
Potential of Single Pulse and Multiplexed Drift-Tube Ion Mobility Spectrometry Coupled to Micropillar Array Column for Proteomics Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chromatographic Optimization
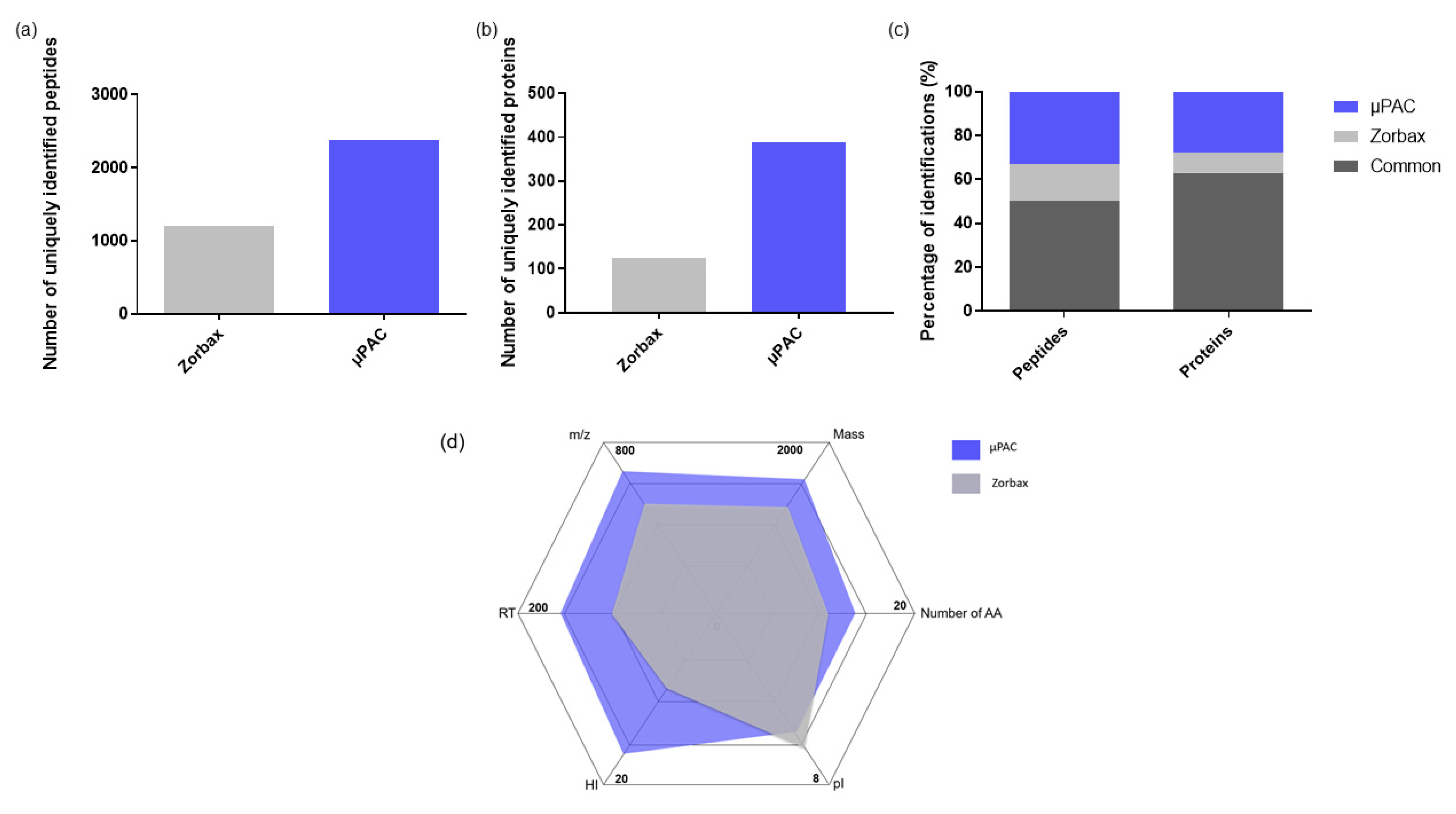
2.1.1. Comparison of Two Trapping Columns
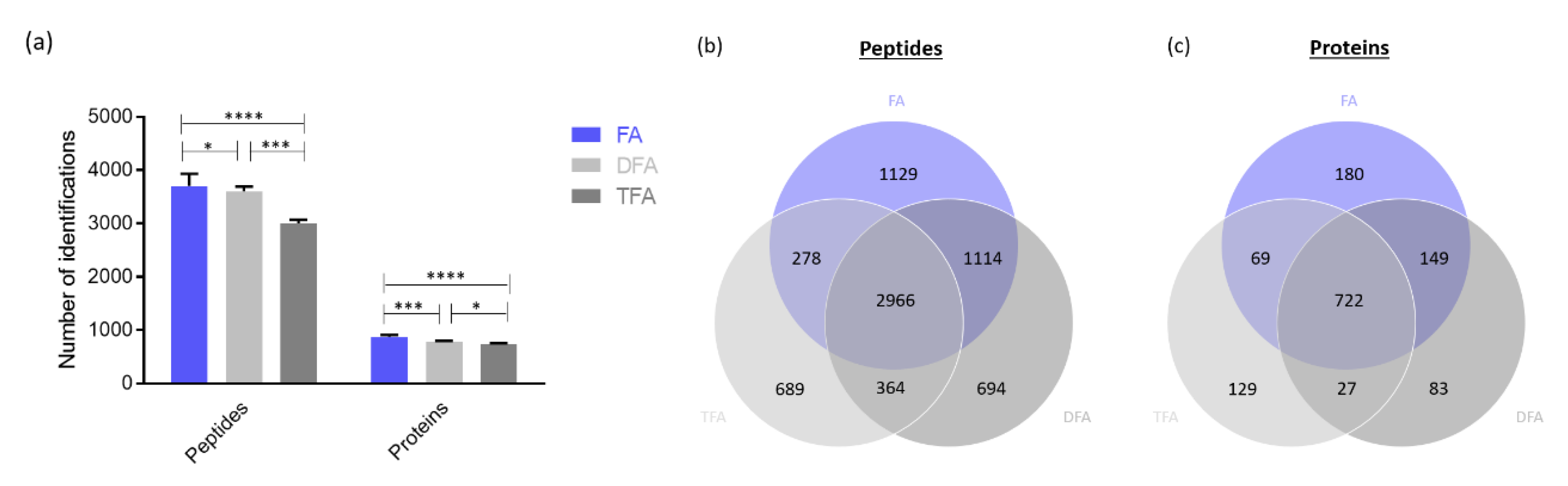
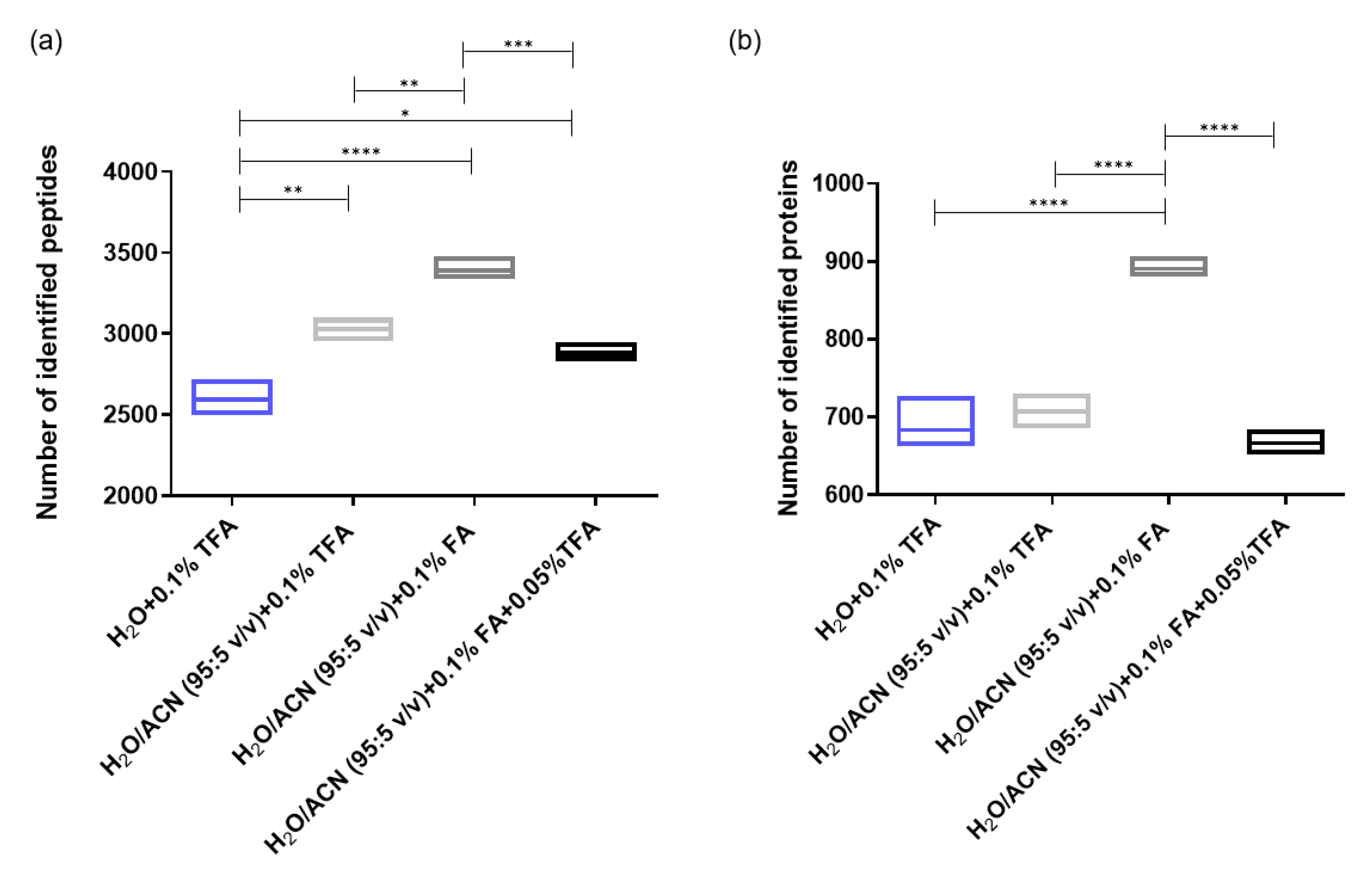
2.1.2. Influence of the Acidic Modifier in Sample Loading Mobile Phase and Sample Resuspending Solvent
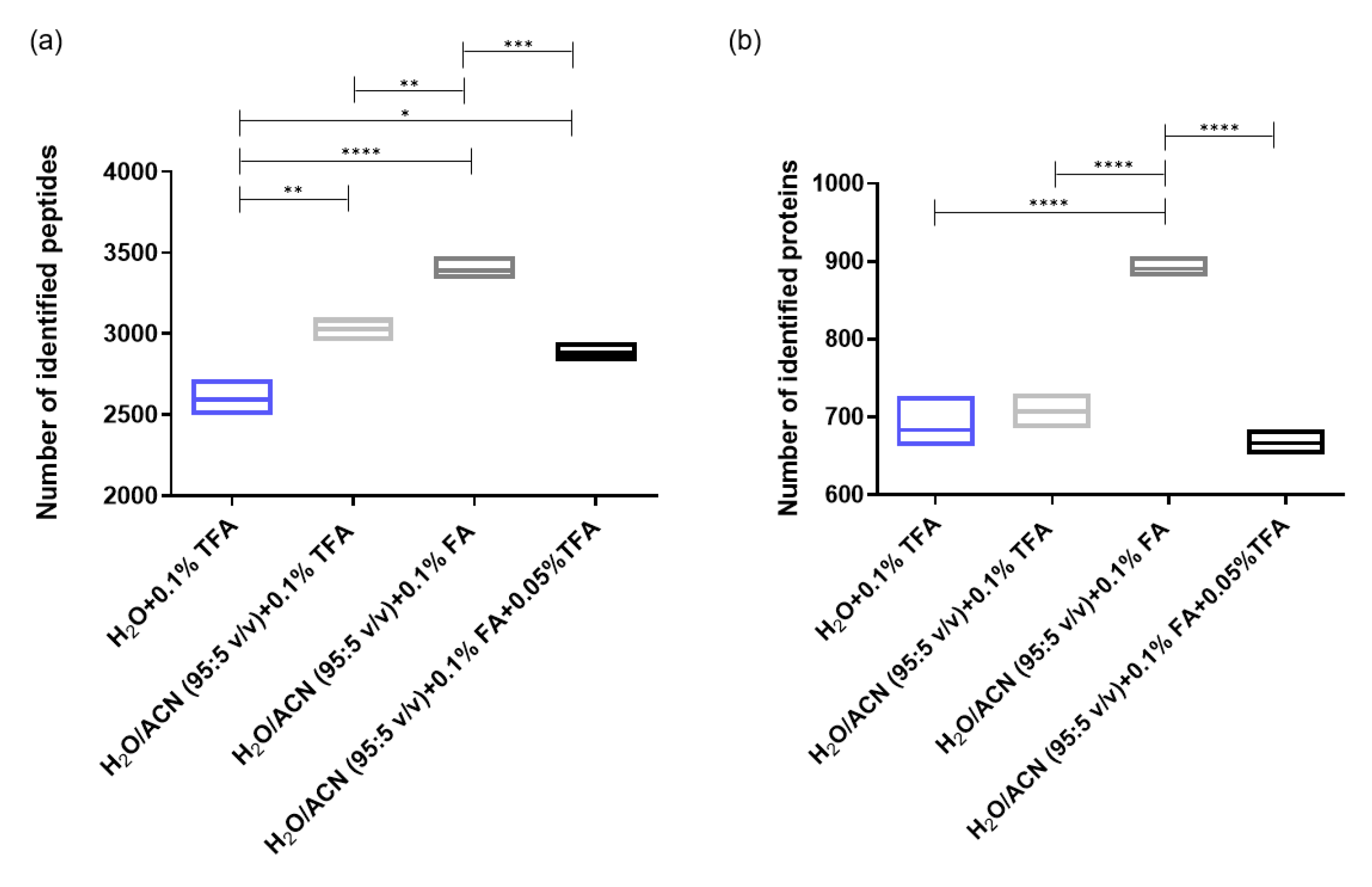
2.1.3. Optimization of the Sample Resuspending Solvent
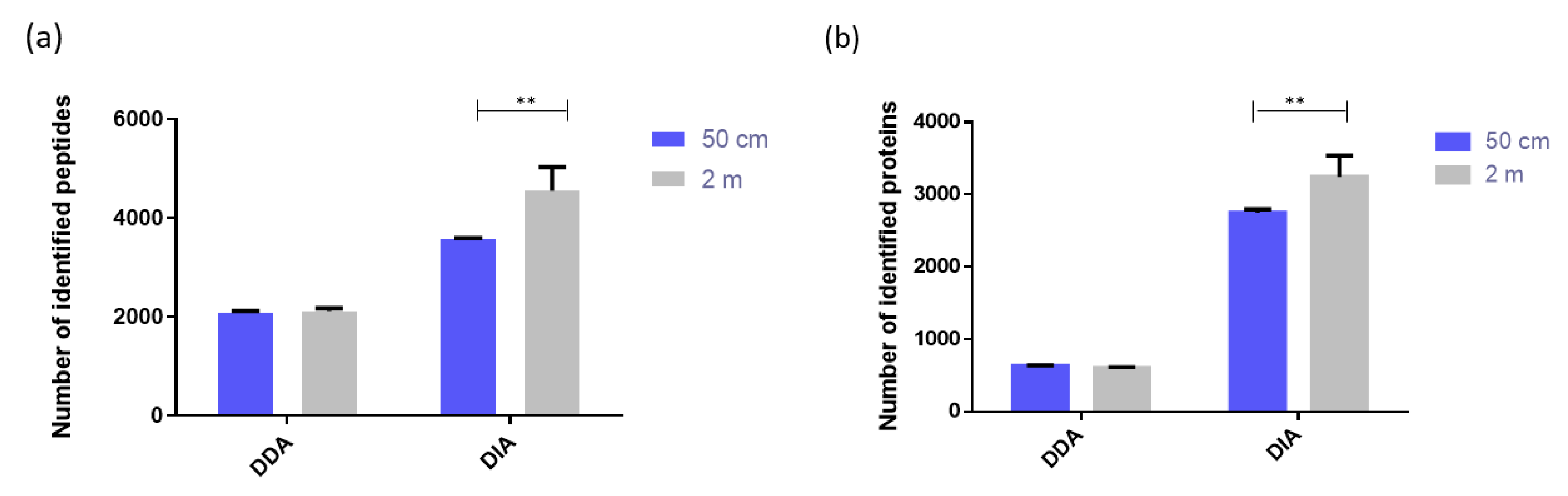
2.1.4. Comparison of Two µPAC Column Lengths
2.2. Interest of Using a Chip as Spray Emitter
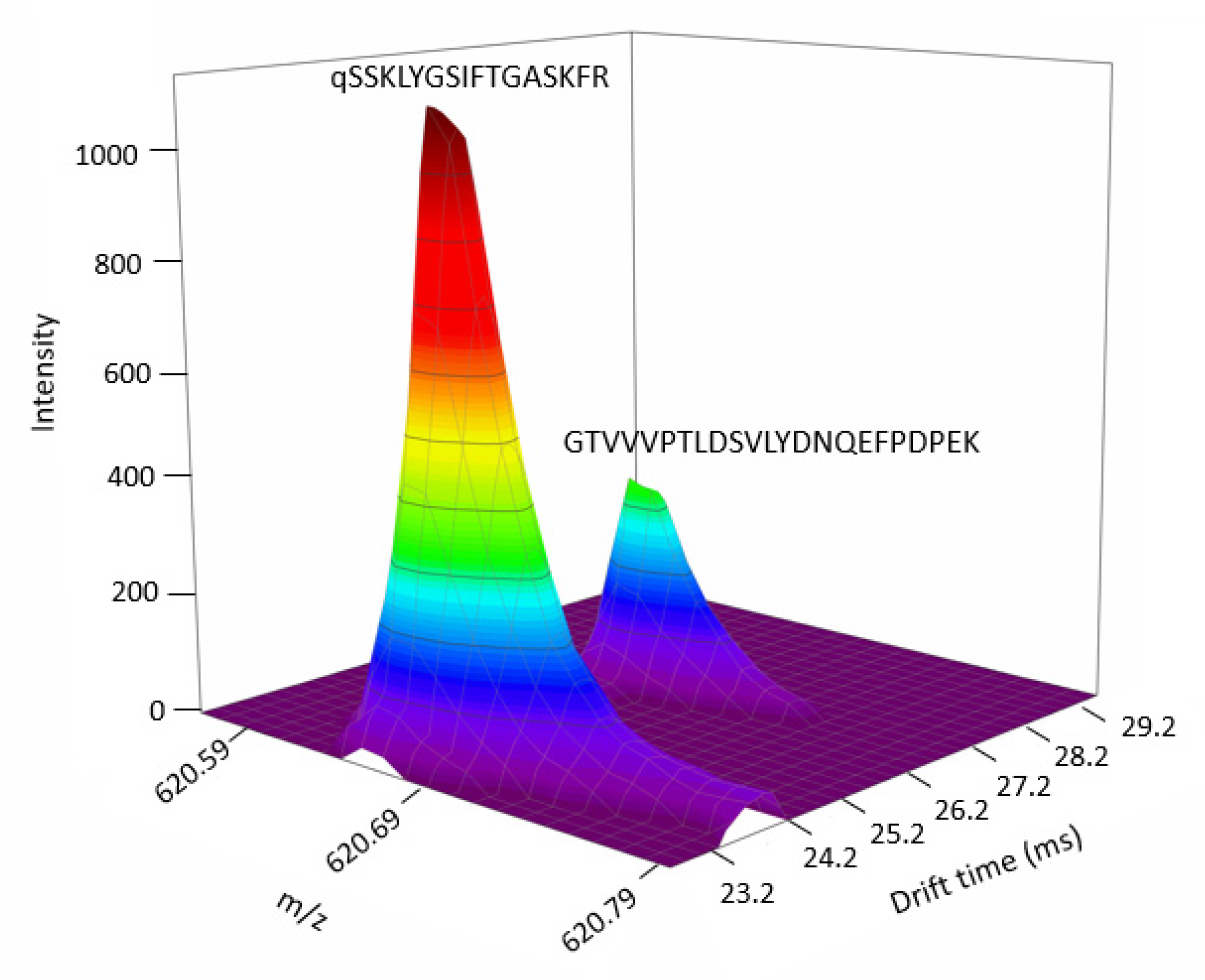
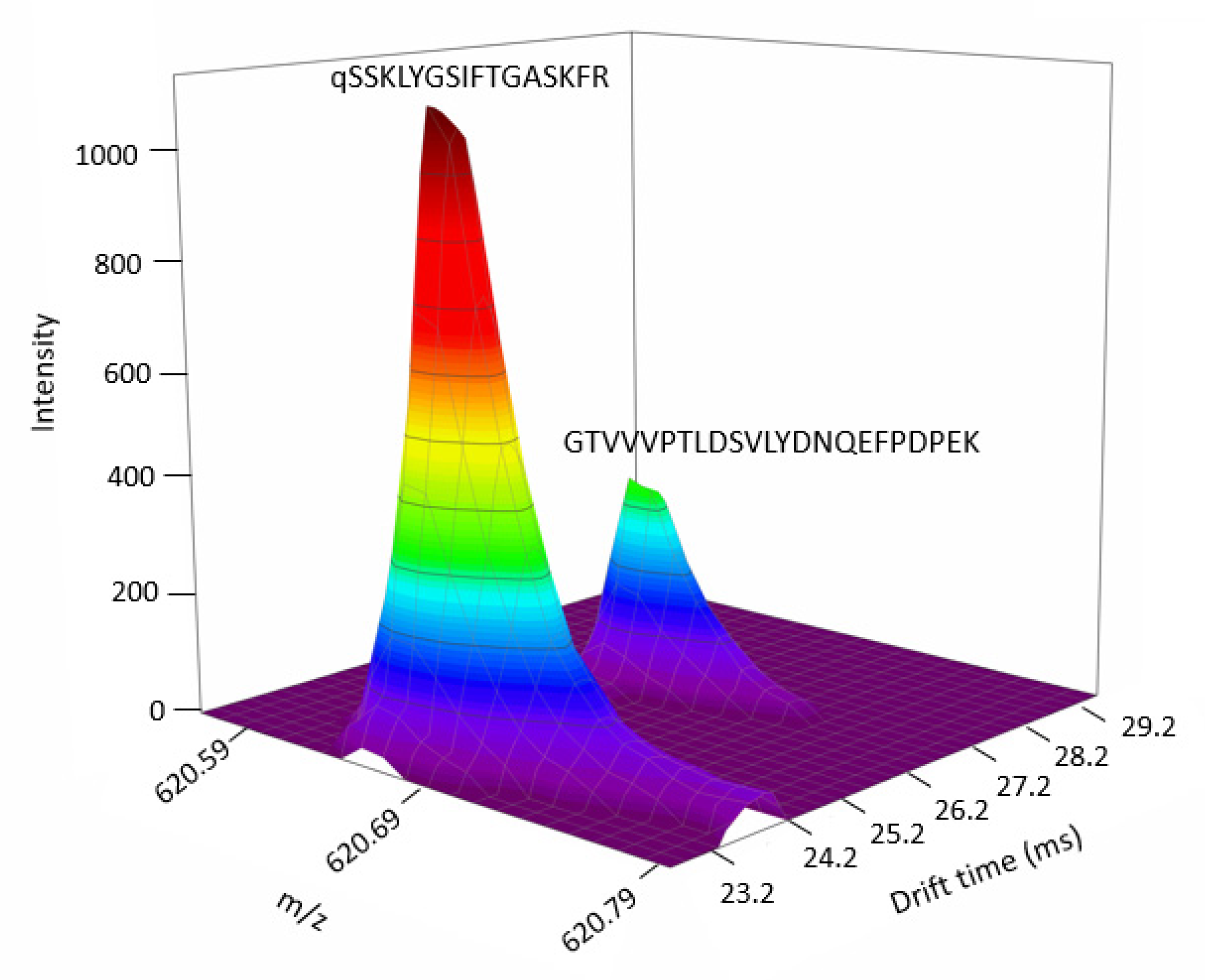
2.3. Data-Dependent Acquisition and Data-Independent Acquisition Driven by DTIMS
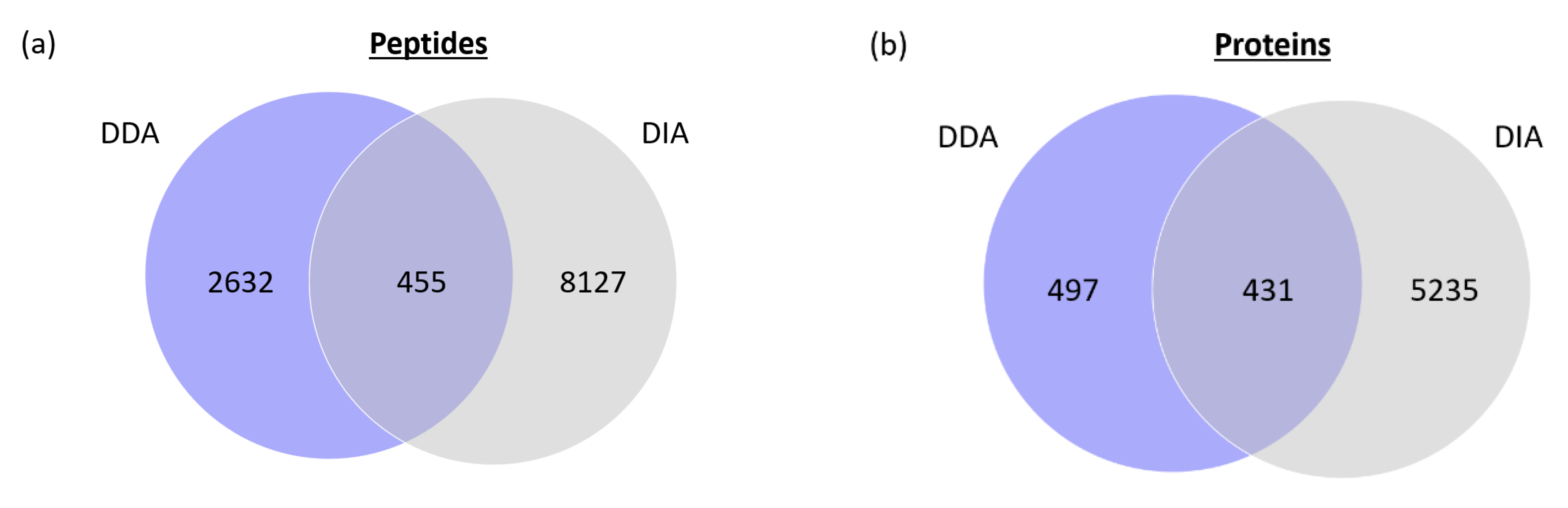
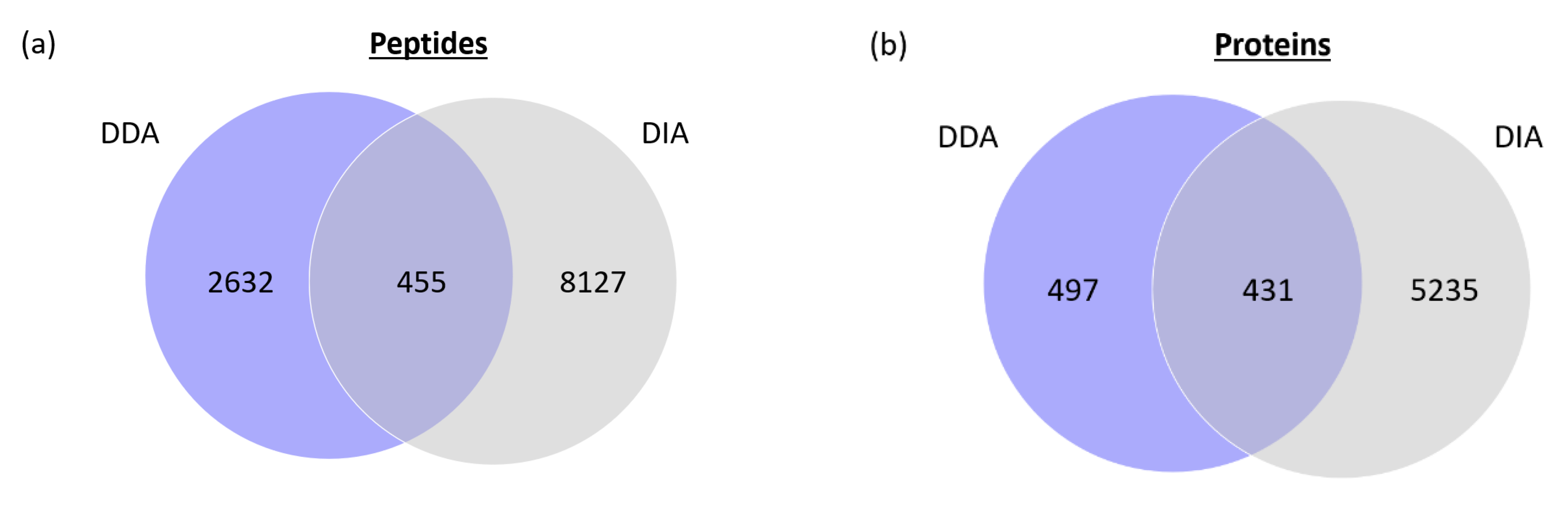
2.4. Interest of Drift-Tube Data-Independent Acquisition in Proteomics
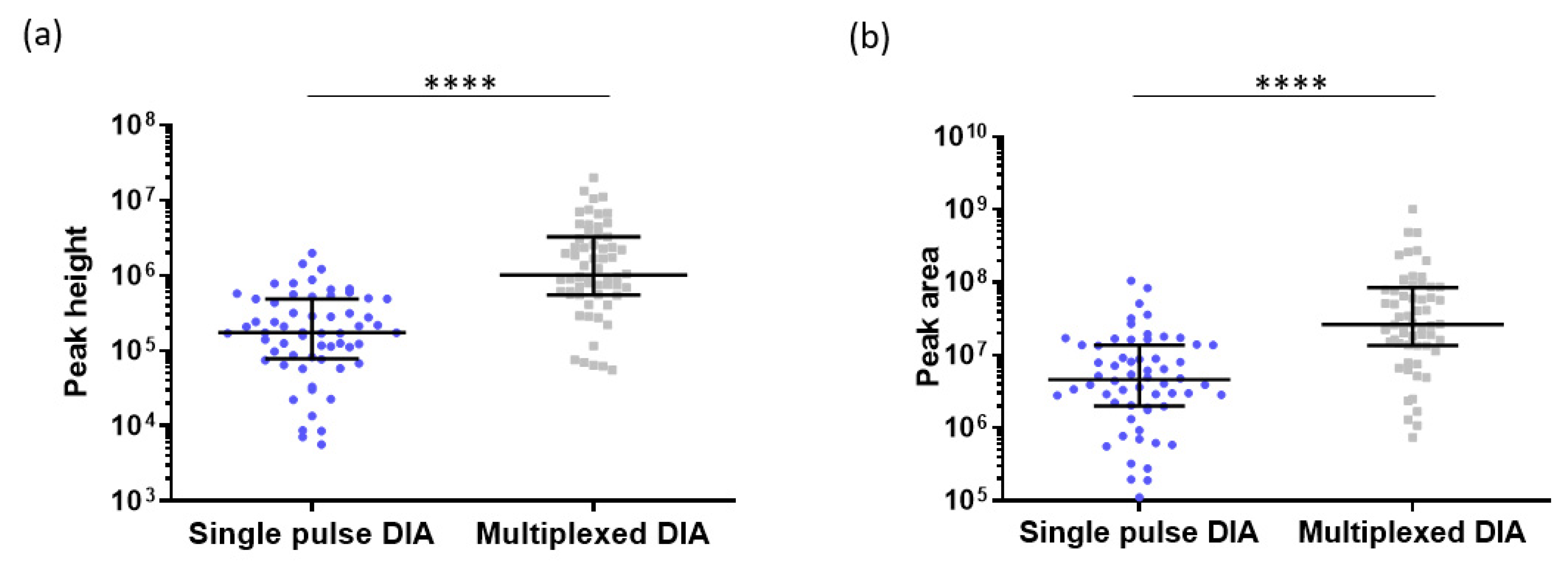
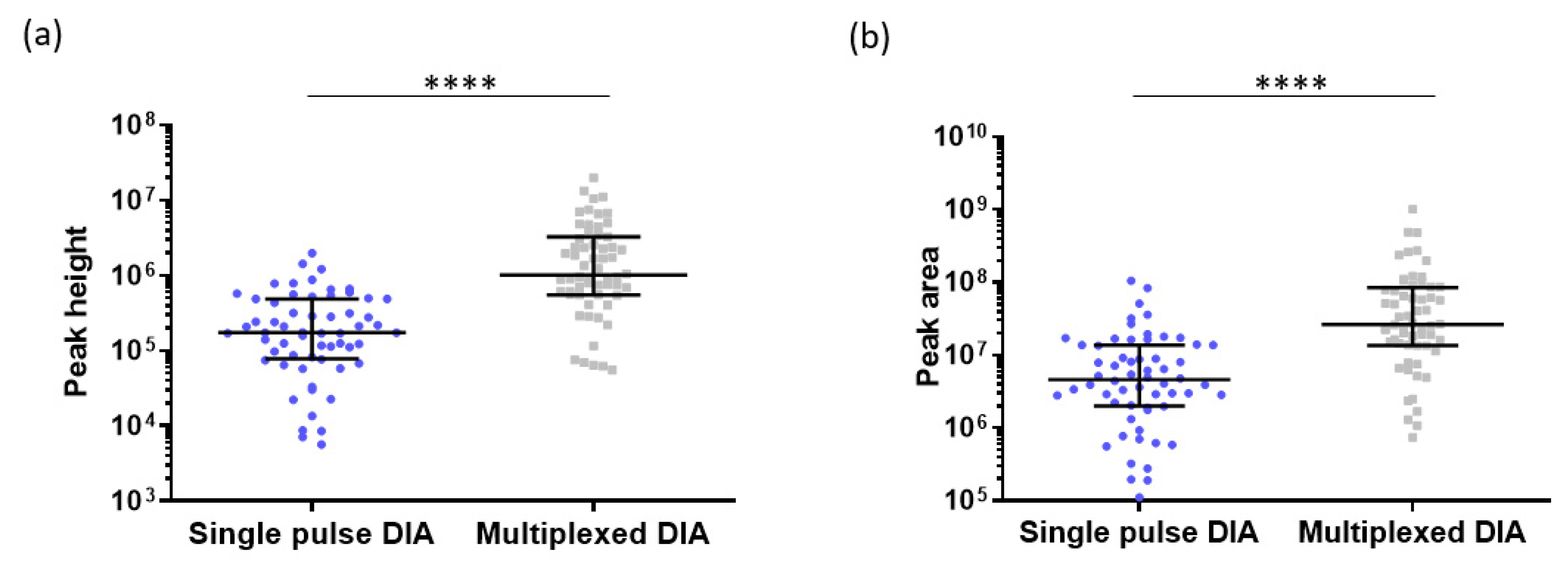
2.5. Interest of Multiplexed DIA in Proteomics
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Nanofluidic Liquid Chromatography
3.3. Mass Spectrometry Analysis
3.4. Data Treatment
3.5. Comparison of Two Trapping Columns
3.6. Influence of the Acidic Modifier in Sample Loading Mobile Phase and Sample Resuspending Solvent
3.7. Optimization of Sample Resuspending Solvent
3.8. Comparison of Two µPAC Column Lengths and Impact of the Temperature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Veenstra, T.D. Omics in Systems Biology: Current Progress and Future Outlook. Proteomics 2021, 21, 2000235. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, T.; Ciborowski, M.; Kisluk, J.; Kretowski, A.; Barbas, C. Mass Spectrometry Based Proteomics and Metabolomics in Personalized Oncology. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165690. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, R.; Loreti, C.; Giovannini, S.; Ricciardi, W.; Padua, L.; Boccia, S. Challenges of Prevention for a Sustainable Personalized Medicine. J. Pers. Med. 2021, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasny, L.; Huang, P.H. Molecular Omics Data-Independent Acquisition Mass Spectrometry (DIA-MS) for Proteomic Applications in Oncology. Mol. Omics 2021, 17, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Nys, G.; Nix, C.; Cobraiville, G.; Servais, A.C.; Fillet, M. Enhancing Protein Discoverability by Data Independent Acquisition Assisted by Ion Mobility Mass Spectrometry. Talanta 2020, 213, 120812. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics 2020, 20, 1900276. [Google Scholar] [CrossRef]
- Ye, Z.; Vakhrushev, S.Y. The Role of Data-Independent Acquisition for Glycoproteomics. Mol. Cell. Proteom. 2021, 20, 100042. [Google Scholar] [CrossRef]
- Burnum-johnson, K.E.; Zheng, X.; Dodds, J.N.; Ash, J.; Fourches, D.; Nicora, C.D.; Wendler, J.P.; Metz, T.O.; Waters, K.M.; Jansson, J.K.; et al. Trends in Analytical Chemistry Ion Mobility Spectrometry and the Omics: Distinguishing Isomers, Molecular Classes and Contaminant Ions in Complex Samples. Trends Anal. Chem. 2019, 116, 292–299. [Google Scholar] [CrossRef]
- Dodds, J.N.; Baker, E.S. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30, 2185–2195. [Google Scholar] [CrossRef]
- Ridgeway, M.E.; Lubeck, M.; Jordens, J.; Mann, M.; Park, M.A. Trapped Ion Mobility Spectrometry: A Short Review. Int. J. Mass Spectrom. 2018, 425, 22–35. [Google Scholar] [CrossRef]
- Meier, F.; Park, M.A.; Mann, M. Trapped Ion Mobility Spectrometry and Parallel Accumulation–Serial Fragmentation in Proteomics. Mol. Cell. Proteom. 2021, 20, 100138. [Google Scholar] [CrossRef]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef] [Green Version]
- D’Atri, V.; Causon, T.; Hernandez-Alba, O.; Mutabazi, A.; Veuthey, J.; Cianferani, S.; Guillarme, D. Adding a new separation dimension to MS and LC-MS: What is the utility of ion mobility spectrometry? J. Sep. Sci. 2018, 41, 20–67. [Google Scholar] [CrossRef]
- Nys, G.; Cobraiville, G.; Fillet, M. Multidimensional Performance Assessment of Micro Pillar Array Column Chromatography Combined to Ion Mobility-Mass Spectrometry for Proteome Research. Anal. Chim. Acta 2019, 1086, 1–13. [Google Scholar] [CrossRef]
- Vargas Medina, D.A.; Maciel, E.V.S.; Lanças, F.M. Miniaturization of Liquid Chromatography Coupled to Mass Spectrometry. 3. Achievements on Chip-Based LC–MS Devices. TrAC Trends Anal. Chem. 2020, 131, 116003. [Google Scholar] [CrossRef]
- Rozing, G. Micropillar Array Columns for Advancing Nanoflow HPLC. Microchem. J. 2021, 170, 106629. [Google Scholar] [CrossRef]
- Røberg-Larsen, H.; Lundanes, E.; Nyman, T.A.; Berven, F.S.; Wilson, S.R. Liquid Chromatography, a Key Tool for the Advancement of Single-Cell Omics Analysis. Anal. Chim. Acta 2021, 1178, 338551. [Google Scholar] [CrossRef]
- Tóth, G.; Panić-Janković, T.; Mitulović, G. Pillar Array Columns for Peptide Separations in Nanoscale Reversed-Phase Chromatography. J. Chromatogr. A 2019, 1603, 426–432. [Google Scholar] [CrossRef]
- Furkert, D.; Hostachy, S.; Nadler-Holly, M.; Fiedler, D. Triplexed Affinity Reagents to Sample the Mammalian Inositol Pyrophosphate Interactome. Cell Chem. Biol. 2020, 27, 1097–1108.e4. [Google Scholar] [CrossRef]
- Kristensen, K.; Münter, R.; Kempen, P.J.; Thomsen, M.E.; Stensballe, A.; Andresen, T.L. Isolation Methods Commonly Used to Study the Liposomal Protein Corona Suffer from Contamination Issues. Acta Biomater. 2021, 130, 460–472. [Google Scholar] [CrossRef]
- Pucci, C.; Marino, A.; Şen, Ö.; De Pasquale, D.; Bartolucci, M.; Iturrioz-Rodríguez, N.; di Leo, N.; de Vito, G.; Debellis, D.; Petretto, A.; et al. Ultrasound-Responsive Nutlin-Loaded Nanoparticles for Combined Chemotherapy and Piezoelectric Treatment of Glioblastoma Cells. Acta Biomater. 2021, 139, 218–236. [Google Scholar] [CrossRef]
- Merckaert, T.; Zwaenepoel, O.; Gevaert, K.; Gettemans, J. An AKT2-Specific Nanobody That Targets the Hydrophobic Motif Induces Cell Cycle Arrest, Autophagy and Loss of Focal Adhesions in MDA-MB-231 Cells. Biomed. Pharmacother. 2021, 133, 111055. [Google Scholar] [CrossRef]
- Prajapati, M.; Conboy, H.L.; Hojyo, S.; Fukada, T.; Budnik, B.; Bartnikas, T.B. Biliary Excretion of Excess Iron in Mice Requires Hepatocyte Iron Import by Slc39a14. J. Biol. Chem. 2021, 297, 100835. [Google Scholar] [CrossRef]
- Hamouda, N.N.; van den Haute, C.; Vanhoutte, R.; Sannerud, R.; Azfar, M.; Mayer, R.; Calabuig, Á.C.; Swinnen, J.V.; Agostinis, P.; Baekelandt, V.; et al. ATP13A3 Is a Major Component of the Enigmatic Mammalian Polyamine Transport System. J. Biol. Chem. 2021, 296, 100182. [Google Scholar] [CrossRef]
- Available online: https://www.thermofisher.com/order/catalog/product/COL-trpnano16G1B2 (accessed on 20 April 2022).
- Available online: https://www.analytics-shop.com/gb/ag5065-9913-gb.html (accessed on 20 April 2022).
- Nshanian, M.; Lakshmanan, R.; Chen, H.; Loo, R.R.O.; Loo, J.A. Enhancing Sensitivity of Liquid Chromatography–Mass Spectrometry of Peptides and Proteins Using Supercharging Agents. Int. J. Mass Spectrom. 2018, 427, 157–164. [Google Scholar] [CrossRef]
- Maráková, K.; Rai, A.J.; Schug, K.A. Effect of Difluoroacetic Acid and Biological Matrices on the Development of a Liquid Chromatography–Triple Quadrupole Mass Spectrometry Method for Determination of Intact Growth Factor Proteins. J. Sep. Sci. 2020, 43, 1663–1677. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Whiteaker, J.R.; Carr, S.A.; Kuhn, E.; Liu, T.; Massoni, S.A.; Thomas, S.N.; Townsend, R.R.; Zimmerman, L.J.; Boja, E.; et al. Recommendations for the generation, quantification, storage and handling of peptides used for mass spectrometry-based assays. Clin. Chem. 2016, 62, 48–69. [Google Scholar] [CrossRef] [Green Version]
- Gussakovsky, D.; Anderson, G.; Spicer, V.; Krokhin, O.V. Peptide Separation Selectivity in Proteomics LC-MS Experiments: Comparison of Formic and Mixed Formic/Heptafluorobutyric Acids Ion-Pairing Modifiers. J. Sep. Sci. 2020, 43, 3830–3839. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.E.; Silveira, J.R.; Treier, K.M.; Tracy, P.B.; Matthews, D.E. Total Retention Liquid Chromatography-Mass Spectrometry to Achieve Maximum Protein Sequence Coverage. Anal. Chem. 2021, 93, 5054–5060. [Google Scholar] [CrossRef] [PubMed]
- Agilent Technologies. Agilent 1260 Infinity HPLC-Chip Cube Interface (G4240A). Available online: https://www.agilent.com/cs/library/usermanuals/Public/G4240-90006_ChipCube_USR_EN.pdf (accessed on 25 April 2022).
- Pino, L.K.; Just, S.C.; MacCoss, M.J.; Searle, B.C. Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries. Mol. Cell. Proteom. 2020, 19, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnum-Johnson, K.E.; Nie, S.; Casey, C.P.; Monroe, M.E.; Orton, D.J.; Ibrahim, Y.M.; Gritsenko, M.A.; Clauss, T.R.W.; Shukla, A.K.; Moore, R.J.; et al. Simultaneous Proteomic Discovery and Targeted Monitoring Using Liquid Chromatography, Ion Mobility Spectrometry, and Mass Spectrometry. Mol. Cell. Proteom. 2016, 15, 3694–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stow, S.M.; Fjeldsted, J.C. Fundamentals of Uniform-Field Drift Tube Ion Mobility and Collision Cross Section. In Ion Mobility-Mass Spectrometry; RSC Publishing: London, UK, 2021. [Google Scholar]
- Furman, C.; Howsam, M.; Lipka, E. Recent Developments in Separation Methods for Enantiomeric Ratio Determination of Amino Acids Specifically Involved in Cataract and Alzheimer’s Disease. TrAC Trends Anal. Chem. 2021, 141, 116287. [Google Scholar] [CrossRef]
- Abdulbagi, M.; Wang, L.; Siddig, O.; Di, B.; Li, B. D-Amino Acids and D-Amino Acid-Containing Peptides: Potential Disease Biomarkers and Therapeutic Targets? Biomolecules 2021, 11, 1716. [Google Scholar] [CrossRef]
- May, J.C.; Knochenmuss, R.; Fjeldsted, J.C.; McLean, J.A. Resolution of Isomeric Mixtures in Ion Mobility Using a Combined Demultiplexing and Peak Deconvolution Technique. Anal. Chem. 2020, 92, 9482–9492. [Google Scholar] [CrossRef]
- Demelenne, A.; Nys, G.; Nix, C.; Fjeldsted, J.C.; Crommen, J.; Fillet, M. Separation of Phosphorothioated Oligonucleotide Diastereomers Using Multiplexed Drift Tube Ion Mobility Mass Spectrometry. Anal. Chim. Acta 2022, 1191, 339297. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nix, C.; Cobraiville, G.; Gou, M.-J.; Fillet, M. Potential of Single Pulse and Multiplexed Drift-Tube Ion Mobility Spectrometry Coupled to Micropillar Array Column for Proteomics Studies. Int. J. Mol. Sci. 2022, 23, 7497. https://doi.org/10.3390/ijms23147497
Nix C, Cobraiville G, Gou M-J, Fillet M. Potential of Single Pulse and Multiplexed Drift-Tube Ion Mobility Spectrometry Coupled to Micropillar Array Column for Proteomics Studies. International Journal of Molecular Sciences. 2022; 23(14):7497. https://doi.org/10.3390/ijms23147497
Chicago/Turabian StyleNix, Cindy, Gael Cobraiville, Marie-Jia Gou, and Marianne Fillet. 2022. "Potential of Single Pulse and Multiplexed Drift-Tube Ion Mobility Spectrometry Coupled to Micropillar Array Column for Proteomics Studies" International Journal of Molecular Sciences 23, no. 14: 7497. https://doi.org/10.3390/ijms23147497
APA StyleNix, C., Cobraiville, G., Gou, M.-J., & Fillet, M. (2022). Potential of Single Pulse and Multiplexed Drift-Tube Ion Mobility Spectrometry Coupled to Micropillar Array Column for Proteomics Studies. International Journal of Molecular Sciences, 23(14), 7497. https://doi.org/10.3390/ijms23147497