
Thiophene α-Chain-End-Functionalized Oligo(2-methyl-2-oxazoline) as Precursor Amphiphilic Macromonomer for Grafted Conjugated Oligomers/Polymers and as a Multifunctional Material with Relevant Properties for Biomedical Applications

 , and
, and
Abstract

1. Introduction
2. Results and Discussion
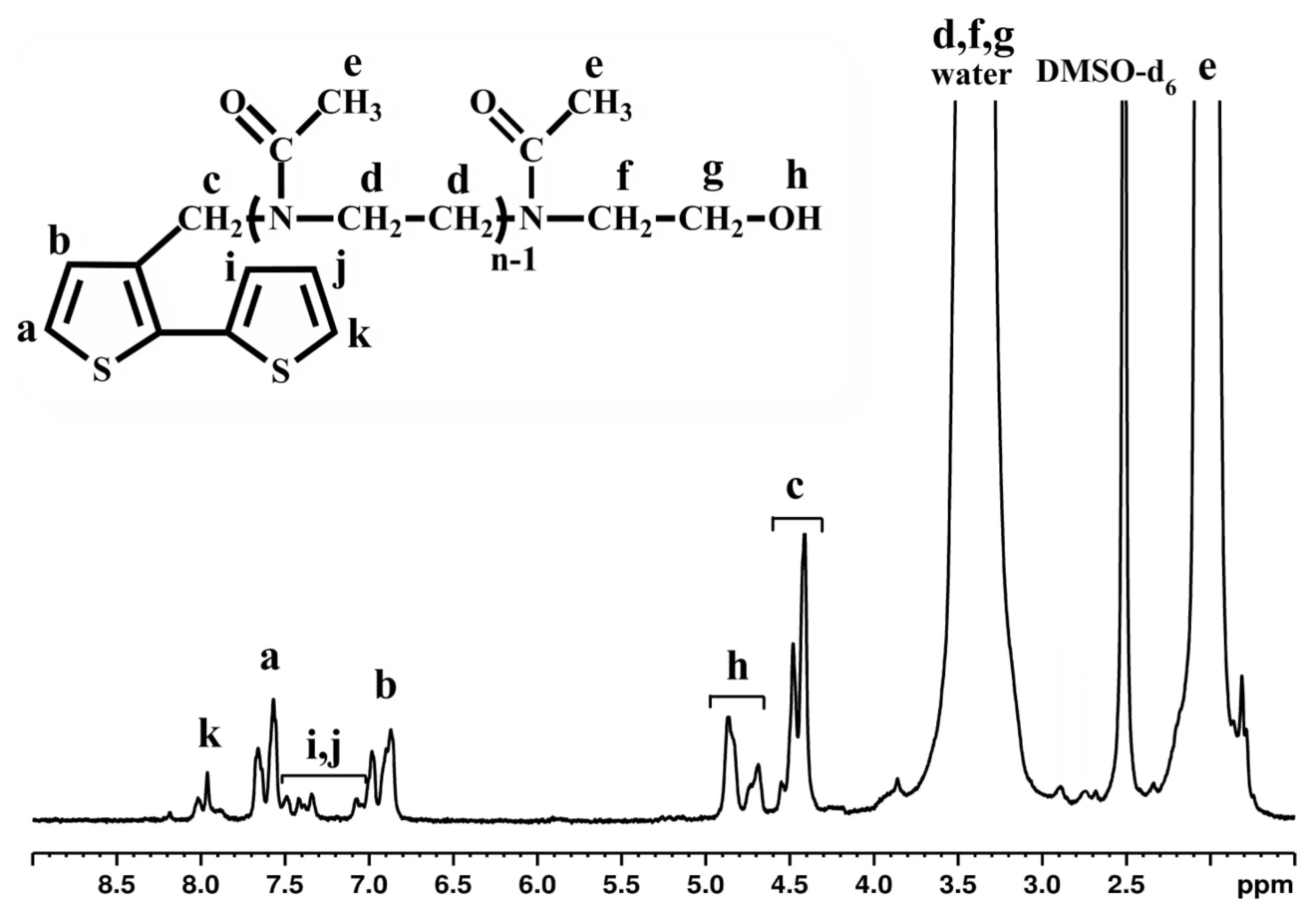
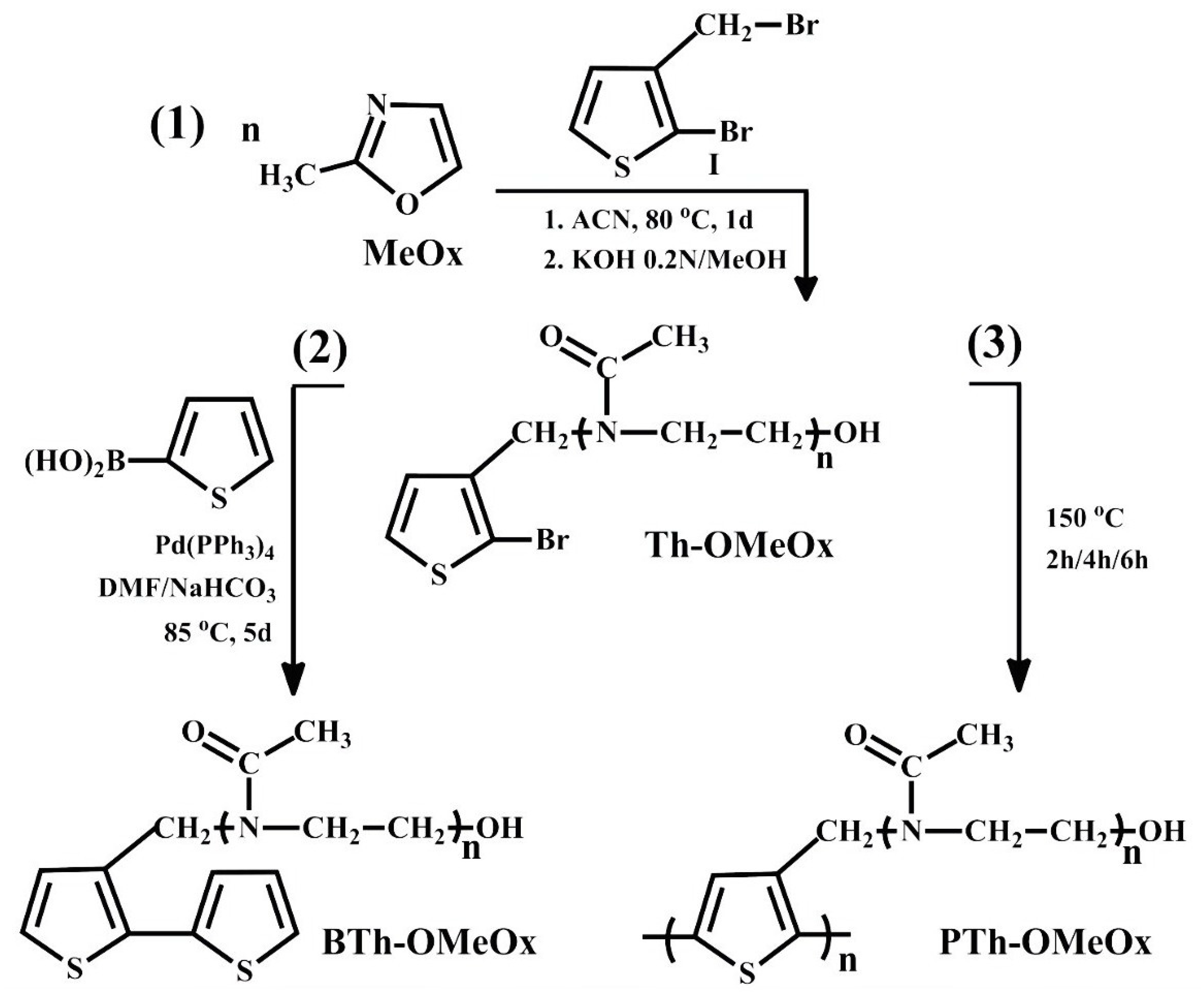
2.1. Design, Synthesis and Structural Characterization of Thiophene-Ended Oligo(2-Methyl-2-Oxazoline) Macromonomer (Th-OMeOx)
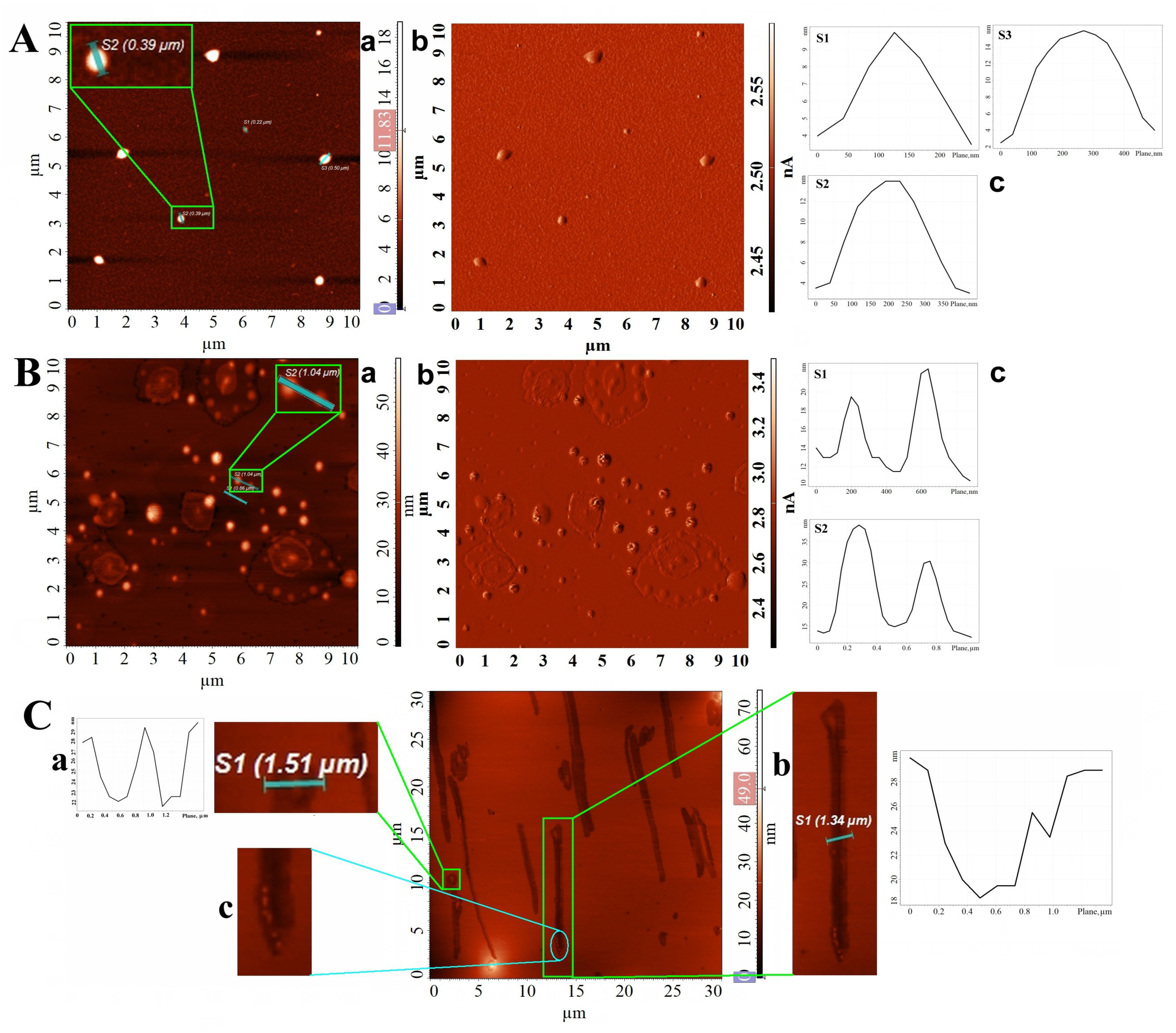
2.2. Properties of Th-OMeOx Macromonomer in Different Solvents and in Thin Films
2.3. Examples of Subsequent Modifications of ThOMeOx
2.3.1. Synthesis of 2,2’-3-OMeOx-Substituted Bithiophene Macromonomer (BTh-OMeOx) by Suzuki Condensation
2.3.2. Self-Acid Assisted Polymerization (SAAP) of Th-OMeOx Macromonomer in Bulk
3. Materials and Methods
3.1. Materials
3.1.1. Synthesis of Thiophene-Ended Oligo(2-methyl-2-oxazoline) Macromonomer (Th-OMeOx)
3.1.2. Synthesis of 2,2’-3-OMeOx-Substituted Bithiophene Macromonomer (BTh-OMeOx) by Suzuki Condensation
3.1.3. SAAP of Th-OMeOx in Bulk
3.2. Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walsha, D.J.; Guironnet, D. Macromolecules with programmable shape, size, and chemistry. Proc. Natl. Acad. Sci. USA 2019, 116, 1538–1542. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, G.; Zapsas, G.; Ntetsikas, K.; Bilalis, P.; Gnanou, Y.; Hadjichristidis, N. 50th Anniversary Perspective: Polymers with Complex Architectures. Macromolecules 2017, 50, 1253–1290. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pispas, S.; Pitsikalis, M. End-functionalized polymers with zwitterionic end-groups. Prog. Polym. Sci. 1999, 24, 875–915. [Google Scholar] [CrossRef]
- Lunn, D.J.; Discekici, E.H.; Read de Alaniz, J.; Gutekunst, W.R.; Hawker, C.J. Established and Emerging Strategies for Polymer Chain-End Modification. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2903–2914. [Google Scholar] [CrossRef]
- Kim, J.; Jung, H.Y.; Park, M.J. End-Group Chemistry and Junction Chemistry in Polymer Science: Past, Present, and Future. Macromolecules 2020, 53, 746–763. [Google Scholar] [CrossRef]
- Zhou, D.; Zhu, L.-W.; Wu, B.-H.; Xu, Z.-K.; Wan, L.-S. End-functionalized polymers by controlled/living radical polymerizations: Synthesis and applications. Polym. Chem. 2022, 13, 300–358. [Google Scholar] [CrossRef]
- Lai, Y.; Lei, Y.; Xu, X.; Li, Y.; He, B.; Gu, Z. Polymeric micelles with π-π conjugated cinnamic acid as lipophilic moieties for doxorubicin delivery. J. Mater. Chem. B 2013, 1, 4289–4296. [Google Scholar] [CrossRef]
- Jiang, Y.; Hadjichristidis, N. Tetraphenylethene-Functionalized Polyethylene-Based Polymers with Aggregation-Induced Emission. Macromolecules 2019, 52, 1955–1964. [Google Scholar] [CrossRef]
- Ito, K. Polymeric Design by Macromonomer Technique. Prog. Polym. Sci. 1998, 23, 581–620. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Pispas, S. The Strength of the Macromonomer Strategy for Complex Macromolecular Architecture: Molecular, Characterization, Properties and Applications of Polymacromonomers. Macromol. Rapid Commun. 2003, 24, 979–1013. [Google Scholar] [CrossRef]
- Rempp, P.; Franta, E.; Masson, P.; Lutz, P. Macromonomers as polymeric intermediates. Synthesis and Applications. Progr. Colloid Polym. Sci. 1986, 72, 112–118. [Google Scholar]
- Yamashita, Y. Synthesis and characterization of functional graft copolymers by macromonomer technique. J. Appl. Polym. Sci.Appl. Polym. Symp. 1981, 36, 193–199. [Google Scholar]
- Neugebauer, D. Macromonomers. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Cianga, I.; Yagci, Y. New polyphenylene-based macromolecular architectures by using well defined macromonomers synthesized via controlled polymerization methods. Prog. Polym. Sci. 2004, 29, 387–399. [Google Scholar] [CrossRef]
- Müllner, M.; Müller, A.H.E. Cylindrical polymer brushes—Anisotropic building blocks, unimolecular templates and particulate nanocarriers. Polymer 2016, 98, 389–401. [Google Scholar] [CrossRef]
- Xie, G.; Martinez, M.R.; Olszewski, M.; Sheiko, S.S.; Matyjaszewski, K. Molecular Bottlebrushes as Novel Materials. Biomacromolecules 2019, 20, 27–54. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, Y. Recent development of brush polymers via polymerization of poly(ethylene glycol)-based macromonomers. Polym. Chem. 2019, 10, 2212–2222. [Google Scholar] [CrossRef]
- Pizzi, D.; Humphries, J.; Morrow, J.P.; Fletcher, N.L.; Bell, C.A.; Thurecht, K.J.; Kempe, K. Poly(2-oxazoline) macromonomers as building blocks for functional and biocompatible polymer architectures. Eur. Polym. J. 2019, 121, 109258. [Google Scholar] [CrossRef]
- Alkan, S.; Toppare, L.; Hepuzer, Y.; Yagci, Y. Block Copolymers of Thiophene-Capped Poly(methyl methacrylate) with Pyrrole. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 4218–4225. [Google Scholar] [CrossRef]
- Mecerreyes, D.; Pomposo, J.A.; Bengoetxea, M.; Grande, H. Novel Pyrrole End-Functional Macromonomers Prepared by Ring-Opening and Atom-Transfer Radical Polymerizations. Macromolecules 2000, 33, 5846–5849. [Google Scholar] [CrossRef]
- Simionescu, C.I.; Grovu-Ivanoiu, M.; Cianga, I.; Grigoras, M.; Duca, A.; Cocarla, I. Electrochemical polymerization of some monomers with Schiff’s base structure. Angew. Makromol. Chem. 1996, 239, 2100341. [Google Scholar] [CrossRef]
- Cianga, I.; Yagci, Y. Polystyrene macromonomer with boronic acid propanediol diester functionality prepared by ATRP for synthesis of comb-like polyphenylenes. Polym. Bull. 2001, 47, 17–24. [Google Scholar] [CrossRef]
- Cianga, I.; Yagci, Y. Synthesis and characterization of comb-like polyphenylenes via Suzuki coupling of polystyrene macromonomers prepared by atom transfer radical polymerization. Eur. Polym. J. 2002, 38, 695–703. [Google Scholar] [CrossRef]
- Papila, O.; Toppare, L.; Yagci, Y.; Cianga, L. Conducting Copolymers of Thiophene-Functionalized Polystyrene. Int. J. Polym. Anal. Charact. 2004, 9, 13–28. [Google Scholar] [CrossRef]
- Sahin, E.; Camurlu, P.; Toppare, L.; Mercore, V.M.; Cianga, I.; Yagci, Y. Synthesis and characterization of thiophene functionalized polystyrene copolymers and their electrochemical properties. Polym. Int. 2005, 54, 1599–1605. [Google Scholar] [CrossRef]
- Colak, D.G.; Cianga, I.; Yagci, Y.; Cirpan, A.; Karasz, F.E. Novel poly(phenylene vinylenes) with well-defined poly(ε-caprolactone) or polystyrene as lateral substituents: Synthesis and characterization. Macromolecules 2007, 40, 5301–5310. [Google Scholar] [CrossRef]
- Wang, Y.; Park, J.S.; Leech, J.P.; Miao, S.; Bunz, U.H.F. Poly(aryleneethynylenes) with Orange, Yellow, Green, and Blue Solid-State Fluorescence. Macromolecules 2007, 40, 1843–1850. [Google Scholar] [CrossRef]
- Molina, B.G.; Bendrea, A.D.; Cianga, L.; Armelin, E.; del Valle, L.J.; Cianga, I.; Alemán, C. The biocompatible polythiophene-g-polycaprolactone copolymer as an efficient dopamine sensor platform. Polym. Chem. 2017, 8, 6112–6122. [Google Scholar] [CrossRef]
- Molina, B.G.; Bendrea, A.-D.; Lanzalaco, S.; Franco, L.; Cianga, L.; del Valle, L.J.; Puiggali, J.; Turon, P.; Armelin, E.; Cianga, I.; et al. Smart design for a flexible, functionalized an electroresponsive hybrid platform based on poly(3,4-ethylenedioxythiophene) derivatives to improve cell viability. J. Mater. Chem. B 2020, 8, 8864–8877. [Google Scholar] [CrossRef]
- Marina, S.; Mantione, D.; Manoj Kumar, K.; Kari, V.; Gutierrez, J.; Tercjak, A.; Sanchez-Sanchez, A.; Mecerreyes, D. New electroactive macromonomers and multi-responsive PEDOT graft copolymers. Polym. Chem. 2018, 9, 3780–3790. [Google Scholar] [CrossRef]
- Dominguez-Alfaro, A.; Gabirondo, E.; Alegret, N.; De León-Almazán, C.M.; Hernandez, R.; Vallejo-Illarramendi, A.; Prato, M.; Mecerreyes, D. 3D Printable Conducting and Biocompatible PEDOT-graft-PLA Copolymers by Direct Ink Writing. Macromol. Rapid Commun. 2021, 42, e2100100. [Google Scholar] [CrossRef]
- Waugaman, M.; Sannigrahi, B.; McGeady, P.; Khan, I.M. Synthesis, characterization and biocompatibility studies of oligosiloxane modified polythiophenes. Eur. Polym. J. 2003, 39, 1405–1412. [Google Scholar] [CrossRef]
- Ohnishi, I.; Hashimoto, K.; Tajima, K. Synthesis of diketopyrrolopyrrole-based polymers with polydimethylsiloxane side chains and their application in organic field-effect transistors. R. Soc. Open Sci. 2018, 5, 172025. [Google Scholar] [CrossRef]
- Fruth, A.; Klapper, M.; Mullen, K. Synthesis and Characterization of Amphiphilic Polyelectrolyte Brush Copolymers Based on Poly(2,7-carbazole). Macromolecules 2010, 43, 467–472. [Google Scholar] [CrossRef]
- Demir, B.; Yilmaz, T.; Guler, E.; Pinar Gumus, Z.; Akbulut, H.; Aldemir, E.; Coskunol, H.; Colak, D.G.; Cianga, I.; Yamada, S.; et al. Polypeptide with electroactive endgroups as sensing platform for the abused drug ‘methamphetamine’ by bioelectrochemical method. Talanta 2016, 161, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, Y.; Huang, X.; Liang, P.; Tang, Y.; Zhang, Y.; Fu, N.; Huang, W.; Dong, X. NIR-Absorbing water-soluble conjugated polymer dots for photoacoustic imaging-guided photothermal/photodynamic synergetic cancer therapy. J. Mater. Chem. B 2018, 6, 7402–7410. [Google Scholar] [CrossRef] [PubMed]
- Molina, B.G.; Cianga, L.; Bendrea, A.-D.; Cianga, I.; del Valle, L.J.; Estrany, F.; Aleman, C.; Armelin, E. Amphiphilic polypyrrole-poly(Schiff base) copolymers with poly(ethylene glycol) side chains: Synthesis, properties and applications. Polym. Chem. 2018, 9, 4218–4232. [Google Scholar] [CrossRef]
- Cianga, L.; Bendrea, A.-D.; Fifere, N.; Nita, L.E.; Doroftei, F.; Ag, D.; Seleci, M.; Timur, S.; Cianga, I. Fluorescent micellar nanoparticles by self assembly of amphiphilic, nonionic and water self-dispersible polythiophenes with “hairy rod” architecture. RSC Adv. 2014, 4, 56385–56405. [Google Scholar] [CrossRef]
- Bendrea, A.-D.; Cianga, L.; Hitruc, E.G.; Titorencu, I.; Cianga, I. Fluorescent Nanoparticles from “Hairy-Rods”, Water-Self Dispersible Amphiphilic Polythiophenes. Mater. Plast. 2013, 50, 71–78. [Google Scholar]
- Bendrea, A.-D.; Fabregat, G.; Cianga, L.; Estrany, F.; del Valle, L.J.; Cianga, I.; Aleman, C. Hybrid materials consisting of an all-conjugated polythiophene backbone and grafted hydrophilic poly(ethylene glycol) chains. Polym. Chem. 2013, 4, 2709–2723. [Google Scholar] [CrossRef]
- Yuksel, M.; Colak, D.G.; Akin, M.; Cianga, I.; Kukut, M.; Medine, E.I.; Can, M.; Sakarya, S.; Unak, P.; Timur, S.; et al. Nonionic, Water Self-Dispersible “Hairy-Rod” Poly(p-phenylene)-g-poly(ethylene glycol) Copolymer/Carbon Nanotube Conjugates for Targeted Cell Imaging. Biomacromolecules 2012, 13, 2680–2691. [Google Scholar] [CrossRef]
- Bendrea, A.-D.; Cianga, L.; Ailiesei, G.-L.; Ursu, E.-L.; Colak, D.G.; Cianga, I. 3,4-Ethylenedioxythiophene (EDOT) End-Group Functionalized Poly-ε-caprolactone (PCL): Self-Assembly in Organic Solvents and Its Coincidentally Observed Peculiar Behavior in Thin Film and Protonated Media. Polymers 2021, 13, 2720. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Z.; Zhang, G.; Zhang, X.; Zhang, D. The Effects of Side Chains on the Charge Mobilities and Functionalities of Semiconducting Conjugated Polymers beyond Solubilities. Adv. Mater. 2019, 31, e1903104. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liao, Y.; Bu, W.; Wang, W.; Sun, J.Z. Going beyond the classical amphiphilicity paradigm: The self-assembly of completely hydrophobic polymers into free-standing sheets and hollow nanostructures in solvents of variable quality. Soft Matter 2016, 12, 5011–5021. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.G.; Cianga, I.; Cianga, L.; Yagci, Y. Synthesis and self-assembly of fluorenevinylene alternating copolymers in “Hairy-Rod” architecture: Side chain-mediated tuning of conformation, microstructure and photophysical properties. Des. Monomers Polym. 2016, 19, 508–534. [Google Scholar] [CrossRef]
- Molina, B.G.; Cianga, L.; Bendrea, A.-D.; Cianga, I.; Alemán, C.; Armelin, E. An amphiphilic, heterografted polythiophene copolymer containing biocompatible/biodegradable side chains for use as an (electro)active surface in biomedical applications. Polym. Chem. 2019, 10, 5010–5022. [Google Scholar] [CrossRef]
- Colak, D.G.; Egbe, D.A.M.; Birckner, E.; Yurteri, S.; Cianga, I.; Tekin, E.; Schubert, U.S.; Yagci, Y. Photophysical properties of PPP and PPV derivatives bearing polystyrene or polycaprolactone as side groups. Eur. Polym. J. 2009, 45, 940–945. [Google Scholar] [CrossRef]
- Uyar, T.; Cianga, I.; Cianga, L.; Besenbacher, F.; Yagci, Y. Self-aligned and bundled electrospun fibers prepared from blends of polystyrene (PS) and poly(methyl methacrylate) (PMMA) with a hairy-rod polyphenylene copolymer. Mater. Lett. 2009, 63, 1638–1641. [Google Scholar] [CrossRef][Green Version]
- Kallitsis, J.K.; Andreopoulou, A.K. Rigid-Flexible and Rod-Coil Copolymers, Macromolecular Architectures and Soft Nano-Objects. In Polymer Science: A Comprehensive Reference, 1st ed.; Moller, M., Matyjaszewski, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Chapter 6.19; Volume 6. [Google Scholar]
- Xu, Z.; Park, K.S.; Diao, Y. What Is the Assembly Pathway of a Conjugated Polymer From Solution to Thin Films? Front. Chem. 2020, 8, 583521. [Google Scholar] [CrossRef]
- Hicks, G.E.J.; Li, S.; Obhi, N.K.; Jarrett-Wilkins, C.N.; Seferos, D.S. Programmable Assembly of π-Conjugated Polymers. Adv. Mater. 2021, 33, 2006287. [Google Scholar] [CrossRef]
- Mullin, W.J.; Sharber, S.A.; Thomas, S.W., III. Optimizing the self-assembly of conjugated polymers and small molecules through structurally programmed non-covalent control. J. Polym. Sci. 2021, 59, 1643–1663. [Google Scholar] [CrossRef]
- Sahin, E.; Camurlu, P.; Toppare, L.; Mercore, V.M.; Cianga, I.; Yagci, Y. Conducting copolymers of thiophene functionalized polystyrenes with thiophene. J. Electroanal. Chem. 2005, 579, 189–197. [Google Scholar] [CrossRef]
- Kurosawa, T.; Chiu, Y.-C.; Zhou, Y.; Gu, X.; Chen, W.-C.; Bao, Z. Impact of Polystyrene Oligomer Side Chains on Naphthalene Diimide–Bithiophene Polymers as n-Type Semiconductors for Organic Field-Effect Transistors. Adv. Funct. Mater. 2016, 26, 1261–1270. [Google Scholar] [CrossRef]
- Wen, H.-F.; Wu, H.-C.; Aimi, J.; Hung, C.-C.; Chiang, Y.-C.; Kuo, C.-C.; Chen, W.-C. Soft Poly(butyl acrylate) Side Chains toward Intrinsically Stretchable Polymeric Semiconductors for Field-Effect Transistor Applications. Macromolecules 2017, 50, 4982–4992. [Google Scholar] [CrossRef]
- Du, W.; Ohayon, D.; Combe, C.; Mottier, L.; Maria, I.P.; Ashraf, R.S.; Fiumelli, H.; Inal, S.; McCulloch, I. Improving the Compatibility of Diketopyrrolopyrrole Semiconducting Polymers for Biological Interfacing by Lysine Attachment. Chem. Mater. 2018, 30, 6164–6172. [Google Scholar] [CrossRef]
- Baek, P.; Aydemir, N.; An, Y.; Chan, E.W.C.; Sokolova, A.; Nelson, A.; Mata, J.P.; McGillivray, D.; Barker, D.; Travas-Sejdic, J. Molecularly Engineered Intrinsically Healable and Stretchable Conducting Polymers. Chem. Mater. 2017, 29, 8850–8858. [Google Scholar] [CrossRef]
- Luo, S.-C. Conducting Polymers as Biointerfaces and Biomaterials: A Perspective for a Special Issue of Polymer Reviews. Polym. Rev. 2013, 53, 303–310. [Google Scholar] [CrossRef]
- Baek, P.; Voorhaar, L.; Barker, D.; Travas-Sejdic, J. Molecular Approach to Conjugated Polymers with Biomimetic Properties. Acc. Chem. Res. 2018, 51, 1581–1589. [Google Scholar] [CrossRef]
- Bendrea, A.-D.; Cianga, L.; Cianga, I. Review paper: Progress in the Field of Conducting Polymers for Tissue Engineering Applications. J. Biomater. Appl. 2011, 26, 3–84. [Google Scholar] [CrossRef]
- Petty, A.J.; Keate, R.L.; Jiang, B.; Ameer, G.A.; Rivnay, J. Conducting Polymers for Tissue Regeneration in vivo. Chem. Mater. 2020, 32, 4095–4115. [Google Scholar] [CrossRef]
- Wei, J.; Liu, Y.; Yu, J.; Chen, L.; Luo, M.; Yang, L.; Li, P.; Li, S.; Zhang, X.-H. Conjugated Polymers: Optical Toolbox for Bioimagingand Cancer Therapy. Small 2021, 17, 2103127. [Google Scholar] [CrossRef]
- Borges-Gonzalez, J.; Kousseff, C.J.; Nielsen, C.B. Organic semiconductors for biological sensing. J. Mater. Chem. C 2019, 7, 1111–1130. [Google Scholar] [CrossRef]
- Sista, P.; Ghosh, K.; Martinez, J.S.; Rocha, R.C. Polythiophenes in Biological Applications. J. Nanosci. Nanotechnol. 2014, 14, 250–272. [Google Scholar] [CrossRef] [PubMed]
- So, R.C.; Carreon-Asok, A.C. Molecular Design, Synthetic Strategies, and Applications of Cationic Polythiophenes. Chem. Rev. 2019, 119, 11442–11509. [Google Scholar] [CrossRef] [PubMed]
- Zangoli, M.; DiMaria, F. Synthesis, characterization, and biological applications of semiconducting polythiophene-based nanoparticles. View 2021, 2, 20200086. [Google Scholar] [CrossRef]
- Kim, H.-C.; Kim, E.; Lee, S.G.; Lee, S.J.; Jeong, S.W.; Ha, T.-L.; Lee, B.; Lee, S.W. Folic Acid-Functionalized Polythiophene for Targeted Cellular Imaging. J. Nanosci. Nanotechnol. 2016, 16, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Guler, E.; Akbulut, H.; Geyik, C.; Yilmaz, T.; Gumus, Z.P.; Barlas, F.B.; Ahan, R.E.; Demirkol, D.O.; Yamada, S.; Endo, T.; et al. Complex Structured Fluorescent Polythiophene Graft Copolymer as a Versatile Tool for Imaging, Targeted Delivery of Paclitaxel, and Radiotherapy. Biomacromolecules 2016, 17, 2399–2408. [Google Scholar] [CrossRef]
- Bendrea, A.-D.; Fabregat, G.; Torras, J.; Maione, S.; Cianga, L.; del Valle, L.J.; Cianga, I.; Aleman, C. Polythiophene-g-poly(ethylene glycol) graft copolymers for electroactive scaffolds. J. Mater. Chem. B 2013, 1, 4135–4145. [Google Scholar] [CrossRef]
- Maione, S.; Fabregat, G.; del Valle, L.J.; Bendrea, A.-D.; Cianga, L.; Cianga, I.; Estrany, F.; Aleman, C. Effect of the Graft Ratio on the Properties of Polythiophene-g-poly(ethylene glycol). J. Polym. Sci. Part B Polym. Phys. 2015, 53, 239–252. [Google Scholar] [CrossRef]
- Perez-Madrigal, M.M.; Cianga, L.; del Valle, L.J.; Cianga, I.; Aleman, C. Electroactive and bioactive films of random copolymers containing terthiophene, carboxyl and Schiff base functionalities in the main chain. Polym. Chem. 2015, 6, 4319–4335. [Google Scholar] [CrossRef]
- Aydin, M.; Aydin, E.B.; Sezginturk, M.K. A Highly Selective Poly(thiophene)-graft-Poly(methacrylamide) Polymer Modified ITO Electrode for Neuron Specific Enolase Detection in Human Serum. Macromol. Biosci. 2019, 19, 1900109. [Google Scholar] [CrossRef]
- Lorson, T.; Lübtow, M.M.; Wegener, E.; Haider, M.S.; Borova, S.; Nahm, D.; Jordan, R.; Sokolski-Papkov, M.; Kabanov, A.V.; Luxenhofer, R. Poly(2-oxazoline)s based biomaterials: A comprehensive and critical update. Biomaterials 2018, 178, 204–280. [Google Scholar] [CrossRef] [PubMed]
- Vlassi, E.; Papagiannopoulos, A.; Pispas, S. Amphiphilic poly(2-oxazoline) copolymers as self-assembled carriers for drug delivery applications. Eur. Polym. J. 2017, 88, 516–523. [Google Scholar] [CrossRef]
- Akbar, M.; Cagli, E.; Erel-Göktepe, I. Layer-By-Layer Modified Superparamagnetic Iron Oxide Nanoparticles with Stimuli Responsive Drug Release Properties. Macromol. Chem. Phys. 2019, 220, 1800422. [Google Scholar] [CrossRef]
- You, Y.; Kobayashi, K.; Colak, B.; Luo, P.; Cozens, E.; Fields, L.; Suzuki, K.; Gautrot, J. Engineered cell-degradable poly(2-alkyl-2-oxazoline) hydrogel for epicardial placement of mesenchymal stem cells for myocardial repair. Biomaterials 2021, 269, 120356. [Google Scholar] [CrossRef] [PubMed]
- Cagli, E.; Ugur, E.; Ulusan, S.; Banerjee, S.; Erel-Goktepe, I. Effect of side chain variation on surface and biological properties of poly(2-alkyl-2-oxazoline) multilayers. Eur. Polym. J. 2019, 114, 452–463. [Google Scholar] [CrossRef]
- Grube, M.; Leiske, M.N.; Schubert, U.S.; Nischang, I. POx as an Alternative to PEG? A Hydrodynamic and Light Scattering Study. Macromolecules 2018, 51, 1905–1916. [Google Scholar] [CrossRef]
- Lubich, C.; Allacher, P.; de la Rosa, M.; Bauer, A.; Prenninger, T.; Horling, F.M.; Siekmann, J.; Oldenburg, J.; Scheiflinger, F.; Reipert, B.M. The Mystery of Antibodies Against Polyethylene Glycol (PEG)—What do we Know? Pharm. Res. 2016, 33, 2239–2249. [Google Scholar] [CrossRef]
- Benski, L.; Tiller, J.C. Telechelic biocidal poly(2-oxazoline)s and polycations. Eur. Polym. J. 2019, 120, 109233. [Google Scholar] [CrossRef]
- Cirpan, A.; Alkan, S.; Toppare, L.; David, G.; Yagci, Y. Synthesis and electroactivity of pyrrole-functionalized poly(2-methy-2-oxazoline). Eur. Polym. J. 2001, 37, 2225–2229. [Google Scholar] [CrossRef]
- Wang, Y.; Wilson, J.N.; Smith, M.D.; Bunz, U.H.F. TEMPO-Substituted PPEs: Polystyrene-PPE Graft Copolymers and Double Graft Copolymers. Macromolecules 2004, 37, 9701–9708. [Google Scholar] [CrossRef]
- Demirel, L.A.; Yurteri, S.; Cianga, I.; Yagci, Y. Layered Morphology of Poly(phenylene)s in Thin Films Induced by Substitution of Well-Defined Poly(ε-caprolactone) Side Chains. Macromolecules 2005, 38, 6402–6410. [Google Scholar] [CrossRef]
- Cianga, I.; Mercore, V.M.; Grigoras, M.; Yagci, Y. Poly(thienyl-phenylene)s with macromolecular side chains by oxidative polymerization of well-defined macromonomers. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 848–865. [Google Scholar] [CrossRef]
- Demirel, L.A.; Yurteri, S.; Cianga, I.; Yagci, Y. Synthesis and Morphological Characterization of Poly(ϵ-caprolactone) and Poly(2-methyl-oxazoline) substituted Phenyl Rings and Phenylene Oligomers. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2091–2104. [Google Scholar] [CrossRef]
- Wang, Y.; Andrew, J.Z.; Wilson, J.N.; Kim, I.-B.; Solntsev, K.M.; Tolbert, L.M.; Bunz, U.H.F. Optical Spectroscopy of Grafted Poly(p-phenyleneethynylene)s in Water and Water-DMF Mixtures. Macromolecules 2008, 41, 1112–1117. [Google Scholar] [CrossRef]
- Alemseghed, M.G.; Servello, J.; Hundt, N.; Sista, P.; Biewer, M.C.; Stefan, M.C. Amphiphilic Block Copolymers Containing Regioregular Poly(3-hexylthiophene) and Poly(2-ethyl-2-oxazoline. Macromol. Chem. Phys. 2010, 211, 1291–1297. [Google Scholar] [CrossRef]
- Chan, E.W.C.; Baek, P.; De la Rosa, V.R.; Barker, D.; Hoogenboom, R.; Travas-Sejdic, J. Thermoresponsive laterally-branched polythiophene phenylene derivative as water-soluble temperature sensor. Polym. Chem. 2017, 8, 4352–4358. [Google Scholar] [CrossRef]
- Creamer, A.; Wood, C.S.; Howes, P.D.; Casey, A.; Cong, S.; Marsh, A.V.; Godin, R.; Panidi, J.; Anthopoulos, T.D.; Burgess, C.H.; et al. Post-polymerisationfunctionalisation of conjugated polymer backbones and its application in multifunctional emissive nanoparticles. Nat. Commun. 2018, 9, 3237. [Google Scholar] [CrossRef]
- Lam, K.H.; Foong, T.R.B.; Ooi, Z.E.; Zhang, J.; Grimsdale, A.C.; Lam, Y.M. Enhancing the Performance of Solution-Processed Bulk-Heterojunction Solar Cells Using Hydrogen-Bonding-Induced Self-Organization of Small Molecules. ACS Appl. Mater. Interfaces 2013, 5, 13265–13274. [Google Scholar] [CrossRef]
- Jin, R.-H.; Motoyoshi, K.-I. Porphyrin-centered Water-soluble Star-shaped Polymers: Poly(N-acetylethylenimine) and Poly(ethylenimine) Arms. J. Porphyr. Phthalocyanines 1999, 3, 60–64. [Google Scholar] [CrossRef]
- Bose, A.; Jana, S.; Saha, A.; Mandal, T.K. Amphiphilic polypeptide-polyoxazoline graft copolymer conjugate with tunable thermoresponsiveness: Synthesis and self-assembly into various micellar structures in aqueous and nonaqueous media. Polymer 2017, 110, 12–24. [Google Scholar] [CrossRef]
- Heller, L.E.; Whitleigh, J.; Roth, D.F.; Oherlein, E.M.; Lucci, F.R.; Kolonko, K.J.; Plass, K.E. Self-Assembly of Isomeric Monofunctionalized Thiophenes. Langmuir 2012, 28, 14855–14859. [Google Scholar] [CrossRef] [PubMed]
- Glassner, M.; Vergaelen, M.; Hoogenboom, R. Poly(2-oxazoline)s: A comprehensive overview of polymer structures and their physical properties. Polym. Int. 2018, 67, 32–45. [Google Scholar] [CrossRef]
- Yuan, W.Z.; Zhang, Y. Nonconventional Macromolecular Luminogens with Aggregation-Induced Emission Characteristics. J.Polym. Sci. Part A Polym. Chem. 2017, 55, 560–574. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Klajnert-Maculewicz, B.; Johnson, K.A.-M.; Brinkman, H.F.; Janaszewska, A.; Hedstrand, D.M. Non-traditional intrinsic luminescence: Inexplicable blue fluorescence observed for dendrimers, macromolecules and small molecular structures lacking traditional/conventional luminophores. Prog. Polym. Sci. 2019, 90, 35–117. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Zhang, H.; Sun, J.Z.; Tang, B.Z. The mysterious blue emission around 440 nm in carbonyl-based aliphatic clusteroluminogens. J. Polym. Sci. 2022, 9. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Li, S.; Lei, D.; Tang, B.Z.; Ye, R. Recent Advances in Clusteroluminescence. Top. Curr. Chem. 2021, 379, 14. [Google Scholar] [CrossRef]
- Liao, P.; Huang, J.; Yan, Y.; Tang, B.Z. Clusterization-triggered emission (CTE): One for all, all for one. Mater. Chem. Front. 2021, 5, 6693–6717. [Google Scholar] [CrossRef]
- Jiang, N.; Zhu, D.; Su, Z.; Bryce, M.R. Recent advances in oligomers/polymers with unconventional chromophores. Mater. Chem. Front. 2021, 5, 60–75. [Google Scholar] [CrossRef]
- Tang, S.; Yang, T.; Zhao, Z.; Zhu, T.; Zhang, Q.; Hou, W.; Yuan, W.Z. Nonconventional luminophores: Characteristics, advancements and perspectives. Chem. Soc. Rev. 2021, 50, 12616–12655. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, B.Z. Through-Space Interactions in Clusteroluminescence. JACS Au 2021, 1, 1805–1814. [Google Scholar] [CrossRef]
- Zhou, X.; Luo, W.; Nie, H.; Xu, L.; Hu, R.; Zhao, Z.; Qin, A.; Tang, B.Z. Oligo(maleic anhydride)s: A platform for unveiling the mechanism of clusteroluminescence of non-aromatic polymers. J. Mater. Chem. C 2017, 5, 4775–4779. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, Z.; Dou, X.; Wang, Y.; Liu, S.; Zhang, Y.; Yuan, W.Z. Emission mechanism understanding and tunable persistent room temperature phosphorescence of amorphous nonaromatic polymers. Mater. Chem. Front. 2019, 3, 257–264. [Google Scholar] [CrossRef]
- Naumann, C.A.; Brooks, C.F.; Fuller, G.G.; Lehmann, T.; Ruhe, J.; Knoll, W.; Kuhn, P.; Nuyken, O.; Frank, C.W. Two-Dimensional Physical Networks of Lipopolymers at the Air/Water Interface: Correlation of Molecular Structure and Surface Rheological Behavior. Langmuir 2001, 17, 2801–2806. [Google Scholar] [CrossRef]
- Kroning, A.; Furchner, A.; Adam, S.; Uhlmann, P.; Hinrichs, K. Probing carbonyl-water hydrogen-bond interactions in thin polyoxazoline brushes. Biointerphases 2016, 11, 019005. [Google Scholar] [CrossRef]
- del Mercato, L.L.; Pompa, P.P.; Maruccio, G.; Torre, A.D.; Sabella, S.; Tamburro, A.M.; Cingolani, R.; Rinaldi, R. Charge transport and intrinsic fluorescence in amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2007, 104, 18019–18024. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Correia, V.G.; Pinho, M.G.; Lima, J.C.; Aguiar-Ricardo, A. Blue emission of carbamic acid oligooxazoline biotags. Mat. Lett. 2012, 81, 205–208. [Google Scholar] [CrossRef]
- Du, J.; Willcock, H.; Patterson, J.P.; Portman, I.; O’Reilly, R.K. Self-Assembly of Hydrophilic Homopolymers: A Matter of RAFT End Groups. Small 2011, 7, 2070–2080. [Google Scholar] [CrossRef]
- Rettler, E.F.-J.; Lambermont-Thijs, H.M.L.; Kranenburg, J.M.; Hoogenboom, R.; Unger, M.V.; Siesler, H.W.; Schubert, U.S. Water uptake of poly(2-N-alkyl-2-oxazoline)s: Influence of crystallinity and hydrogen-bonding on the mechanical properties. J. Mater. Chem. 2011, 21, 17331–17337. [Google Scholar] [CrossRef]
- Kolodziejczyk, B.; Mayevsky, D.; Winther-Jensen, B. Enhanced absorption spectra of conducting polymers co-polymerised from thiophene derivatives. RSC Adv. 2013, 3, 4568–4573. [Google Scholar] [CrossRef]
- Hoffmann, K.J.; Bakken, E.; Samuelsen, E.J.; Carlsen, P.H.J. Synthesis and polymerization of 3,3′-dialkyl-2,2′-bithiophenes. Synth. Met. 2000, 113, 39–44. [Google Scholar] [CrossRef]
- Ogoshi, T.; Kim, K.-M.; Chujo, Y. Synthesis and characterization of transparent poly(2-methyl-2-oxazoline) (POZO)-vanadium oxide (V2O5) hybrids with reversible formation. J. Mater. Chem. 2003, 13, 2202–2207. [Google Scholar] [CrossRef]
- Available online: https://www.sigmaaldrich.com/RO/en/product/aldrich/241636 (accessed on 29 May 2022).
- Pisuchpena, T.; Keaw-on, N.; Kitikulvarakorn, K.; Kusonsong, S.; Sritana-ananta, Y.; Supaphol, P.; Hovena, V.P. Electrospinning and solid state polymerization: A simple and versatile route to conducting PEDOT composite films. Eur. Polym. J. 2017, 96, 452–462. [Google Scholar] [CrossRef]
- Meng, H.; Perepichka, D.F.; Wudl, F. Facile Solid-State Synthesis of Highly Conducting Poly(ethylenedioxythiophene). Angew. Chem. Int. Ed. 2003, 42, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Spencer, H.J.; Berridge, R.; Crouch, D.J.; Wright, S.P.; Giles, M.; McCulloch, I.; Coles, S.J.; Hursthouse, M.B.; Skabara, P.J. Further evidence for spontaneous solid-state polymerization reactions in 2,5-dibromothiophene derivatives. J. Mater. Chem. 2003, 13, 2075–2077. [Google Scholar] [CrossRef]
- Patra, A.; Wijsboom, Y.H.; Leitus, G.; Bendikov, M. Tuning the Band Gap of Low-Band-Gap Polyselenophenes and Polythiophenes: The Effect of the Heteroatom. Chem. Mater. 2011, 23, 896–906. [Google Scholar] [CrossRef]
- Gulprasertrat, N.; Chapromma, J.; Aree, T.; Sritana-anant, Y. Synthesis of functionalizable derivatives of 3,4 ethylenedioxythiophene and their solid-state polymerizations. J. Appl. Polym. Sci. 2015, 132, 42233. [Google Scholar] [CrossRef]
- Wagner, P.; Jolley, K.W.; Officer, D.L. Why Do Some Alkoxybromothiophenes Spontaneously Polymerize? Aust. J. Chem. 2011, 64, 335–338. [Google Scholar] [CrossRef]
- Yin, Y.; Li, Z.; Jin, J.; Tusy, C.; Xia, J. Facile synthesis of poly(3,4-ethylenedioxythiophene) by acid-assisted polycondensation of 5-bromo-2,3-dihydro-thieno[3,4-b][1,4]dioxine. Synth. Met. 2013, 175, 97–102. [Google Scholar] [CrossRef]
- Bonillo, B.; Swager, T.M. Chain-Growth Polymerization of 2-Chlorothiophenes Promoted by Lewis Acids. J. Am. Chem. Soc. 2012, 134, 18916–18919. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Ku, T.-C.; Shih, H.-P.; Suman, A.; Lin, H.-J.; Shih, T.-W.; Han, C.-C. Chain-growth cationic polymerization of 2-halogenated thiophenes promoted by Brønsted acids. Polym. Chem. 2014, 5, 5928–5941. [Google Scholar] [CrossRef]
- Tusy, C.; Jiang, K.; Peng, K.; Huang, L.; Xia, J. Effect of monomers’ structure on self-acid assisted polycondensation for the synthesis of poly(3,4 ethylenedioxythiophene) and homopolythiophene. Polym. Chem. 2015, 6, 1014–1022. [Google Scholar] [CrossRef]
- Jiang, K.; Cai, X.; Liu, X.; Xia, J. Exploring functionalized polythiophene derivatives based on thiophene-linker-thiophene platform, analysis of prototype monomer crystal for C-Br/C-H bulk polycondensation and its application for acid detection. Polymer 2019, 168, 70–76. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Sun, H.-S.; Yang, H.-R.; Lai, Y.-Y.; Hou, K.-Y.; Liu, Y.-H. Aqueous Palladium-Catalyzed Direct Arylation Polymerization of 2 Bromothiophene Derivatives. Macromol. Rapid Commun. 2020, 41, e2000021. [Google Scholar] [CrossRef] [PubMed]
- ACCU DYNE TEST. Available online: https://www.accudynetest.com/solubility_table.html?sortby=sort_h_h#007 (accessed on 9 May 2022).
- Hansen, C.M. Hansen Solubility Parameteres: A User’s Handbook, 2nd ed.; Taylor & Francis Group, LLC; CRC Press: Boca Raton, FL, USA, 2007; p. 361. [Google Scholar]
- Lübtow, M.M.; Haider, M.S.; Kirsch, M.; Klisch, S.; Luxenhofer, R. Like Dissolves Like? A Comprehensive Evaluation of Partial Solubility Parameters to Predict Polymer-Drug Compatibility in Ultrahigh Drug-Loaded Polymer Micelles. Biomacromolecules 2019, 20, 3041–3056. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yuan, Y.; Jiang, R.; Fu, N.; Lu, X.; Tian, C.; Hu, W.; Fan, Q.; Huang, W. Homogeneous near-infrared emissive polymeric nanoparticles based on amphiphilic diblock copolymers with perylene diimide and PEG pendants: Self-assembly behavior and cellular imaging application. Polym. Chem. 2014, 5, 1372–1380. [Google Scholar] [CrossRef]
- Boccia, A.C.; Lukes, V.; Eckstein-Andicsova, A.; Kozma, E. Solvent- and concentration-induced self-assembly of an amphiphilic perylene dye. New J. Chem. 2020, 44, 892–899. [Google Scholar] [CrossRef]
- Walderhaug, H.; Söderman, O. NMR studies of block copolymer micelles. Curr. Opin. Colloid Interface Sci. 2009, 14, 171–177. [Google Scholar] [CrossRef]
- Heald, C.R.; Stolnik, S.; Kujawinski, K.S.; De Matteis, C.; Garnett, M.C.; Illum, L.; Davis, S.S.; Purkiss, S.C.; Barlow, R.J.; Gellert, P.R. Poly(lactic acid)-Poly(ethylene oxide) (PLA-PEG) Nanoparticles: NMR Studies of the Central Solid-like PLA Core and the Liquid PEG Corona. Langmuir 2002, 18, 3669–3675. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, Y.J.; Jang, W.D. Multimodal Stimuli-Responsive Fluorophore-Functionalized Heterotelechelic Poly(2-isopropyl-2-oxazoline). ACS Appl. Polym. Mater. 2020, 2, 3535–3542. [Google Scholar] [CrossRef]
- Rayeroux, D.; Travelet, C.; Lapinte, V.; Borsali, R.; Robin, J.J.; Bouilhac, C. Tunable amphiphilic graft copolymers bearing fatty chains and polyoxazoline: Synthesis and self-assembly behavior in solution. Polym. Chem. 2017, 8, 4246–4263. [Google Scholar] [CrossRef]
- Volet, G.; Auvray, L.; Amiel, C. Monoalkyl Poly(2-methyl-2-oxazoline) Micelles. A Small-Angle Neutron Scattering Study. J. Phys. Chem. B 2009, 113, 13536–13544. [Google Scholar] [CrossRef] [PubMed]
- Volet, G.; Chanthavong, V.; Wintgens, V.; Amiel, C. Synthesis of Monoalkyl End-Capped Poly(2-methyl-2-oxazoline) and Its Micelle Formation in Aqueous Solution. Macromolecules 2005, 38, 5190–5197. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent (conc., mg/mL) | Size of the Self-Assembled Structures (nm) a | λmaxem (nm) b (λex = 315 nm) | λmaxem (nm) b (λex = 330 nm) |
|---|---|---|---|
| water (2) | 496 | 360 (sh); 405 | 381; 413 |
| water (1.5) | 278 | 356 (sh); 407 | 378; 417 |
| water (1) | 251 | 358; 408 | 378; 418 |
| water (0.7) | 248 | 354; 410 | 373; 422 |
| water (0.5) | 520 | 357; 407 | 375; 417 |
| Chl (1) | 627 | 396; 490 (sh); 540 (sh) | 400; 420; 450 (sh); 476 (sh) |
| THF (1) | 1.05 (20%) 875 (70%) 4428 (10%) | 349 (sh); 365; 373 (sh) | 370; 382 (sh) |
| Sample No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Temperature (°C) | 65 | 80 | 90 | 100 | 130 | 150 | 150 | 150 | 150 |
| Reaction time (h) | 24 | 24 | 24 | 24 | 4 | 2 | 4 | 4 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bendrea, A.-D.; Cianga, L.; Ailiesei, G.-L.; Göen Colak, D.; Popescu, I.; Cianga, I. Thiophene α-Chain-End-Functionalized Oligo(2-methyl-2-oxazoline) as Precursor Amphiphilic Macromonomer for Grafted Conjugated Oligomers/Polymers and as a Multifunctional Material with Relevant Properties for Biomedical Applications. Int. J. Mol. Sci. 2022, 23, 7495. https://doi.org/10.3390/ijms23147495
Bendrea A-D, Cianga L, Ailiesei G-L, Göen Colak D, Popescu I, Cianga I. Thiophene α-Chain-End-Functionalized Oligo(2-methyl-2-oxazoline) as Precursor Amphiphilic Macromonomer for Grafted Conjugated Oligomers/Polymers and as a Multifunctional Material with Relevant Properties for Biomedical Applications. International Journal of Molecular Sciences. 2022; 23(14):7495. https://doi.org/10.3390/ijms23147495
Chicago/Turabian StyleBendrea, Anca-Dana, Luminita Cianga, Gabriela-Liliana Ailiesei, Demet Göen Colak, Irina Popescu, and Ioan Cianga. 2022. "Thiophene α-Chain-End-Functionalized Oligo(2-methyl-2-oxazoline) as Precursor Amphiphilic Macromonomer for Grafted Conjugated Oligomers/Polymers and as a Multifunctional Material with Relevant Properties for Biomedical Applications" International Journal of Molecular Sciences 23, no. 14: 7495. https://doi.org/10.3390/ijms23147495
APA StyleBendrea, A.-D., Cianga, L., Ailiesei, G.-L., Göen Colak, D., Popescu, I., & Cianga, I. (2022). Thiophene α-Chain-End-Functionalized Oligo(2-methyl-2-oxazoline) as Precursor Amphiphilic Macromonomer for Grafted Conjugated Oligomers/Polymers and as a Multifunctional Material with Relevant Properties for Biomedical Applications. International Journal of Molecular Sciences, 23(14), 7495. https://doi.org/10.3390/ijms23147495