
Quantitative Trait Locus Mapping of Marsh Spot Disease Resistance in Cranberry Common Bean (Phaseolus vulgaris L.)


 and
and 
Abstract
:1. Introduction
2. Results
2.1. SNP Identification
2.2. Genetic Map
2.3. Genomic Heritability
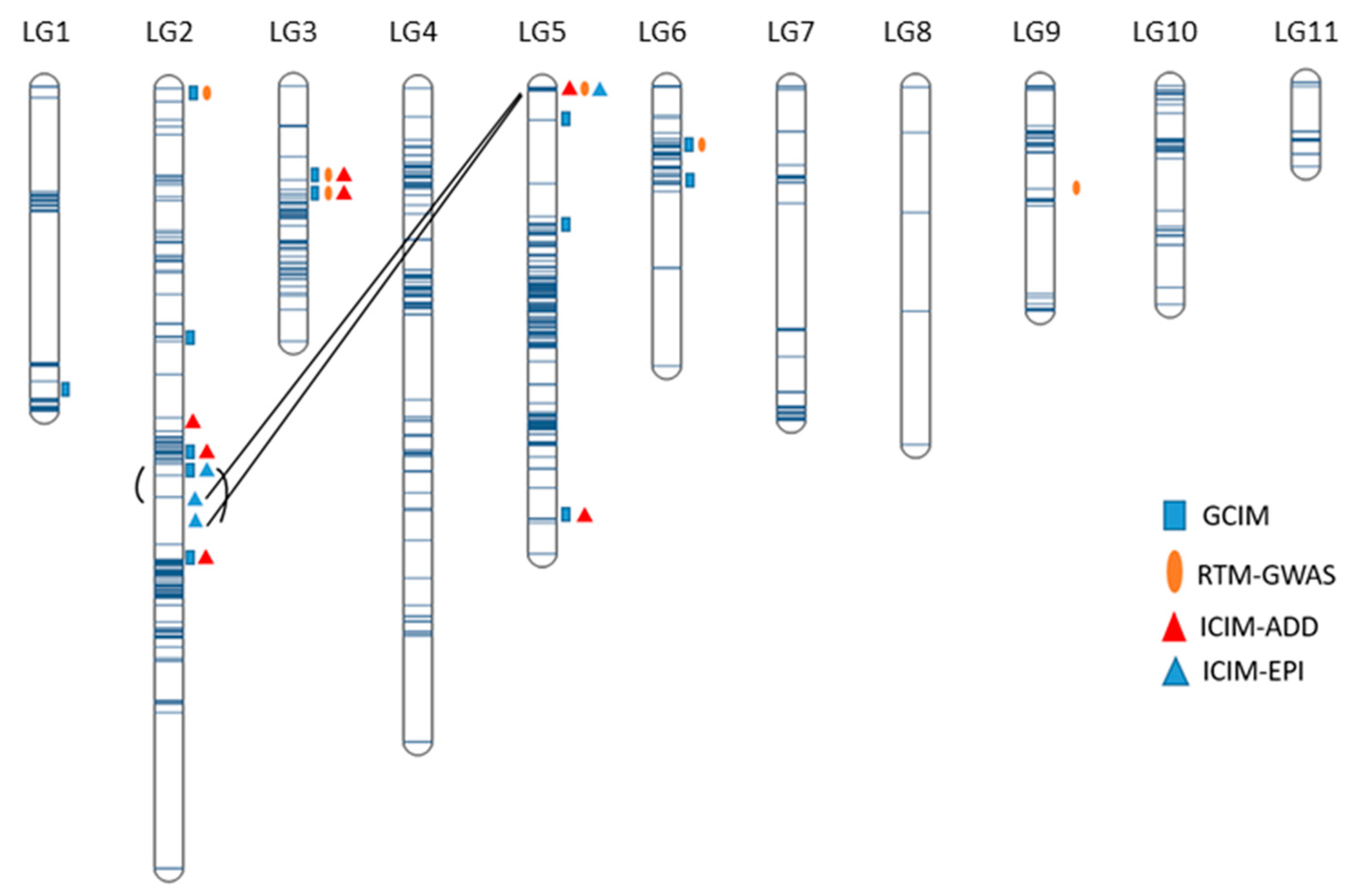
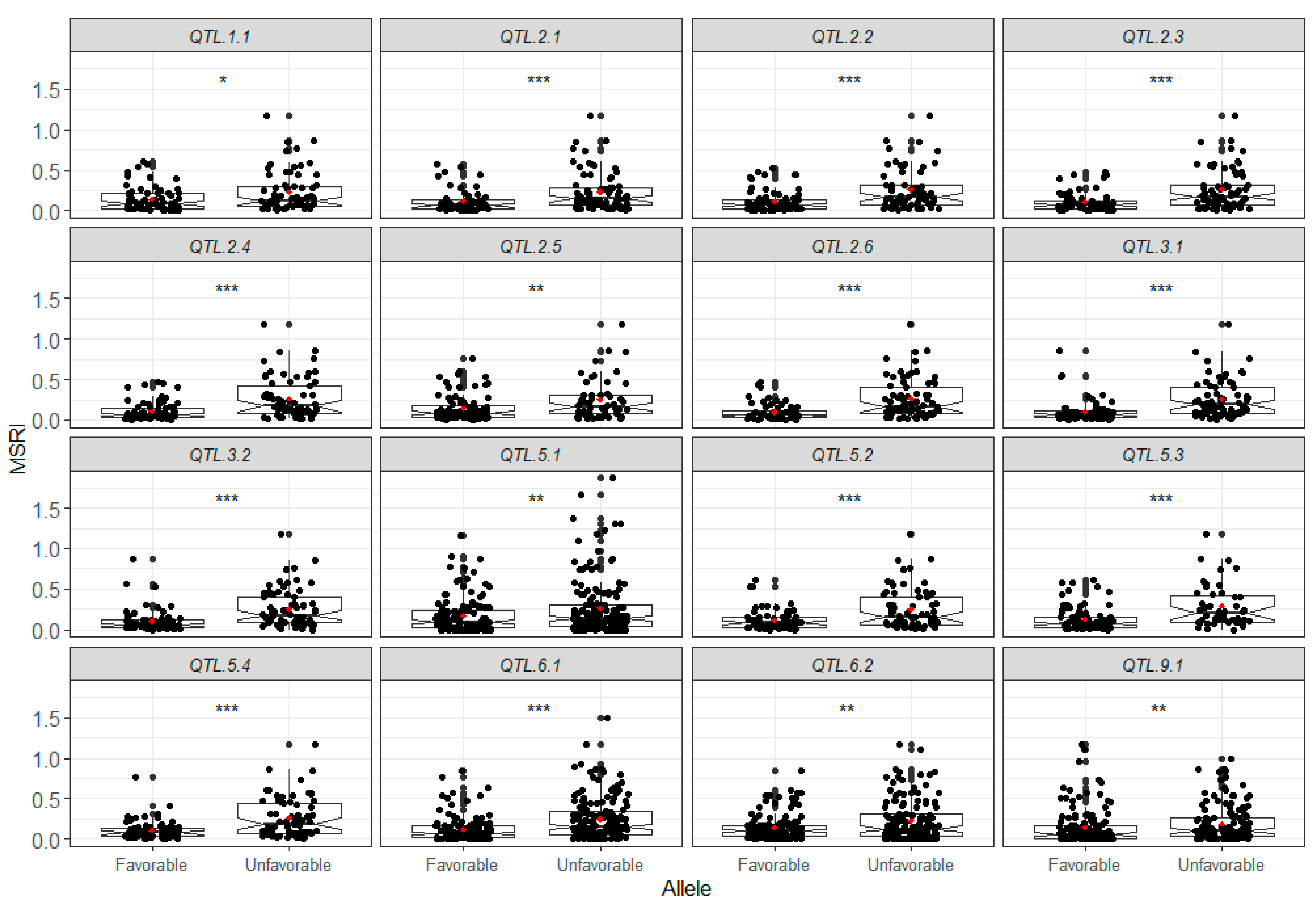
2.4. Mapping of Additive QTL
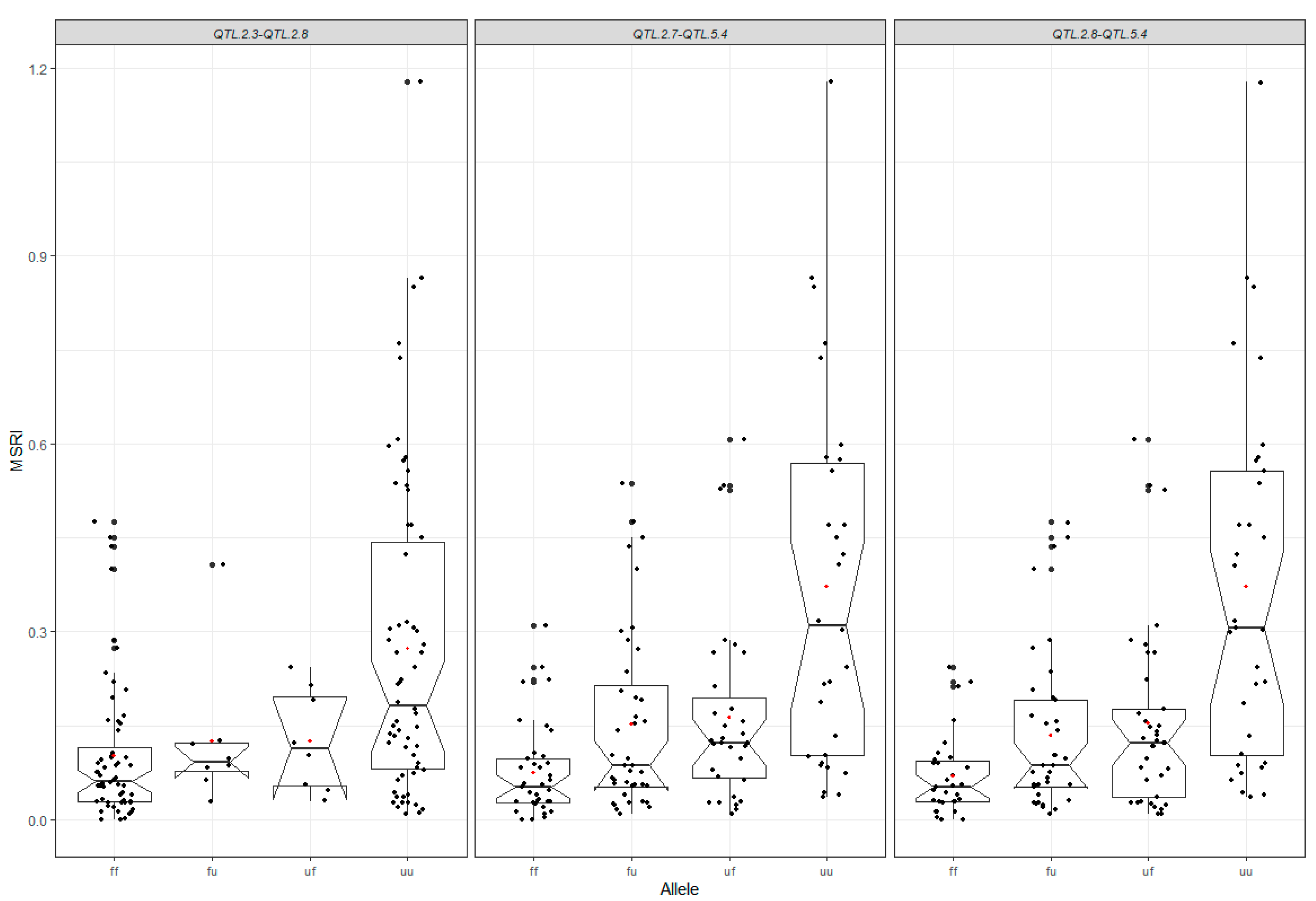
2.5. Mapping of Epistatic QTL
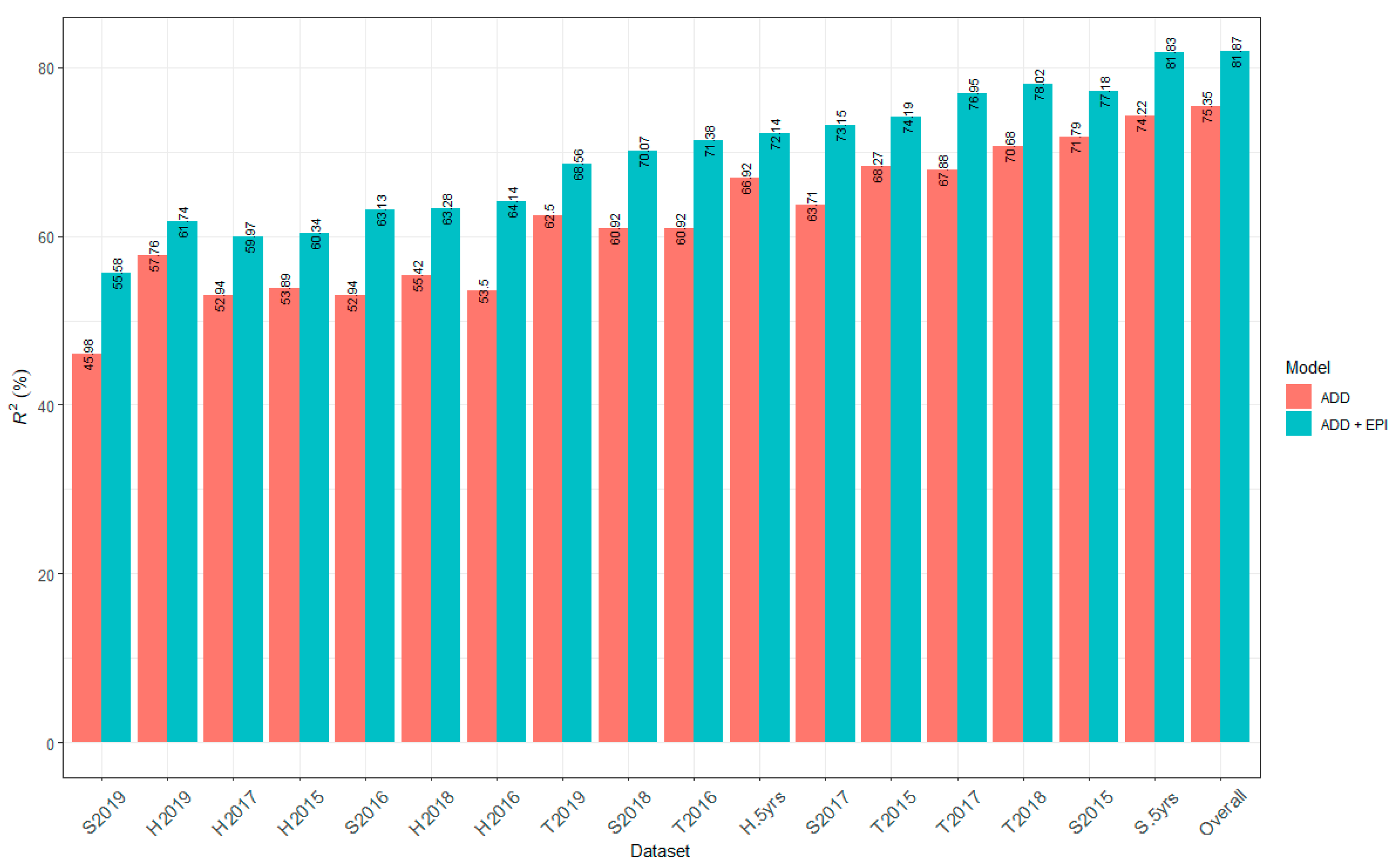
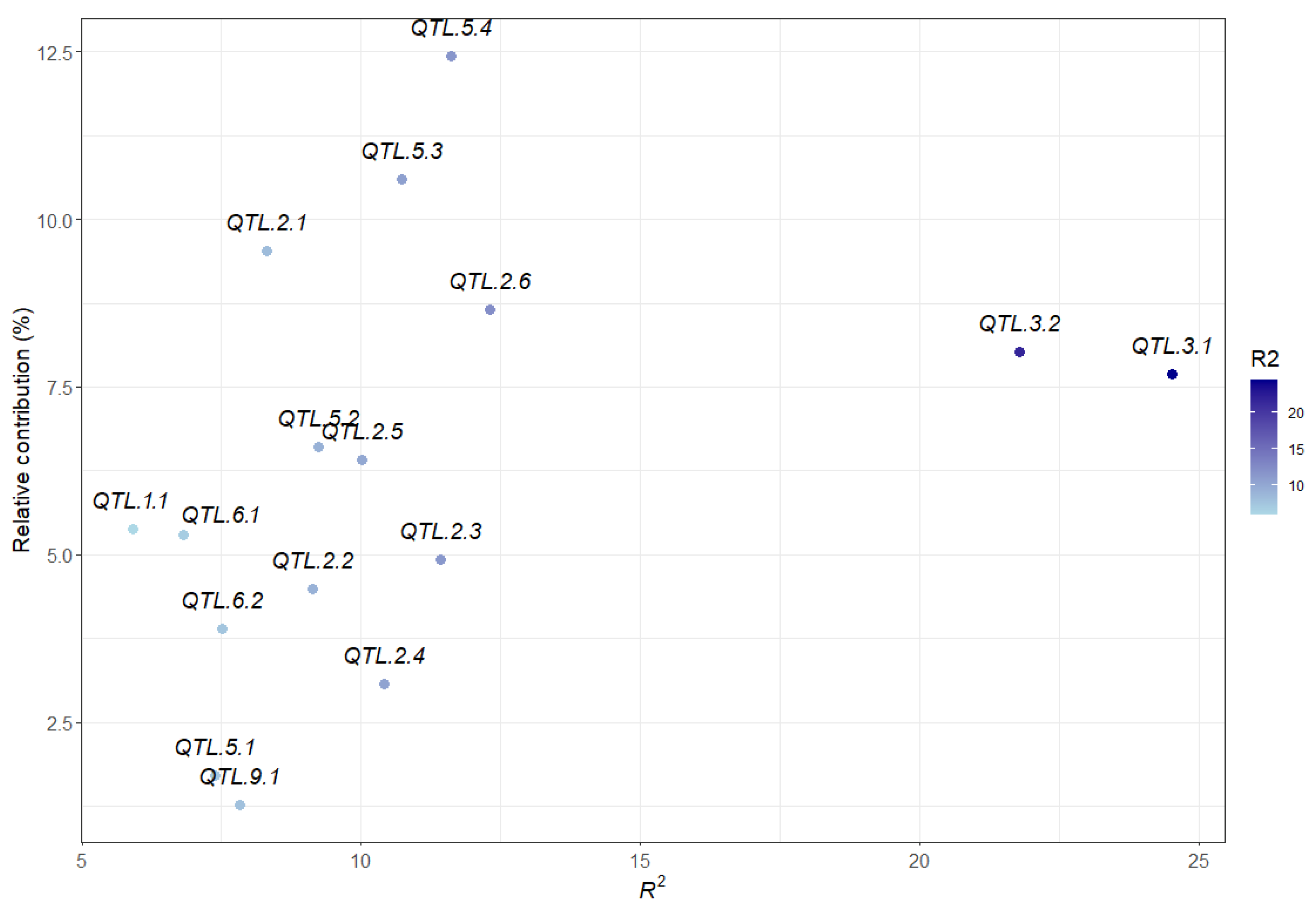
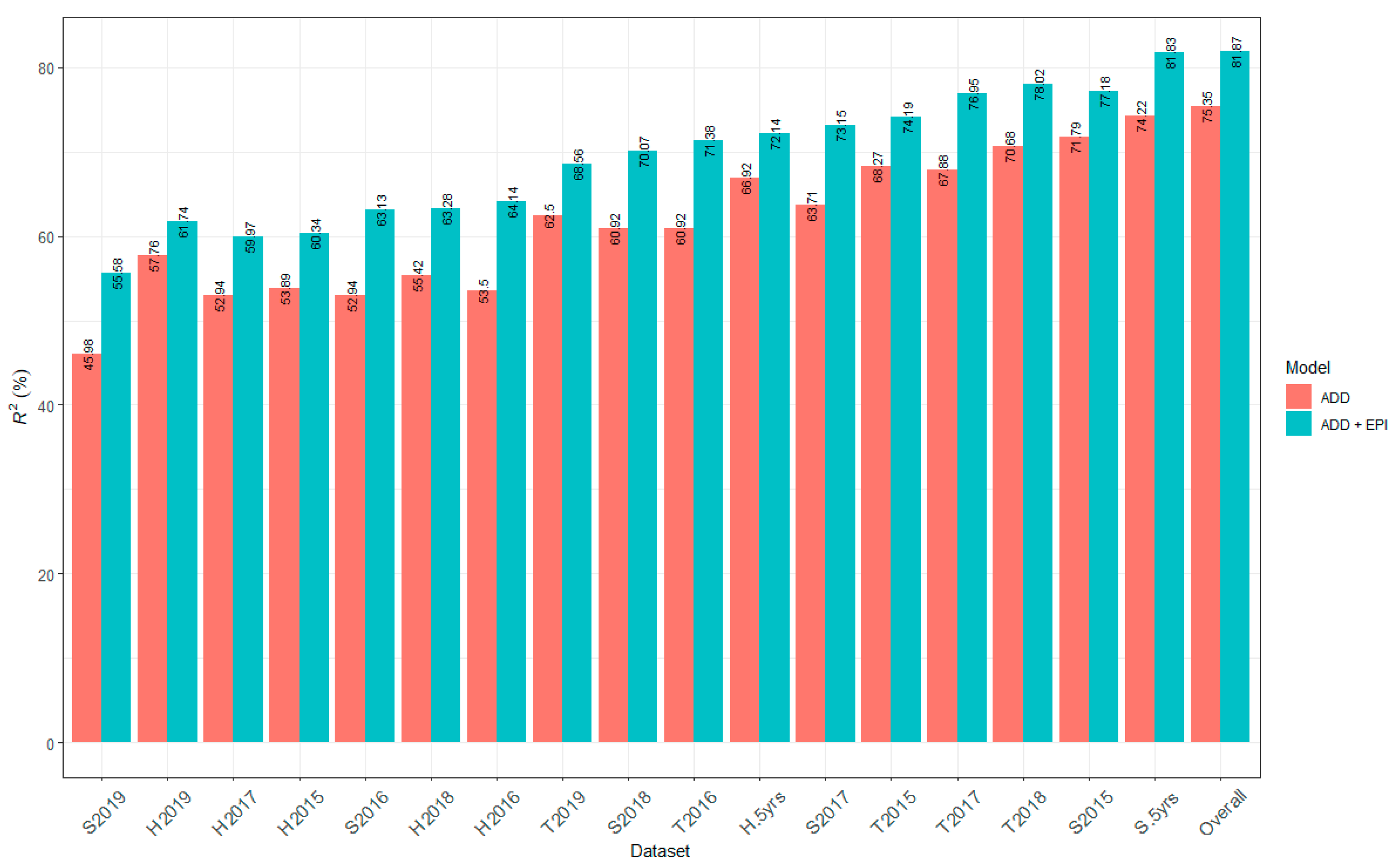
2.6. Contribution of All Detected QTL to Marsh Spot Resistance
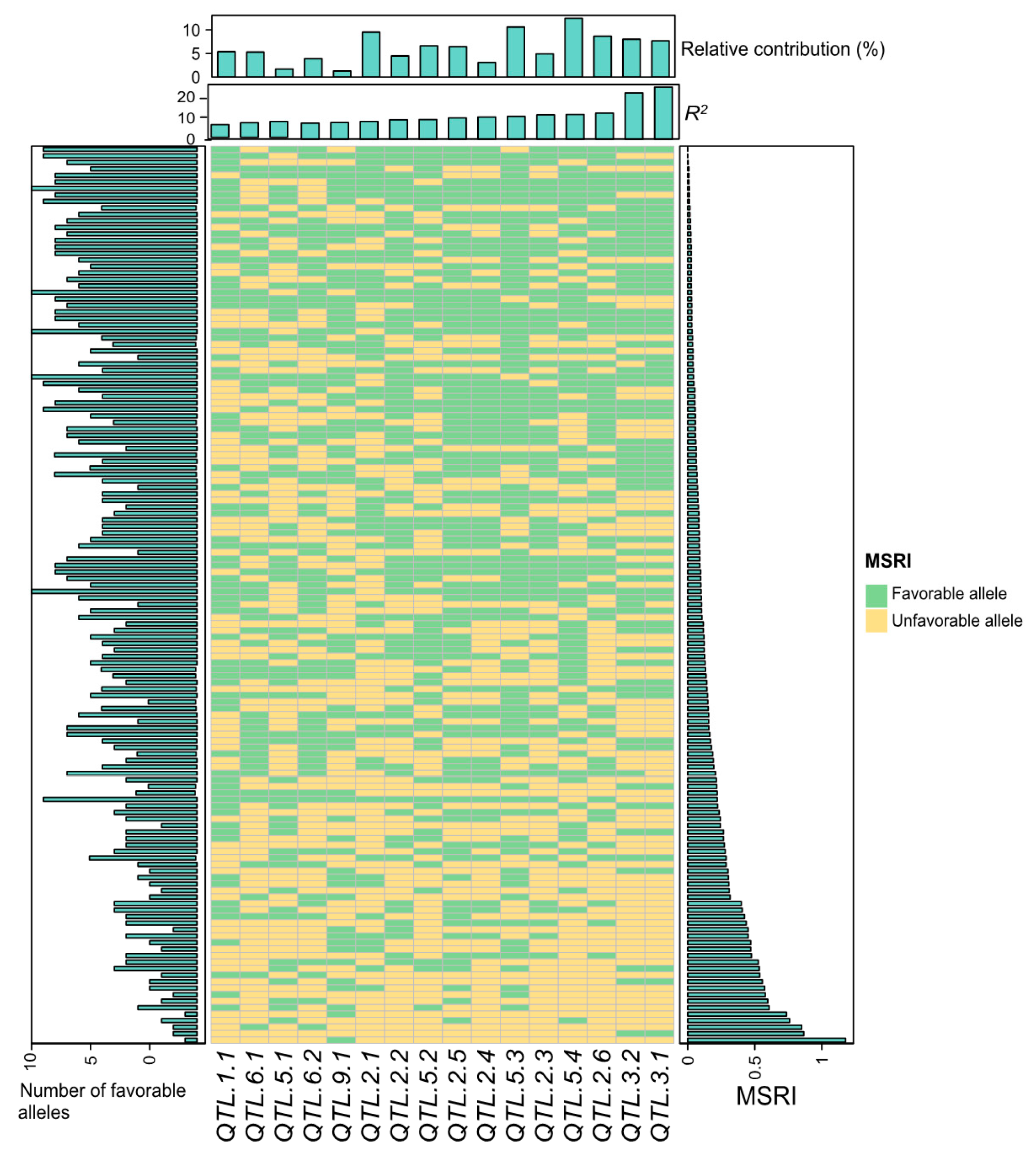
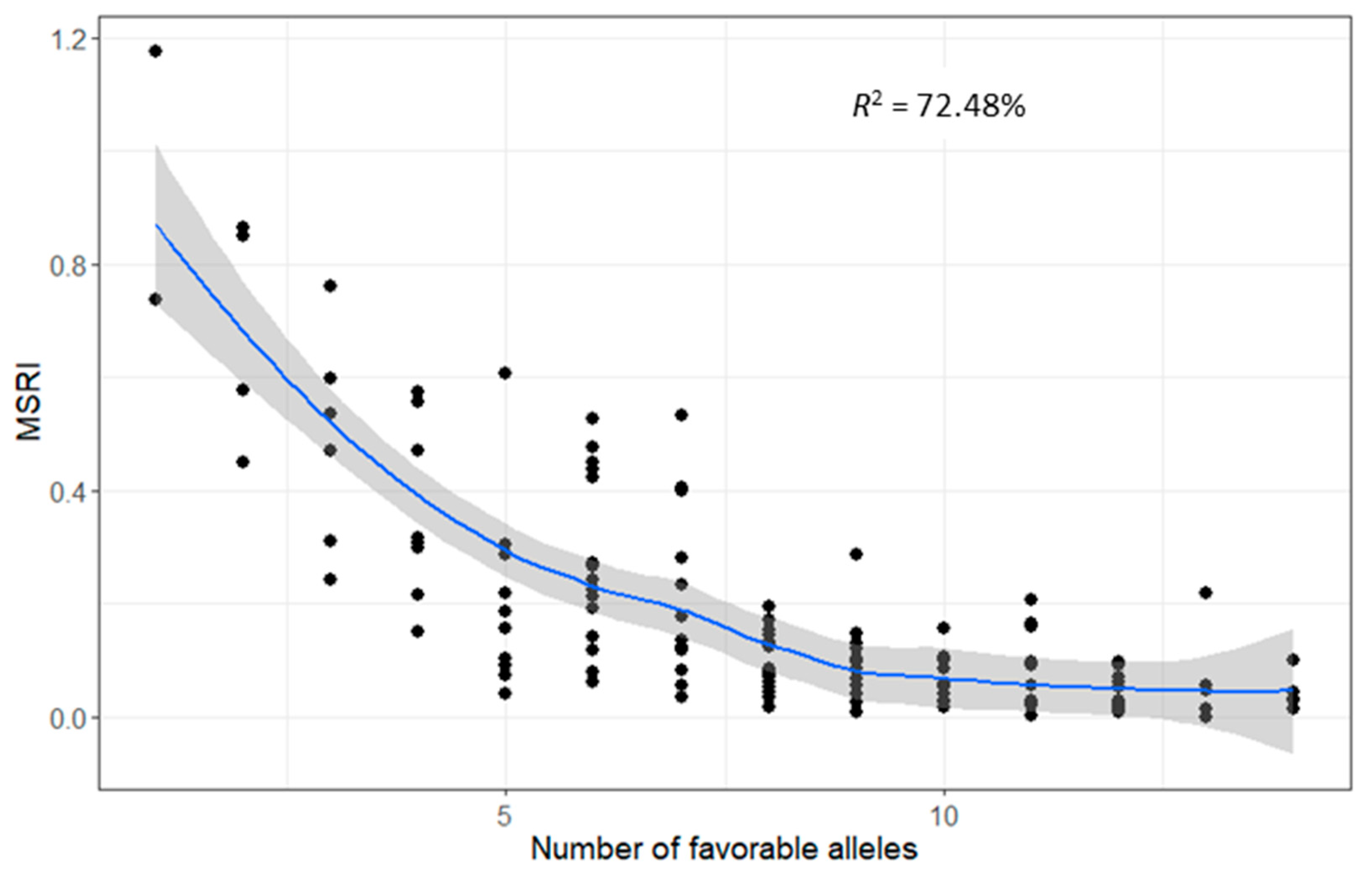
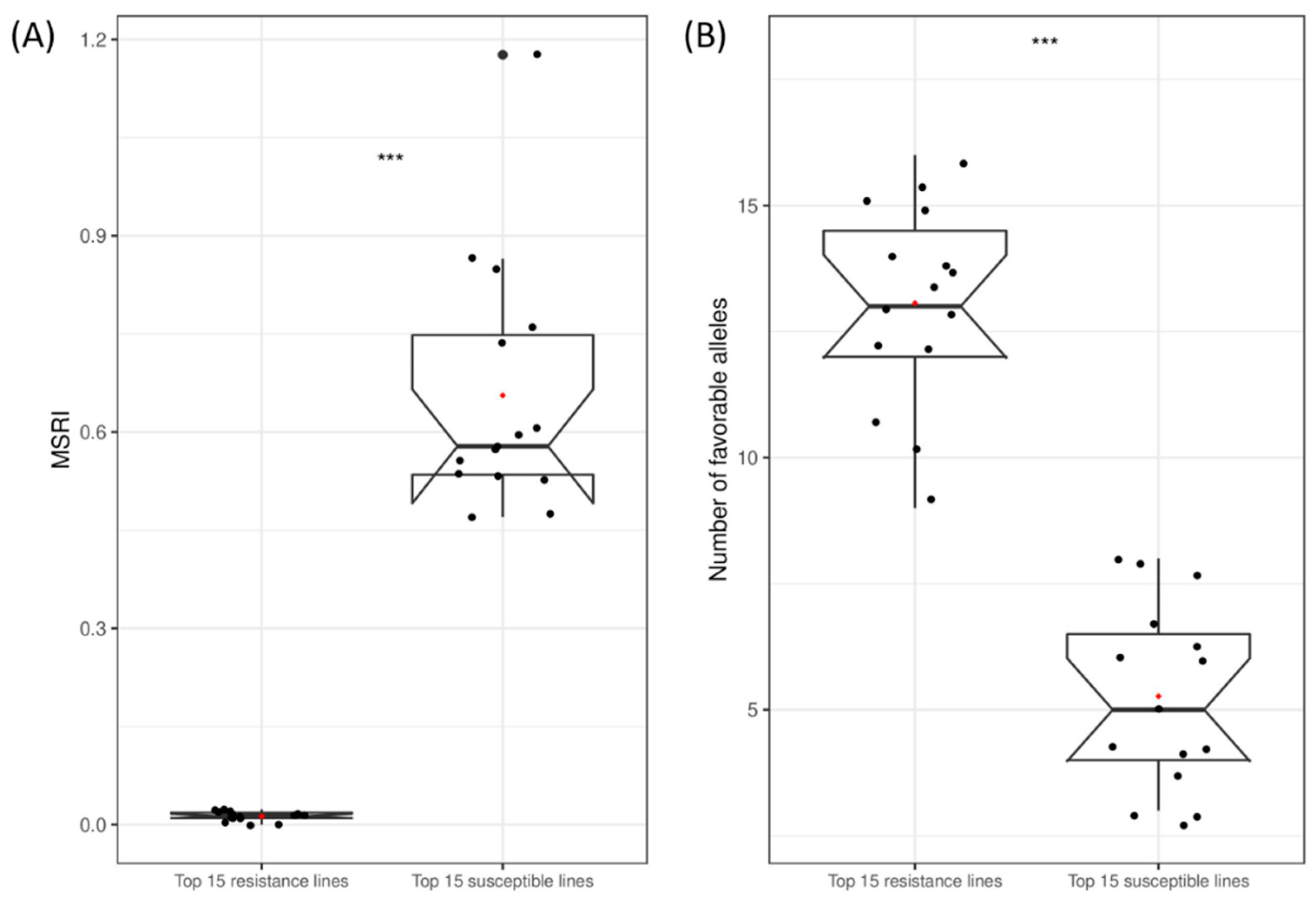
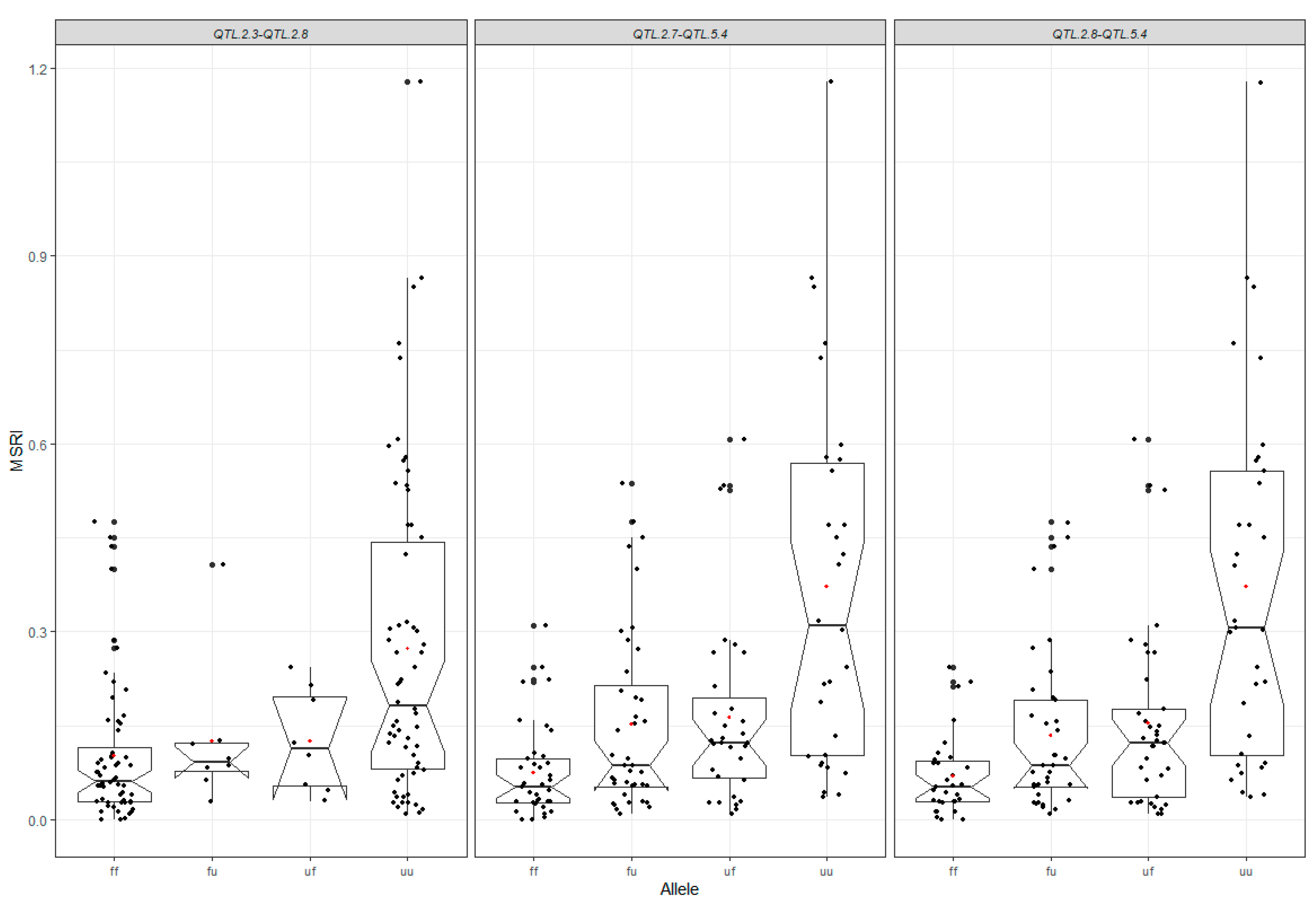
2.7. Favorable Alleles of QTL in RILs
2.8. Candidate Genes of Major QTL
3. Discussion
3.1. Genomic Heritability and Contribution of QTL to Marsh Spot Resistance
3.2. Statistical Models for QTL Identification and QTL Validation
3.3. Additive/Epistatic QTL and Genomics-Assisted Selection
3.4. Candidate Gene Prediction
4. Methods and Materials
4.1. Recombinant Inbred Lines (RILs)
4.2. Phenotyping of Marsh Spot Resistance
4.3. Genotyping by Sequencing and SNP Identification
4.4. Genomic Heritability
4.5. Construction of Linkage Map
4.6. QTL Identification
4.7. Favorable Alleles
4.8. Candidate Gene Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Statistics Canada Table 32-10-0007-01 Stocks of Grain and Oilseeds at March 31, July 31 and December 31 (×1000) 2020. Available online: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=3210000701 (accessed on 21 March 2020).
- FAO. FAOSTAT Statistical Database. 2019. Available online: https://www.fao.org/faostat/en/#home (accessed on 19 October 2021).
- Ndakidemi, P.A.; Dakora, F.D.; Nkonya, E.M.; Ringo, D.; Mansoor, H. Yield and economic benefits of common bean (Phaseolus vulgaris) and soybean (Glycine max) inoculation in northern Tanzania. Aust J. Exp. Agric. 2006, 46, 571–577. [Google Scholar] [CrossRef]
- Rosales-Serna, R.; Kohashi-Shibata, J.; Acosta-Gallegos, J.A.; Trejo-López, C.; Ortiz-Cereceres, J.n.; Kelly, J.D. Biomass distribution, maturity acceleration and yield in drought-stressed common bean cultivars. Field Crops Res. 2004, 85, 203–211. [Google Scholar] [CrossRef]
- Blair, M.W.; Iriarte, G.; Beebe, S. QTL analysis of yield traits in an advanced backcross population derived from a cultivated Andean × wild common bean (Phaseolus vulgaris L.) cross. Theor. Appl. Genet. 2006, 112, 1149–1163. [Google Scholar] [CrossRef]
- Celmeli, T.; Sari, H.; Canci, H.; Sari, D.; Adak, A.; Eker, T.; Toker, C. The nutritional content of common bean (Phaseolus vulgaris L.) landraces in comparison to modern varieties. Agronomy 2018, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Biddle, A.J.; Cattlin, N.D. Pests, Diseases, and Disorders of Peas and Beans: A Colour Handbook; CRC Press: London, UK, 2007. [Google Scholar]
- Heintze, S.G. Readily soluble manganese of soils and marsh spot of peas. J. Agric. Sci. 1938, 28, 175–186. [Google Scholar] [CrossRef]
- Henkens, C.H. The prevention of marsh spot in peas by spraying with manganese sulphate. Landbouwvoorlichting 1958, 15, 262–265. [Google Scholar]
- Howard, R.J.; Garland, J.A.; Seaman, W.L. Diseases and Pests of Vegetable Crops in Canada; Canadian Phytopathological Society: Ottawa, ON, Canada, 1994. [Google Scholar]
- Lacey, M.S. Studies in bacteriosis: Xxi. An investigation of marsh spot of peas: With a note on the morphological structure. Ann. Appl. Biol. 1934, 21, 621–640. [Google Scholar] [CrossRef]
- Piper, C.S. Marsh spot of peas: A manganese deficiency disease. J. Agric. Sci. 1941, 31, 448–453. [Google Scholar] [CrossRef]
- Reynolds, J.D. Marsh spot of peas: A review of present knowledge. J. Sci. Food Agric. 1955, 6, 725–734. [Google Scholar] [CrossRef]
- Hewitt, E.J. ‘Marsh spot’ in beans. Nature 1945, 155, 22–23. [Google Scholar] [CrossRef]
- De Bruyn, H.L.G. Kwade harten van de erwten. Tijdschr. Over Plantenziekten 1933, 39, 281–318. [Google Scholar]
- Jia, B.; Waldo, P.; Conner, R.L.; Khan, N.; Hou, A.; Xia, X.; You, F.M. Marsh spot disease and its causal factor, manganese deficiency in plants: A historical and prospective review. Agric. Sci. 2021, 12, 928–948. [Google Scholar] [CrossRef]
- Koopman, C. Invloed van mangaansulfaatbespuiting tegen kwaadhartigheid bij schokkererwten. Tijdschr. Over Plantenziekten 1937, 43, 64–66. [Google Scholar] [CrossRef]
- Lewis, A.H. Manganese deficiencies in crops. I. Spraying pea crops with solutions of manganese salts to eliminate marsh-spot. J. Exp. Agric. Int. 1939, 7, 150–154. [Google Scholar]
- Jia, B.; Conner, R.L.; Khan, N.; Hou, A.; Xia, X.; You, F.M. Inheritance of marsh spot disease resistance in cranberry common bean (Phaseolus vulgaris L.). Crop J. 2021, 10, 456–467. [Google Scholar] [CrossRef]
- Araki, R.; Murata, J.; Murata, Y. A novel barley Yellow Stripe 1-like Transporter (HvYSL2) localized to the root endodermis transports metal–phytosiderophore complexes. Plant Cell Physiol. 2011, 52, 1931–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruyn, H.L.G. Mn-deficiency as the cause of marsh spot of pea seeds. Tijdschr. Over Plantenziekten 1939, 45, 106–120. [Google Scholar]
- Pethybridge, G.H. Marsh spot in pea seeds: Is it a deficiency disease. J. Minist. Agric. 1936, 43, 55–58. [Google Scholar]
- Moraghan, J.G.; Afton, K. Marsh spot in cranberry bean seed. Ann. Rep. 2000, 43, 9–10. [Google Scholar]
- Guerinot, M.L. The ZIP family of metal transporters. Biochim. Biophys. Acta (BBA) 2000, 1465, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Milner, M.J.; Seamon, J.; Craft, E.; Kochian, L.V. Transport properties of members of the ZIP family in plants and their role in Zn and Mn homeostasis. J. Exp. Bot. 2012, 64, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Inoue, H.; Mizuno, D.; Takahashi, M.; Nakanishi, H.; Mori, S.; Nishizawa, N.K. OsYSL2 is a rice metal-nicotianamine transporter that is regulated by iron and expressed in the phloem. Plant J. 2004, 39, 415–424. [Google Scholar] [CrossRef]
- Thomine, S.; Lelièvre, F.; Debarbieux, E.; Schroeder, J.I.; Barbier-Brygoo, H. Cadmium and iron transport by members of a plant metal transporter family in Arabidopsis with homology to Nramp genes. Proc. Natl. Acad. Sci. USA 2000, 97, 4991–4996. [Google Scholar] [CrossRef] [Green Version]
- Castaings, L.; Caquot, A.; Loubet, S.; Curie, C. The high-affinity metal transporters NRAMP1 and IRT1 team up to take up iron under sufficient metal provision. Sci. Rep. 2016, 6, 37222. [Google Scholar] [CrossRef] [Green Version]
- Edmond, C.; Shigaki, T.; Ewert, S.; Nelson, M.D.; Connorton, J.M.; Chalova, V.; Noordally, Z.; Pittman, J.K. Comparative analysis of CAX2-like cation transporters indicates functional and regulatory diversity. Biochem. J. 2009, 418, 145–154. [Google Scholar] [CrossRef]
- Cheng, N.-h.; Pittman, J.K.; Shigaki, T.; Hirschi, K.D. Characterization of CAX4, an Arabidopsis H+/cation antiporter. Plant Physiol. 2002, 128, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnham, K.P.; Anderson, D.R. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach, 2nd ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Zhang, Y.; GAI, J.; Yang, Y. The EIM algorithm in the joint segregation analysis of quantitative traits. Genet. Res. 2003, 81, 157–163. [Google Scholar]
- Gai, J.; Wang, Y.; Wu, X.; Chen, S. A comparative study on segregation analysis and QTL mapping of quantitative traits in plants—with a case in soybean. Front. Agric. China 2007, 1, 1–7. [Google Scholar] [CrossRef]
- Lan, S.; Zheng, C.; Hauck, K.; McCausland, M.; Duguid, S.D.; Booker, H.M.; Cloutier, S.; You, F.M. Genomic prediction accuracy of seven breeding selection traits improved by QTL identification in flax. Int. J. Mol. Sci. 2020, 21, 1577. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Li, M.; Hu, X.; Ma, Q.; Mu, Y.; Tan, Z.; Xia, Q.; Zhang, G.; Nian, H. Construction of high-density genetic map and QTL mapping of yield-related and two quality traits in soybean RILs population by RAD-sequencing. BMC Genom. 2017, 18, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poveda, A.; Chen, Y.; Brändström, A.; Engberg, E.; Hallmans, G.; Johansson, I.; Renström, F.; Kurbasic, A.; Franks, P.W. The heritable basis of gene-environment interactions in cardiometabolic traits. Diabetologia 2017, 60, 442–452. [Google Scholar] [CrossRef] [Green Version]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Kumar, S.; Soto-Cerda, B.; Duguid, S.D.; Booker, H.M.; et al. Genome-wide association study and selection signatures detect genomic regions associated with seed yield and oil quality in flax. Int. J. Mol. Sci. 2018, 19, 2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.-J.; Zhang, Y.-W.; Zhang, J.; Feng, J.-Y.; Dunwell, J.M.; Zhang, Y.-M. An efficient multi-locus mixed model framework for the detection of small and linked QTLs in F2. Brief. Bioinform. 2018, 20, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Mueen Alam, K.; Fei, T.; Wubin, W.; Jianbo, H.; Tuanjie, Z.; Junyi, G.; Christian, W. Using the RTM-GWAS procedure to detect the drought tolerance QTL-allele system at the seedling stage under sand culture in a half-sib population of soybean [Glycine max (L.) Merr.]. Can. J. Plant Sci. 2019, 99, 801–814. [Google Scholar]
- Bernardo, R. Genomewide markers as cofactors for precision mapping of quantitative trait loci. Theor. Appl. Genet. 2013, 126, 999–1009. [Google Scholar] [CrossRef]
- Li, H.; Ye, G.; Wang, J. A modified algorithm for the improvement of composite interval mapping. Genetics 2007, 175, 361–374. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Meng, S.; Zhao, T.; Xing, G.; Yang, S.; Li, Y.; Guan, R.; Lu, J.; Wang, Y.; Xia, Q.; et al. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theor. Appl. Genet. 2017, 130, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Fan, T.; Chen, S.; Ou, X.; Chen, Y.; Jiang, Q.; Diao, Y.; Sun, Z.; Peng, W.; Ren, Z.; et al. QTL mapping and validation for kernel area and circumference in common wheat via high-density SNP-based genotyping. Front. Plant Sci. 2021, 12, 713890. [Google Scholar] [CrossRef]
- Su, J.; Wang, C.; Ma, Q.; Zhang, A.; Shi, C.; Liu, J.; Zhang, X.; Yang, D.; Ma, X. An RTM-GWAS procedure reveals the QTL alleles and candidate genes for three yield-related traits in upland cotton. BMC Plant Biol. 2020, 20, 416. [Google Scholar] [CrossRef]
- Shi, H.; Guan, W.; Shi, Y.; Wang, S.; Fan, H.; Yang, J.; Chen, W.; Zhang, W.; Sun, D.; Jing, R. QTL mapping and candidate gene analysis of seed vigor-related traits during artificial aging in wheat (Triticum aestivum). Sci. Rep. 2020, 10, 22060. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R Soc. Lond. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Xiao, J.; Rashid, K.Y.; Jia, G.; Li, P.; Yao, Z.; Wang, X.; Cloutier, S.; You, F.M. Evaluation of genomic prediction for pasmo resistance in flax. Int. J. Mol. Sci. 2019, 20, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, B.; Ariza-Suarez, D.; de la Hoz, J.; Aparicio, J.S.; Portilla-Benavides, A.E.; Buendia, H.F.; Mayor, V.M.; Studer, B.; Raatz, B. Genomic prediction of agronomic traits in common bean (Phaseolus vulgaris L.) under environmental stress. Front. Plant Sci. 2020, 11, 1001. [Google Scholar] [CrossRef]
- Delfini, J.; Moda-Cirino, V.; Dos Santos Neto, J.; Ruas, P.M.; Sant’Ana, G.C.; Gepts, P.; Goncalves, L.S.A. Population structure, genetic diversity and genomic selection signatures among a Brazilian common bean germplasm. Sci. Rep. 2021, 11, 2964. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; You, F.M.; Duguid, S.; Booker, H.; Rowland, G.; Cloutier, S. QTL for fatty acid composition and yield in linseed (Linum usitatissimum L.). Theor. Appl. Genet. 2015, 128, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Sertse, D.; You, F.M.; Ravichandran, S.; Cloutier, S. The complex genetic architecture of early root and shoot traits in flax revealed by genome-wide association analyses. Front. Plant Sci. 2019, 10, 1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, F.M.; Cloutier, S. Mapping quantitative trait loci onto chromosome-scale pseudomolecules in flax. Methods Protoc. 2020, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.D.; Hannam, R.J.; Uren, N.C. Manganese in Soils and Plants; Springer: Dordrecht, The Netherlands, 1988; Volume 33. [Google Scholar]
- Humphries, J.; Stangoulis, J.; Graham, R. Manganese. In Handbook of Plant Nutrition; Barker, A.V., Pilbeam, D.J., Eds.; Routledge Taylor and Francis: New York, NY, USA, 2007; pp. 351–366. [Google Scholar]
- Schmidt, S.B.; Jensen, P.E.; Husted, S. Manganese deficiency in plants: The impact on photosystem II. Trends Plant Sci. 2016, 21, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Boswell, F.C.; Parker, M.B.; Shuman, L.M.; Wilson, D.O. Critical manganese deficiency level of soybean related to leaf position. Agron. J. 1979, 71, 233–234. [Google Scholar] [CrossRef]
- Pedas, P.; Ytting, C.K.; Fuglsang, A.T.; Jahn, T.P.; Schjoerring, J.K.; Husted, S. Manganese efficiency in barley: Identification and characterization of the metal ion transporter HvIRT1. Plant Physiol. 2008, 148, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Enrico, M.; Stefan, M.; Alexis, D.A.; Réka, N. Vacuolar transporters in their physiological context. Annu. Rev. Plant Biol. 2012, 63, 183–213. [Google Scholar]
- Colston-Nepali, L.; Tigano, A.; Boyle, B.; Friesen, V. Hybridization does not currently pose conservation concerns to murres in the Atlantic. Conserv. Genet. 2019, 20, 1465–1470. [Google Scholar] [CrossRef]
- Gioia, T.; Logozzo, G.; Marzario, S.; Spagnoletti Zeuli, P.; Gepts, P. Evolution of SSR diversity from wild types to U.S. advanced cultivars in the Andean and Mesoamerican domestications of common bean (Phaseolus vulgaris). PLoS ONE 2019, 14, e0211342. [Google Scholar] [CrossRef] [Green Version]
- You, F.M.; Huo, N.; Deal, K.R.; Gu, Y.Q.; Luo, M.C.; McGuire, P.E.; Dvorak, J.; Anderson, O.D. Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence. BMC Genom. 2011, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Bonfield, J.K.; Marshall, J.; Danecek, P.; Li, H.; Ohan, V.; Whitwham, A.; Keane, T.; Davies, R.M. HTSlib: C library for reading/writing high-throughput sequencing data. GigaScience 2021, 10, giab007. [Google Scholar] [CrossRef]
- You, F.M.; Deal, K.R.; Wang, J.; Britton, M.T.; Fass, J.N.; Lin, D.; Dandekar, A.M.; Leslie, C.A.; Aradhya, M.; Luo, M.C.; et al. Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms. BMC Genom. 2012, 13, 354. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; You, F.M.; Cloutier, S. Genome wide SNP discovery in flax through next generation sequencing of reduced representation libraries. BMC Genom. 2012, 13, 684. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.L.; Zhu, J.; Li, Z.K.L.; Paterson, A.H. Mapping QTLs with epistatic effects and QTL×environment interactions by mixed linear model approaches. Theor. Appl. Genet. 1999, 99, 1255–1264. [Google Scholar] [CrossRef]
- Kosambi, D.D. The estimation of map distance from recombination values. Ann. Eugen. 1944, 12, 172–175. [Google Scholar] [CrossRef]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Madden, T.L. PowerBLAST: A new network BLAST application for interactive or automated sequence analysis and annotation. Genome Res. 1997, 7, 649–656. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linage Group (Chromosome) | No. of Markers | Total Distance (cm) | No. of Recombination Intervals | |
|---|---|---|---|---|
| 1 | 101 | 131 | 27 | 4.86 ± 13.40 |
| 2 | 263 | 305 | 96 | 3.18 ± 7.17 |
| 3 | 115 | 105 | 41 | 2.56 ± 3.75 |
| 4 | 160 | 257 | 69 | 3.73 ± 7.10 |
| 5 | 360 | 186 | 76 | 2.44 ± 4.39 |
| 6 | 71 | 114 | 28 | 4.08 ± 9.60 |
| 7 | 72 | 135 | 21 | 6.41 ± 11.52 |
| 8 | 9 | 144 | 4 | 36.11 ± 14.80 |
| 9 | 59 | 93 | 26 | 3.59 ± 7.73 |
| 10 | 44 | 91 | 26 | 3.50 ± 5.40 |
| 11 | 19 | 38 | 9 | 4.20 ± 6.32 |
| Total | 1273 | 1599 | 434 | 3.78 ± 8.13 |
| Phenotypic Dataset | Genomic Heritability (h2 ± s) (%) | Phenotypic Dataset | Genomic Heritability (h2 ± s) (%) |
|---|---|---|---|
| H2015 | 24.22 ± 0.09 | S2019 | 12.07 ± 0.07 |
| H2016 | 18.43 ± 0.08 | T2015 | 32.48 ± 0.10 |
| H2017 | 32.96 ± 0.10 | T2016 | 28.02 ± 0.01 |
| H2018 | 33.13 ± 0.10 | T2017 | 45.53 ± 0.11 |
| H2019 | 32.82 ± 0.10 | T2018 | 45.48 ± 0.10 |
| S2015 | 27.03 ± 0.10 | T2019 | 41.12 ± 0.11 |
| S2016 | 24.76 ± 0.10 | H-5 yrs | 46.46 ± 0.11 |
| S2017 | 30.37 ± 0.10 | S-5 yrs | 47.14 ± 0.11 |
| S2018 | 16.67 ± 0.08 | Overall | 55.91 ± 0.10 |
| QTL | Flanking Markers and Position | LG Pos (cm) | Additive Effect | No. of Datasets (a) | Significant Datasets (b) | Average R2 (%) (c) | Model |
|---|---|---|---|---|---|---|---|
| QTL.1.1 | Chr1_48339634–Chr1_50146614 | 119.26–126.33 | 0.10 | 1 | 3 | 5.92 | GCIM |
| QTL.2.1 | Chr2_872663–Chr2_1135128 | 0.06–5.22 | 0.05–0.11 | 6 | 11 | 8.30 | GCIM, RTM-GWAS |
| QTL.2.2 | Chr2_32113326 | 97.46 | 0.07 | 1 | 17 | 9.12 | GCIM |
| QTL.2.3 | Chr2_34070996–Chr2_35065692 | 147.11–151.45 | 0.15 | 1 | 16 | 11.43 | GCIM |
| QTL.2.4 | Chr2_35130486–Chr2_35289581 | 143.04–142.69 | 0.07–0.10 | 5 | 18 | 10.42 | GCIM, ICIM-ADD |
| QTL.2.5 | Chr2_35344261–Chr2_36750706 | 128.94–134.16 | 0.14 | 1 | 17 | 10.02 | ICIM-ADD |
| QTL.2.6 | Chr2_37937595–Chr2_38452857 | 187.14–188.25 | 0.05–0.51 | 5 | 18 | 12.30 | ICIM-ADD, GCIM |
| QTL.3.1 | Chr3_11944447–Chr3_19043093 | 47.42–51.49 | −0.08 | 5 | 17 | 24.52 | GCIM, RTM-GWAS, ICIM-ADD |
| QTL.3.2 | Chr3_19701297–Chr3_30221015 | 48.9–50.4 | −0.57–0.17 | 13 | 17 | 21.78 | ICIM-ADD, GCIM, RTM-GWAS |
| QTL.5.1 | Chr5_11498360–Chr5_19238819 | 56.2–57.31 | −0.46 | 1 | 4 | 7.39 | GCIM |
| QTL.5.2 | Chr5_1647320–Chr5_31681432 | 12.94–38.3 | 0.06–0.10 | 6 | 18 | 9.23 | GCIM |
| QTL.5.3 | Chr5_38536162–Chr5_38536272 | 171.77–171.75 | 0.06–0.13 | 10 | 17 | 10.72 | GCIM, ICIM-ADD |
| QTL.5.4 | Chr5_623370–Chr5_673021 | 0–1.13 | −0.10–−0.07 | 12 | 17 | 11.61 | ICIM-ADD, RTM-GWAS |
| QTL.6.1 | Chr6_1374720–Chr6_1504675 | 23.18–23.92 | 0.04–0.08 | 4 | 13 | 6.81 | GCIM, RTM-GWAS |
| QTL.6.2 | Chr6_13598278–Chr6_14124318 | 39.58–38.48 | 0.05–0.07 | 2 | 5 | 7.52 | GCIM |
| QTL.9.1 | Chr9_17827630–Chr9_20865151 | 42.84–46.75 | 0.20 | 2 | 3 | 7.82 | GCIM, RTM-GWAS |
| QTL 1 | QTL 2 | No. Datasets with QTL (a) | No. Datasets with Significant Effect (b) | Average of R2 (%) (c) | Additive Effect of QTL 1 | Additive Effect of QTL 2 | Epistatic Effect | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QTL 1 | Left Marker | Right Marker | LG Pos (cm) | QTL 2 | Left Marker | Right Marker | LG Pos (cm) | ||||||
| QTL.5.4 | Chr5_673021 | Chr5_1647342 | 1.13–12.92 | QTL.2.7 | Chr2_36996368 | Chr2_37937763 | 160.01–178.45 | 1 | 18 | 30.64 | −0.05 | 0.04 | −0.11 |
| QTL.5.4 | Chr5_673021 | Chr5_1647342 | 1.13–12.92 | QTL.2.8 | Chr2_37937763 | Chr2_37531627 | 178.45–184.57 | 2 | 18 | 30.69 | −0.05 | 0.04 | −0.11 |
| QTL.2.3 | Chr2_34070996 | Chr2_35065692 | 147.11–151.45 | QTL.2.8 | Chr2_37937763 | Chr2_37531627 | 178.45–184.57 | 1 | 9 | 16.31 | −0.06 | 0.04 | −0.10 |
| QTL | Gene | Chr | Gene Coordinates (a) | Annotation |
|---|---|---|---|---|
| QTL.1.1 | Phvul.001G250300 | 1 | 50100939–50102262 | Heavy metal transport/detoxification superfamily protein |
| Phvul.001G247400 | 1 | 49894723–49895275 | Heavy metal transport/detoxification superfamily protein | |
| QTL.2.3 | Phvul.002G184200 | 2 | 34468080–34481795 | ZIP metal ion transporter family |
| QTL.3.1 | Phvul.003G086300 | 3 | 16877488–16879137 | Heavy metal transport/detoxification superfamily protein |
| QTL.3.2 | Phvul.003G092500 | 3 | 21113140–21115717 | Heavy metal transport/detoxification superfamily protein |
| Phvul.003G104900 | 3 | 23350962–23352673 | Heavy metal transport/detoxification superfamily protein | |
| Phvul.003G099700 | 3 | 25708027–25708491 | Heavy metal transport/detoxification superfamily protein | |
| Phvul.003G108900 | 3 | 27536901–27538128 | Heavy metal transport/detoxification superfamily protein | |
| QTL.5.2 | Phvul.005G095400 | 5 | 29797811–29799135 | Heavy metal transport/detoxification superfamily protein |
| Phvul.005G049300 | 5 | 5742223–5745905 | Cation efflux family protein | |
| Phvul.005G048900 | 5 | 5682746–5684903 | Zinc transporter 1 precursor | |
| QTL.9.1 | Phvul.009G137100 | 9 | 20653368–20662253 | Manganese tracking factor for mitochondrial SOD2 |
| Phvul.009G127900 | 9 | 19429094–19433403 | NRAMP metal ion transporter 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, B.; Conner, R.L.; Penner, W.C.; Zheng, C.; Cloutier, S.; Hou, A.; Xia, X.; You, F.M. Quantitative Trait Locus Mapping of Marsh Spot Disease Resistance in Cranberry Common Bean (Phaseolus vulgaris L.). Int. J. Mol. Sci. 2022, 23, 7639. https://doi.org/10.3390/ijms23147639
Jia B, Conner RL, Penner WC, Zheng C, Cloutier S, Hou A, Xia X, You FM. Quantitative Trait Locus Mapping of Marsh Spot Disease Resistance in Cranberry Common Bean (Phaseolus vulgaris L.). International Journal of Molecular Sciences. 2022; 23(14):7639. https://doi.org/10.3390/ijms23147639
Chicago/Turabian StyleJia, Bosen, Robert L. Conner, Waldo C. Penner, Chunfang Zheng, Sylvie Cloutier, Anfu Hou, Xuhua Xia, and Frank M. You. 2022. "Quantitative Trait Locus Mapping of Marsh Spot Disease Resistance in Cranberry Common Bean (Phaseolus vulgaris L.)" International Journal of Molecular Sciences 23, no. 14: 7639. https://doi.org/10.3390/ijms23147639
APA StyleJia, B., Conner, R. L., Penner, W. C., Zheng, C., Cloutier, S., Hou, A., Xia, X., & You, F. M. (2022). Quantitative Trait Locus Mapping of Marsh Spot Disease Resistance in Cranberry Common Bean (Phaseolus vulgaris L.). International Journal of Molecular Sciences, 23(14), 7639. https://doi.org/10.3390/ijms23147639