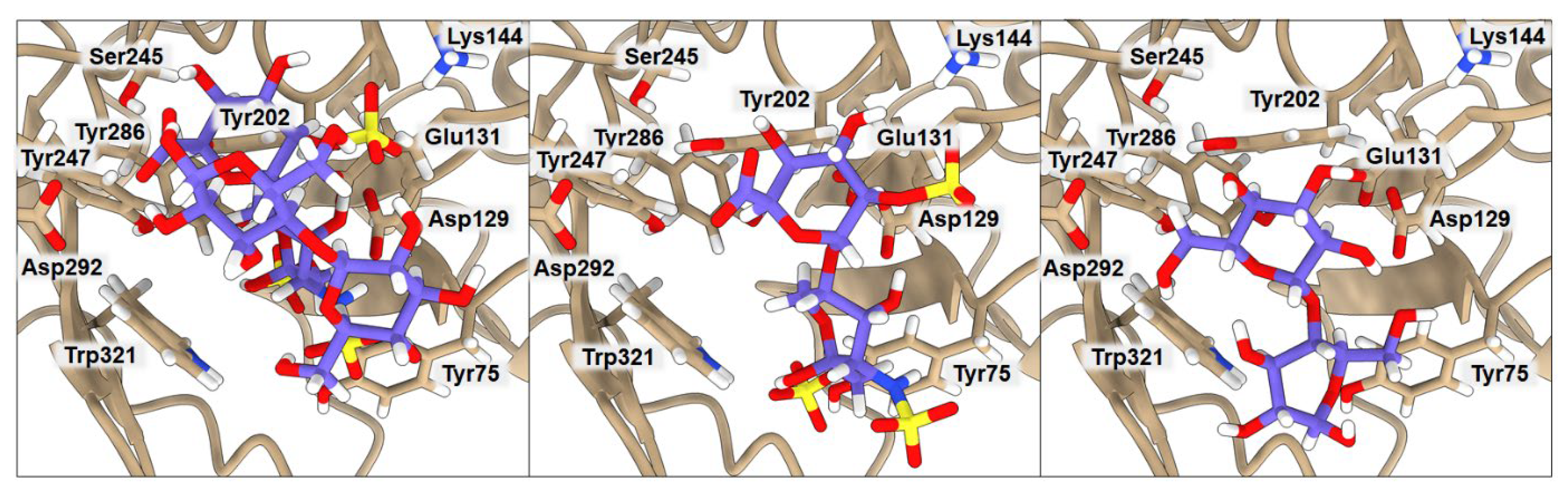
2.3. Binding of Isolated Ligands and Complexes HA/CHI, HA/CB, and HP/CB to hHyal-1
3. Computational Methods
3.1. Quantum-Mechanical Calculations
3.2. Protein-Systems Setup and Interaction Analysis
3.3. Docking Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greish, K.; Fang, J.; Inutsuka, T.; Nagamitsu, A.; Maeda, H. Macromolecular therapeutics: Advantages and prospects with special emphasis on solid tumour targeting. Clin. Pharmacokinet. 2003, 42, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Pitakchatwong, C.; Schlegel, I.; Landfester, K.; Crespy, D.; Chirachanchai, S. Chitosan nanocapsules for pH-triggered dual release based on corrosion inhibitors as model study. Part. Part. Syst. Charact. 2018, 35, 1800086. [Google Scholar] [CrossRef]
- Ishihara, M.; Kishimoto, S.; Nakamura, S.; Sato, Y.; Hattori, H. Polyelectrolyte complexes of natural polymers and their biomedical applications. Polymers 2019, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Milosavljevic, V.; Jamroz, E.; Gagic, M.; Haddad, Y.; Michalkova, H.; Balkova, R.; Tesarova, B.; Moulick, A.; Heger, Z.; Richtera, L.; et al. Encapsulation of doxorubicin in furcellaran/chitosan nanocapsules by layer-by-layer technique for selectively controlled drug delivery. Biomacromolecules 2020, 21, 418–434. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Pedregal, J.R.-G.; Sciortino, G.; Guasp, J.; Municoy, M.; Maréchal, J.-D. GaudiMM: A modular multi-objective platform for molecular modeling. J. Comput. Chem. 2017, 38, 2118–2126. [Google Scholar] [CrossRef]
- Jedrzejas, M.J.; Stern, R. Structures of vertebrate hyaluronidases and their unique enzymatic mechanism of hydrolysis. Proteins 2005, 61, 227–238. [Google Scholar] [CrossRef]
- Davies, G.J.; Gloster, T.M.; Henrissat, B. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 2005, 15, 637–645. [Google Scholar] [CrossRef]
- Rye, C.S.; Withers, S.G. Glycosidase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 573–580. [Google Scholar] [CrossRef]
- Rathee, V.S.; Sidky, H.; Sikora, B.J.; Whitmer, J.K. Role of associative charging in the entropy–energy balance of polyelectrolyte complexes. J. Am. Chem. Soc. 2018, 140, 15319–15328. [Google Scholar] [CrossRef]
- Laaser, J.E.; McGovern, M.; Jiang, Y.; Lohmann, E.; Reineke, T.M.; Morse, D.C.; Dorfman, K.D.; Lodge, T.P. Equilibration of micelle–polyelectrolyte complexes: Mechanistic differences between static and annealed charge distributions. J. Phys. Chem. B 2017, 121, 4631–4641. [Google Scholar] [CrossRef]
- Ranok, A.; Wongsantichon, J.; Robinson, R.C.; Suginta, W. Structural and thermodynamic insights into chitooligosaccharide binding to human cartilage chitinase 3-like protein 2 (CHI3L2 or YKL-39). J. Biol. Chem. 2015, 290, 2617–2629. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation theory approach to intermolecular potential energy surfaces of van der Waals complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An open-source ab initio electronic structure program. WIREs Comput. Mol. Sci. 2012, 2, 556. [Google Scholar] [CrossRef]
- Chao, K.L.; Herzberg, O. Structure of human hyaluronidase 1, a hyaluronan hydrolyzing enzyme involved in tumor growth and angiogenesis. Biochemistry 2007, 46, 6911–6920. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Case, D.H.M.; Belfon, I.Y.; Ben-Shalom, S.R.; Brozell, D.S.; Cerutti, T.E.; Cheatham, G.A., III; Cisneros, V.W.D.; Cruzeiro, T.A.; Darden, R.E.; Duke, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Gagne, M.; Rapppe, A.K. pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar]
- Hubbard, R.E.; Haider, K. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Zhang, L.; Bharadwaj, A.G.; Casper, A.; Barkley, J.; Barycki, J.J.; Simpson, M.A. Hyaluronidase activity of human hyal1 requires active site acidic and tyrosine residues. J. Biol. Chem. 2009, 284, 9433–9442. [Google Scholar] [CrossRef]
- Yadav, S.; Sharma, A.K.; Kumar, P. Nanoscale self-assembly for therapeutic delivery. Front. Bioeng. Biotechnol. 2020, 8, 127. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disaccharide | Net Charge | # of NH3+ Groups | # of OH Groups | # of SO3− Groups | # of COO− Groups | Complex | Net Charge | # of H-Bonds |
|---|---|---|---|---|---|---|---|---|
| HP | −4 | 0 | 4 | 3 | 1 | HP/CB | −4 | 6 |
| FUR | −1 | 0 | 4 | 1 | 0 | HA/CB | −1 | 2 |
| HA | −1 | 0 | 5 | 0 | 1 | HA/CHI | +1 | 3 |
| CB | 0 | 0 | 8 | 0 | 0 | CHI/FUR | +1 | 4 |
| CHI | 2 | 2 | 6 | 0 | 0 |
| Energy Component | HA/CHI | HA/CB | HP/CB | CHI/FUR | ||||
|---|---|---|---|---|---|---|---|---|
| electrostatic | −322.6 | 27.0% | −46.5 | 32.6% | −139.7 | 38.3% | −324.9 | 29.9% |
| exchange | 541.8 | 45.4% | 49.3 | 34.6% | 124.1 | 34.1% | 508.1 | 46.7% |
| induction | −193.9 | 16.3% | −20.5 | 14.3% | −60.2 | 16.5% | −139.7 | 12.8% |
| dispersion | −134.5 | 11.3% | −26.3 | 18.4% | −40.3 | 11.1% | −114.8 | 10.6% |
| total energy | −109.2 | - | −43.9 | - | −116.0 | - | −71.3 | - |
| BA | Tyr104 | Leu105 | Leu210 | Asp213 | Tyr269 | Phe301 | Trp360 | |
|---|---|---|---|---|---|---|---|---|
| GlcNAc2 | −5.7 | 3.17 | 3.02 | 4.08 | 2.59 | 3.70 | 3.91 | 3.45 |
| CHI | −7.2 | 3.61 | 4.55 | 4.21 | 2.40 | 3.32 | 4.86 | 3.85 |
| FUR | −7.2 | 3.22 | 3.72 | 6.81 | 2.97 | 4.84 | 3.30 | 4.02 |
| CHI/FUR | −8.3 | 3.77 | 2.65 | 6.23 | 2.89 | 4.29 | 3.27 | 2.84 |
| BA | Ser76 | Asp129 | Glu131 | Tyr202 | Tyr247 | Trp321 | Trp324 | |
|---|---|---|---|---|---|---|---|---|
| HA | −6.3 | 7.93 | 2.20 | 2.05 | 3.02 | 2.99 | 3.53 | 3.46 |
| CHI | −6.3 | 3.19 | 3.75 | 2.91 | 8.15 | 6.16 | 3.41 | 5.30 |
| HA/CHI | −7.2 | 4.71 | 3.03 | 2.52 | 2.47 | 2.35 | 3.46 | 5.27 |
| BA | Asp129 | Glu131 | Tyr202 | Tyr247 | Asp292 | Trp321 | Trp324 | Glu325 | |
|---|---|---|---|---|---|---|---|---|---|
| HA | −6.3 | 2.20 | 2.05 | 3.02 | 2.99 | 2.96 | 3.53 | 3.46 | 6.37 |
| CB | −6.4 | 3.04 | 1.87 | 2.08 | 3.54 | 2.84 | 3.19 | 2.60 | 4.29 |
| HA/CB | −7.2 | 2.87 | 2.60 | 2.54 | 3.27 | 3.86 | 4.15 | 3.68 | 3.15 |
| BA | Tyr75 | Asp129 | Glu131 | Lys144 | Tyr202 | Tyr247 | Tyr286 | Asp292 | Trp321 | |
|---|---|---|---|---|---|---|---|---|---|---|
| HP | −6.3 | 2.54 | 3.46 | 2.20 | 4.00 | 5.79 | 6.32 | 8.56 | 5.01 | 3.23 |
| CB | −6.4 | 4.04 | 3.04 | 1.87 | 5.30 | 2.08 | 3.54 | 8.58 | 2.84 | 3.19 |
| HP/CB | −7.9 | 3.39 | 2.67 | 1.91 | 3.61 | 3.09 | 3.72 | 2.52 | 3.03 | 2.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzemińska, A.; Sánchez-Aparicio, J.-E.; Maréchal, J.-D.; Paneth, A.; Paneth, P. Influence of Association on Binding of Disaccharides to YKL-39 and hHyal-1 Enzymes. Int. J. Mol. Sci. 2022, 23, 7705. https://doi.org/10.3390/ijms23147705
Krzemińska A, Sánchez-Aparicio J-E, Maréchal J-D, Paneth A, Paneth P. Influence of Association on Binding of Disaccharides to YKL-39 and hHyal-1 Enzymes. International Journal of Molecular Sciences. 2022; 23(14):7705. https://doi.org/10.3390/ijms23147705
Chicago/Turabian StyleKrzemińska, Agnieszka, José-Emilio Sánchez-Aparicio, Jean-Didier Maréchal, Agata Paneth, and Piotr Paneth. 2022. "Influence of Association on Binding of Disaccharides to YKL-39 and hHyal-1 Enzymes" International Journal of Molecular Sciences 23, no. 14: 7705. https://doi.org/10.3390/ijms23147705
APA StyleKrzemińska, A., Sánchez-Aparicio, J.-E., Maréchal, J.-D., Paneth, A., & Paneth, P. (2022). Influence of Association on Binding of Disaccharides to YKL-39 and hHyal-1 Enzymes. International Journal of Molecular Sciences, 23(14), 7705. https://doi.org/10.3390/ijms23147705