
NRN1 Gene as a Potential Marker of Early-Onset Schizophrenia: Evidence from Genetic and Neuroimaging Approaches

 , , , , , , , ,
, , , , , , , ,
Abstract
1. Introduction
2. Results
2.1. Genetic Association Analyses
2.1.1. Family-Based
2.1.2. Case–Control
2.2. Neuroimaging Genetic Association Analyses
2.2.1. N-Back Functional Response
2.2.2. N-Back Behavioural Response
3. Discussion
4. Materials and Methods
4.1. Sample
4.2. Genotyping
4.3. fMRI Task Description and Acquisition Parameters
4.3.1. N-Back Task
4.3.2. N-Back Performance Data
4.3.3. fMRI Acquisition Parameters
4.4. Statistical Analyses
4.4.1. Design
4.4.2. Genetic Association Analyses
4.4.3. Neuroimaging Association Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Heritability of schizophrenia and schizophrenia spectrum based on the nationwide Danish Twin register. Biol. Psychiatry 2018, 83, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic Loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R. Biological insights from 108 schizophrenia-associated genetic Loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Liang, S.G.; Greenwood, T.A. The impact of clinical heterogeneity in schizophrenia on genomic analyses. Schizophr. Res. 2015, 161, 490–495. [Google Scholar] [CrossRef]
- Glahn, D.C.; Nimgaonkar, V.L.; Raventós, H.; Contreras, J.; McIntosh, A.M.; Thomson, P.A.; Jablensky, A.; McCarthy, N.S.; Charlesworth, J.C.; Blackburn, N.B.; et al. Rediscovering the value of families for psychiatric genetics research. Mol. Psychiatry 2019, 24, 523–535. [Google Scholar] [CrossRef]
- Lange, E.M.; Sun, J.; Lange, L.A.; Zheng, S.L.; Duggan, D.; Carpten, J.D.; Gronberg, H.; Isaacs, W.B.; Xu, J.; Chang, B.-L. Family-based samples can play an important role in genetic association studies. Cancer Epidemiol. Prev. Biomark. 2008, 17, 2208–2214. [Google Scholar] [CrossRef][Green Version]
- Hare, E.; Glahn, D.C.; Dassori, A.; Raventos, H.; Nicolini, H.; Ontiveros, A.; Medina, R.; Mendoza, R.; Jerez, A.; Muñoz, R.; et al. Heritability of age of onset of psychosis in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 298–302. [Google Scholar] [CrossRef]
- Esterberg, M.L.; Trotman, H.D.; Holtzman, C.; Compton, M.T.; Walker, E.F. The impact of a family history of psychosis on age-at-onset and positive and negative symptoms of schizophrenia: A meta-analysis. Schizophr. Res. 2010, 120, 121–130. [Google Scholar] [CrossRef]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Is an early age at illness onset in schizophrenia associated with increased genetic susceptibility? Analysis of data from the nationwide Danish Twin register. EBioMedicine 2017, 18, 320–326. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hollis, C. Adult outcomes of child- and adolescent-onset schizophrenia: Diagnostic stability and predictive validity. Am. J. Psychiatry 2000, 157, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, J.K.; Asarnow, R.F. Genetics of childhood-onset schizophrenia: 2019 update. Child Adolesc. Psychiatr. Clin. N. Am. 2020, 29, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.K.; Friis, S.; Haahr, U.; Johannessen, J.O.; Melle, I.; Opjordsmoen, S.; Rund, B.R.; Simonsen, E.; Vaglum, P.; McGlashan, T.H. Premorbid adjustment in first-episode non-affective psychosis: Distinct patterns of pre-onset course. Br. J. Psychiatry 2004, 185, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Rajji, T.K.; Ismail, Z.; Mulsant, B.H. Age at onset and cognition in schizophrenia: Meta-analysis. Br. J. Psychiatry 2009, 195, 286–293. [Google Scholar] [CrossRef]
- Clemmensen, L.; Vernal, D.L.; Steinhausen, H.-C. A systematic review of the long-term outcome of early onset schizophrenia. BMC Psychiatry 2012, 12, 150. [Google Scholar] [CrossRef]
- Rapoport, J.L.; Addington, A.M.; Frangou, S.; Psych, M.R.C. The neurodevelopmental model of schizophrenia: Update. Mol. Psychiatry 2005, 10, 434–449. [Google Scholar] [CrossRef]
- Rapoport, J.L.; Gogtay, N. Childhood onset schizophrenia: Support for a progressive neurodevelopmental disorder. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2011, 29, 251–258. [Google Scholar] [CrossRef]
- Bergen, S.E.; O’Dushlaine, C.T.; Lee, P.H.; Fanous, A.H.; Ruderfer, D.M.; Ripke, S.; Sullivan, P.F.; Smoller, J.W.; Purcell, S.M.; Corvin, A. Genetic modifiers and subtypes in schizophrenia: Investigations of age at onset, severity, sex and family history. Schizophr. Res. 2014, 154, 48–53. [Google Scholar] [CrossRef]
- Guo, S.; Liu, J.; Li, W.; Yang, Y.; Lv, L.; Xiao, X.; Li, M.; Guan, F.; Luo, X.-J. Genome wide association study identifies four loci for early onset schizophrenia. Transl. Psychiatry 2021, 11, 248. [Google Scholar] [CrossRef]
- Wang, K.-S.; Liu, X.; Zhang, Q.; Aragam, N.; Pan, Y. Genome-wide association analysis of age at onset in schizophrenia in a European-American sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Woolston, A.L.; Hsiao, P.-C.; Kuo, P.-H.; Wang, S.-H.; Lien, Y.-J.; Liu, C.-M.; Hwu, H.-G.; Lu, T.-P.; Chuang, E.Y.; Chang, L.-C.; et al. Genetic Loci associated with an earlier age at onset in multiplex schizophrenia. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; An, S.S.; Shugart, Y.Y.; Rapoport, J.L. Common polygenic variation and risk for childhood-onset schizophrenia. Mol. Psychiatry 2016, 21, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhou, J. Neuritin, a Neurotrophic factor in nervous system physiology. Curr. Med. Chem. 2014, 21, 1212–1219. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Nedivi, E.; Fieldust, S.; Theill, L.E.; Hevron, D. A Set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc. Natl. Acad. Sci. USA 1996, 93, 2048–2053. [Google Scholar] [CrossRef]
- Nedivi, E.; Hevroni, D.; Naot, D.; Israeli, D.; Citri, Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 1993, 363, 718–722. [Google Scholar] [CrossRef]
- Tongiorgi, E. Activity-Dependent expression of brain-derived neurotrophic factor in dendrites: Facts and open questions. Neurosci. Res. 2008, 61, 335–346. [Google Scholar] [CrossRef]
- Lu, J.-M.; Liu, D.-D.; Li, Z.-Y.; Ling, C.; Mei, Y.-A. Neuritin enhances synaptic transmission in medial prefrontal cortex in mice by increasing CaV3.3 surface expression. Cereb. Cortex 2017, 27, 3842–3855. [Google Scholar] [CrossRef]
- Yao, J.-J.; Gao, X.-F.; Chow, C.-W.; Zhan, X.-Q.; Hu, C.-L.; Mei, Y.-A. Neuritin activates insulin receptor pathway to up-regulate Kv4.2-mediated transient outward K + current in rat cerebellar granule neurons. J. Biol. Chem. 2012, 287, 41534–41545. [Google Scholar] [CrossRef]
- An, K.; Jung, J.H.; Jeong, A.Y.; Kim, H.G.; Jung, S.Y.; Lee, K.; Kim, H.J.; Kim, S.-J.; Jeong, T.-Y.; Son, Y.; et al. Neuritin can normalize neural deficits of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1523. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Banasr, M.; Choi, M.; Chae, S.Y.; Licznerski, P.; Lee, B.; Voleti, B.; Li, N.; Lepack, A.; Fournier, N.M.; et al. Neuritin produces antidepressant actions and blocks the neuronal and behavioral deficits caused by chronic stress. Proc. Natl. Acad. Sci. USA 2012, 109, 11378–11383. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, K.; Ryu, J.; Kim, H.G.; Jeong, A.Y.; Woo, R.-S.; Lee, J.-H.; Hyun, J.W.; Hahn, S.; Kim, J.-H.; et al. Neuritin attenuates cognitive function impairments in Tg2576 mouse model of Alzheimer’s Disease. PLoS ONE 2014, 9, e104121. [Google Scholar] [CrossRef]
- Park, K.W.; Kim, I.H.; Sun, W.; Kim, H. Sustained expression of neuritin MRNA after repeated electroconvulsive stimulations in the rat hippocampal formation. Exp. Neurobiol. 2009, 18, 62. [Google Scholar] [CrossRef]
- Chandler, D.; Dragović, M.; Cooper, M.; Badcock, J.C.; Mullin, B.H.; Faulkner, D.; Wilson, S.G.; Hallmayer, J.; Howell, S.; Rock, D.; et al. Impact of neuritin 1 (NRN1) polymorphisms on fluid intelligence in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 428–437. [Google Scholar] [CrossRef]
- Fatjó-Vilas, M.; Prats, C.; Pomarol-Clotet, E.; Lázaro, L.; Moreno, C.; González-Ortega, I.; Lera-Miguel, S.; Miret, S.; Muñoz, M.J.; Ibáñez, I.; et al. Involvement of NRN1 gene in schizophrenia-spectrum and bipolar disorders and its impact on age at onset and cognitive functioning. World J. Biol. Psychiatry 2016, 17, 129–139. [Google Scholar] [CrossRef]
- Birnbaum, R.; Weinberger, D.R. Functional neuroimaging and schizophrenia: A view towards effective connectivity modeling and polygenic risk. Dialogues Clin. Neurosci. 2013, 15, 279–289. [Google Scholar] [CrossRef]
- Bora, E.; Lin, A.; Wood, S.J.; Yung, A.R.; McGorry, P.D.; Pantelis, C. Cognitive deficits in youth with familial and clinical high risk to psychosis: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2014, 130, 1–15. [Google Scholar] [CrossRef]
- Zhang, R.; Picchioni, M.; Allen, P.; Toulopoulou, T. Working memory in unaffected relatives of patients with schizophrenia: A meta-analysis of functional magnetic resonance imaging studies. Schizophr. Bull. 2016, 42, 1068–1077. [Google Scholar] [CrossRef]
- Repovš, G.; Barch, D.M. Working memory related brain network connectivity in individuals with schizophrenia and their siblings. Front. Hum. Neurosci. 2012, 6, 137. [Google Scholar] [CrossRef]
- Silver, H.; Feldman, P.; Bilker, W.; Gur, R.C. Working memory deficit as a core neuropsychological dysfunction in schizophrenia. Am. J. Psychiatry 2003, 160, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, T.G.M.; Walton, E.; Hibar, D.P.; Schmaal, L.; Jiang, W.; Glahn, D.C.; Pearlson, G.D.; Yao, N.; Fukunaga, M.; Hashimoto, R.; et al. Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the enhancing neuro imaging genetics through meta analysis (ENIGMA) consortium. Biol. Psychiatry 2018, 84, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Bittner, R.A.; Linden, D.E.J.; Roebroeck, A.; Härtling, F.; Rotarska-Jagiela, A.; Maurer, K.; Goebel, R.; Singer, W.; Haenschel, C. The when and where of working memory dysfunction in early-onset schizophrenia—A functional magnetic resonance imaging study. Cereb. Cortex 2015, 25, 2494–2506. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulos, M.; Dima, D.; Roiser, J.P.; Corrigall, R.; Barker, G.J.; Frangou, S. Abnormal functional activation and connectivity in the working memory network in early-onset schizophrenia. J. Am. Acad. Child Adolesc. Psychiatry 2012, 51, 911–920.e2. [Google Scholar] [CrossRef] [PubMed]
- Loeb, F.F.; Zhou, X.; Craddock, K.E.S.; Shora, L.; Broadnax, D.D.; Gochman, P.; Clasen, L.S.; Lalonde, F.M.; Berman, R.A.; Berman, K.F.; et al. Reduced functional brain activation and connectivity during a working memory task in childhood-onset schizophrenia. J. Am. Acad. Child Adolesc. Psychiatry 2018, 57, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Pauly, K.; Seiferth, N.Y.; Kellermann, T.; Backes, V.; Vloet, T.D.; Shah, N.J.; Schneider, F.; Habel, U.; Kircher, T.T. Cerebral dysfunctions of emotion—Cognition interactions in adolescent-onset schizophrenia. J. Am. Acad. Child Adolesc. Psychiatry 2008, 47, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Thormodsen, R.; Jensen, J.; Holmèn, A.; Juuhl-Langseth, M.; Emblem, K.E.; Andreassen, O.A.; Rund, B.R. Prefrontal hyperactivation during a working memory task in early-onset schizophrenia spectrum disorders: An FMRI study. Psychiatry Res. Neuroimaging 2011, 194, 257–262. [Google Scholar] [CrossRef]
- White, T.; Hongwanishkul, D.; Schmidt, M. Increased anterior cingulate and temporal lobe activity during visuospatial working memory in children and adolescents with schizophrenia. Schizophr. Res. 2011, 125, 118–128. [Google Scholar] [CrossRef]
- Solmi, M.; Radua, J.; Olivola, M.; Croce, E.; Soardo, L.; Salazar de Pablo, G.; Il Shin, J.; Kirkbride, J.B.; Jones, P.; Kim, J.H.; et al. Age at onset of mental disorders worldwide: Large-scale meta-analysis of 192 epidemiological studies. Mol. Psychiatry 2021, 27, 1–15. [Google Scholar] [CrossRef]
- Lindholm, E.; Ekholm, B.; Shaw, S.; Jalonen, P.; Johansson, G.; Pettersson, U.; Sherrington, R.; Adolfsson, R.; Jazin, E. A schizophrenia-susceptibility locus at 6q25, in one of the world’s largest reported pedigrees. Am. J. Hum. Genet. 2001, 69, 96–105. [Google Scholar] [CrossRef]
- Maziade, M.; Roy, M.-A.; Rouillard, É.; Bissonnette, L.; Fournier, J.-P.; Roy, A.; Garneau, Y.; Montgrain, N.; Potvin, A.; Cliche, D.; et al. A search for specific and common susceptibility loci for schizophrenia and bipolar disorder: A linkage study in 13 target chromosomes. Mol. Psychiatry 2001, 6, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Moises, H.W.; Yang, L.; Kristbjarnarson, H.; Wiese, C.; Byerley, W.; Macciardi, F.; Arolt, V.; Blackwood, D.; Liu, X.; Sjögren, B.; et al. An international two–stage genome–wide search for schizophrenia susceptibility genes. Nat. Genet. 1995, 11, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.E.; MacLean, C.J.; Ma, Y.; Webb, B.T.; Myakishev, M.V.; Harris-Kerr, C.; Wormley, B.; Sadek, H.; Kadambi, B.; O’Neill, F.A.; et al. Genome-wide scans of three independent sets of 90 Irish Multiplex schizophrenia families and follow-up of selected regions in all families provides evidence for multiple susceptibility genes. Mol. Psychiatry 2002, 7, 542–559. [Google Scholar] [CrossRef] [PubMed]
- Fanous, A.H.; Neale, M.C.; Webb, B.T.; Straub, R.E.; Amdur, R.L.; O’Neill, F.A.; Walsh, D.; Riley, B.P.; Kendler, K.S. A genome-wide scan for modifier Loci in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144, 589–595. [Google Scholar] [CrossRef]
- Pidsley, R.; Viana, J.; Hannon, E.; Spiers, H.; Troakes, C.; Al-Saraj, S.; Mechawar, N.; Turecki, G.; Schalkwyk, L.C.; Bray, N.J.; et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 2014, 15, 483. [Google Scholar] [CrossRef]
- Bähner, F.; Meyer-Lindenberg, A. Hippocampal–prefrontal connectivity as a translational phenotype for schizophrenia. Eur. Neuropsychopharmacol. 2017, 27, 93–106. [Google Scholar] [CrossRef]
- Riley, M.R.; Constantinidis, C. Role of prefrontal persistent activity in working memory. Front. Syst. Neurosci. 2016, 9, 181. [Google Scholar] [CrossRef]
- Thomas, M.L.; Duffy, J.R.; Swerdlow, N.; Light, G.A.; Brown, G.G. Detecting the inverted-U in FMRI studies of schizophrenia: A comparison of three analysis methods. J. Int. Neuropsychol. Soc. 2022, 28, 258–269. [Google Scholar] [CrossRef]
- Prats, C.; Arias, B.; Ortet, G.; Ibáñez, M.I.; Moya, J.; Pomarol-Clotet, E.; Fañanás, L.; Fatjó-Vilas, M. Role of neurotrophins in depressive symptoms and executive function: Association analysis of NRN1 gene and its interaction with BDNF gene in a non-clinical sample. J. Affect. Disord. 2017, 211, 92–98. [Google Scholar] [CrossRef]
- Lenroot, R.K.; Giedd, J.N. Brain development in children and adolescents: Insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 2006, 30, 718–729. [Google Scholar] [CrossRef]
- Fair, D.A.; Dosenbach, N.U.F.; Church, J.A.; Cohen, A.L.; Brahmbhatt, S.; Miezin, F.M.; Barch, D.M.; Raichle, M.E.; Petersen, S.E.; Schlaggar, B.L. Development of distinct control networks through segregation and integration. Proc. Natl. Acad. Sci. USA 2007, 104, 13507–13512. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.S.; Sheridan, M.A.; Kam, C.L.H.; Hinshaw, S.; D’Esposito, M. Longitudinal evidence for functional specialization of the neural circuit supporting working memory in the human brain. J. Neurosci. 2010, 30, 11062–11067. [Google Scholar] [CrossRef] [PubMed]
- Andre, J.; Picchioni, M.; Zhang, R.; Toulopoulou, T. Working memory circuit as a function of increasing age in healthy adolescence: A systematic review and meta-analyses. NeuroImage Clin. 2015, 12, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Hartshorne, J.K.; Germine, L.T. When does cognitive functioning peak? The asynchronous rise and fall of different cognitive abilities across the lifespan. Psychol. Sci. 2015, 26, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Bowen, E.F.W.; Burgess, J.L.; Granger, R.; Kleinman, J.E.; Rhodes, C.H. DLPFC Transcriptome defines two molecular subtypes of schizophrenia. Transl. Psychiatry 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Singh-Taylor, A.; Molet, J.; Jiang, S.; Korosi, A.; Bolton, J.L.; Noam, Y.; Simeone, K.; Cope, J.; Chen, Y.; Mortazavi, A.; et al. NRSF-Dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol. Psychiatry 2018, 23, 648–657. [Google Scholar] [CrossRef]
- Ohi, K.; Hashimoto, R.; Yasuda, Y.; Kiribayashi, M.; Iike, N.; Yoshida, T.; Azechi, M.; Ikezawa, K.; Takahashi, H.; Morihara, T.; et al. TATA box-binding protein gene is associated with risk for schizophrenia, age at onset and prefrontal function. Genes Brain Behav. 2009, 8, 473–480. [Google Scholar] [CrossRef]
- Zhang, J.; Chang, L.; Pu, Y.; Hashimoto, K. Abnormal expression of colony stimulating factor 1 receptor (CSF1R) and transcription factor PU.1 (SPI1) in the spleen from patients with major psychiatric disorders: A role of brain–spleen axis. J. Affect. Disord. 2020, 272, 110–115. [Google Scholar] [CrossRef]
- Lee, S.H.; DeCandia, T.R.; Ripke, S.; Yang, J.; Sullivan, P.F.; Goddard, M.E.; Keller, M.C.; Visscher, P.M.; Wray, N.R. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat. Genet. 2012, 44, 247–250. [Google Scholar] [CrossRef]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022, 604, 509–516. [Google Scholar] [CrossRef]
- Fusar-Poli, P. Voxel-wise meta-analysis of FMRI studies in patients at clinical high risk for psychosis. J. Psychiatry Neurosci. 2012, 37, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Radua, J.; Borgwardt, S.; Crescini, A.; Mataix-Cols, D.; Meyer-Lindenberg, A.; McGuire, P.K.; Fusar-Poli, P. Multimodal meta-analysis of structural and functional brain changes in first episode psychosis and the effects of antipsychotic medication. Neurosci. Biobehav. Rev. 2012, 36, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Geller, B.; Zimerman, B.; Williams, M.; Bolhofner, K.; Craney, J.L.; Delbello, M.P.; Soutullo, C. Reliability of the Washington University in St. Louis kiddie schedule for affective disorders and schizophrenia (WASH-U-KSADS) mania and rapid cycling sections. J. Am. Acad. Child Adolesc. Psychiatry 2001, 40, 450–455. [Google Scholar] [CrossRef]
- Andreasen, N.C. The comprehensive assessment of symptoms and history (CASH): An instrument for assessing diagnosis and psychopathology. Arch. Gen. Psychiatry 1992, 49, 615. [Google Scholar] [CrossRef]
- First, M.; Spitzer, R.; Gibbon, M.; Williams, J. Structured clinical interview for DSM-IV-TR Axis I disorders, research version, non-patient edition. In (SCID-I/P); New York State Psychiatric Institute: New York, NY, USA, 2002. [Google Scholar]
- Perkins, D.O.; Leserman, J.; Jarskog, L.F.; Graham, K.; Kazmer, J.; Lieberman, J.A. Characterizing and dating the onset of symptoms in psychotic illness: The symptom onset in schizophrenia (SOS) inventory. Schizophr. Res. 2000, 44, 1–10. [Google Scholar] [CrossRef]
- Gershon, E.S.; DeLisi, L.E.; Hamovit, J.; Nurnberger, J.I., Jr.; Maxwell, M.E.; Schreiber, J.; Dauphinais, D.; Dingman, C.W., II; Guroff, J.J. A controlled family study of chronic psychoses: Schizophrenia and schizoaffective disorder. Arch. Gen. Psychiatry 1988, 45, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Gomar, J.J.; Ortiz-Gil, J.; McKenna, P.J.; Salvador, R.; Sans-Sansa, B.; Sarró, S.; Guerrero, A.; Pomarol-Clotet, E. Validation of the word accentuation test (TAP) as a means of estimating premorbid IQ in spanish speakers. Schizophr. Res. 2011, 128, 175–176. [Google Scholar] [CrossRef]
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef]
- Fullard, J.F.; Giambartolomei, C.; Hauberg, M.E.; Xu, K.; Voloudakis, G.; Shao, Z.; Bare, C.; Dudley, J.T.; Mattheisen, M.; Robakis, N.K.; et al. Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia Loci. Hum. Mol. Genet. 2017, 26, 1942–1951. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Boyle, A.P. Predicting functional variants in enhancer and promoter elements using regulomeDB. Hum. Mutat. 2019, 40, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Jenkinson, M.; Woolrich, M.W.; Beckmann, C.F.; Behrens, T.E.J.; Johansen-Berg, H.; Bannister, P.R.; De Luca, M.; Drobnjak, I.; Flitney, D.E.; et al. Advances in functional and structural mr image analysis and implementation as FSL. NeuroImage 2004, 23, S208–S219. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Worsley, K.J. Statistical Analysis of Activation Images. In Functional Magnetic Resonance Imaging; Oxford University Press: Oxford, UK, 2001; ISBN 978-0-19-263071-1. [Google Scholar]
- Egli, T.; Coynel, D.; Spalek, K.; Fastenrath, M.; Freytag, V.; Heck, A.; Loos, E.; Auschra, B.; Papassotiropoulos, A.; de Quervain, D.J.-F.; et al. Identification of two distinct working memory-related brain networks in healthy young adults. eNeuro 2018, 5, ENEURO.0222-17.2018. [Google Scholar] [CrossRef]
- Wilhelm, O.; Hildebrandt, A.; Oberauer, K. What is working memory capacity, and how can we measure it? Front. Psychol. 2013, 4, 433. [Google Scholar] [CrossRef]



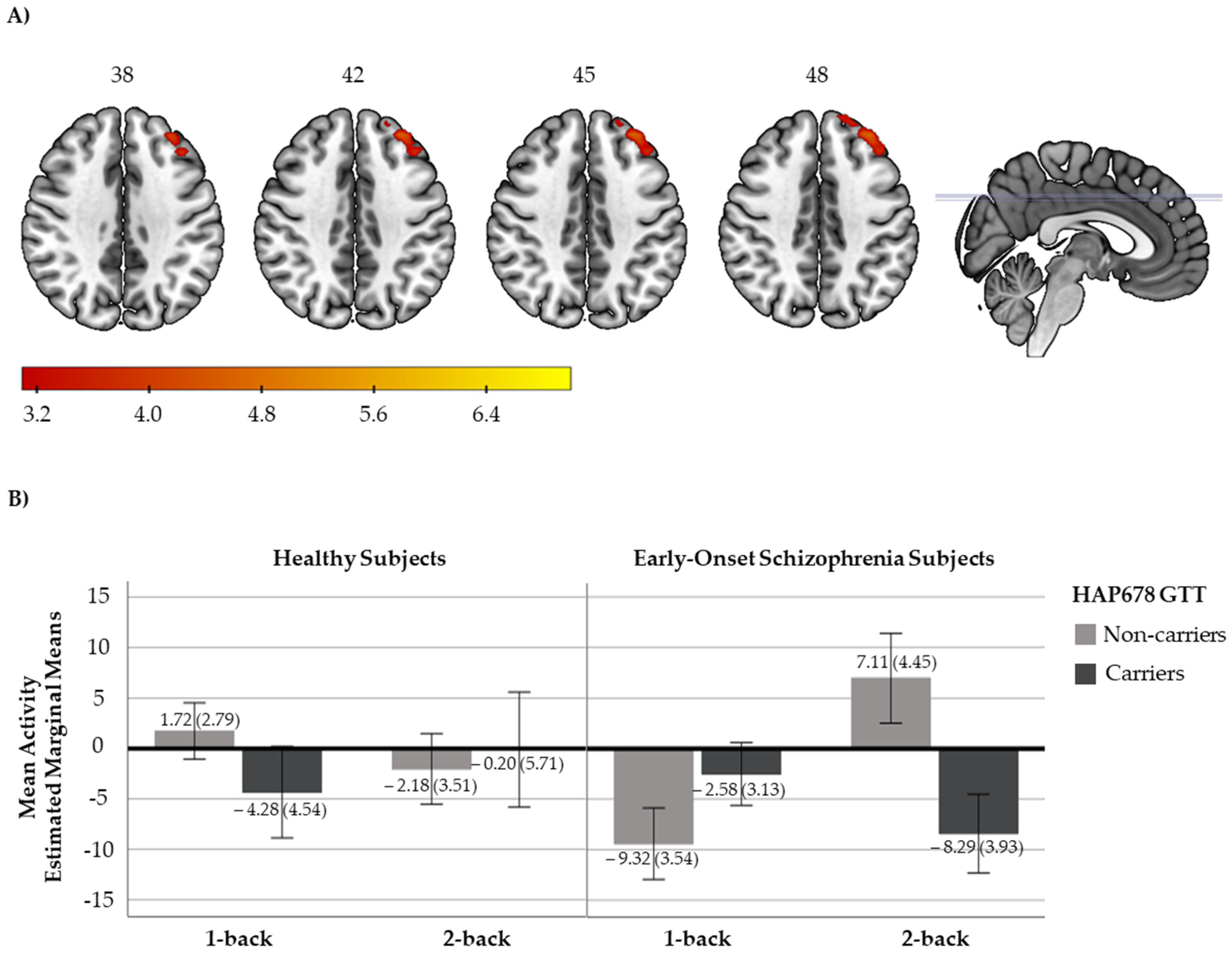{kind=link}
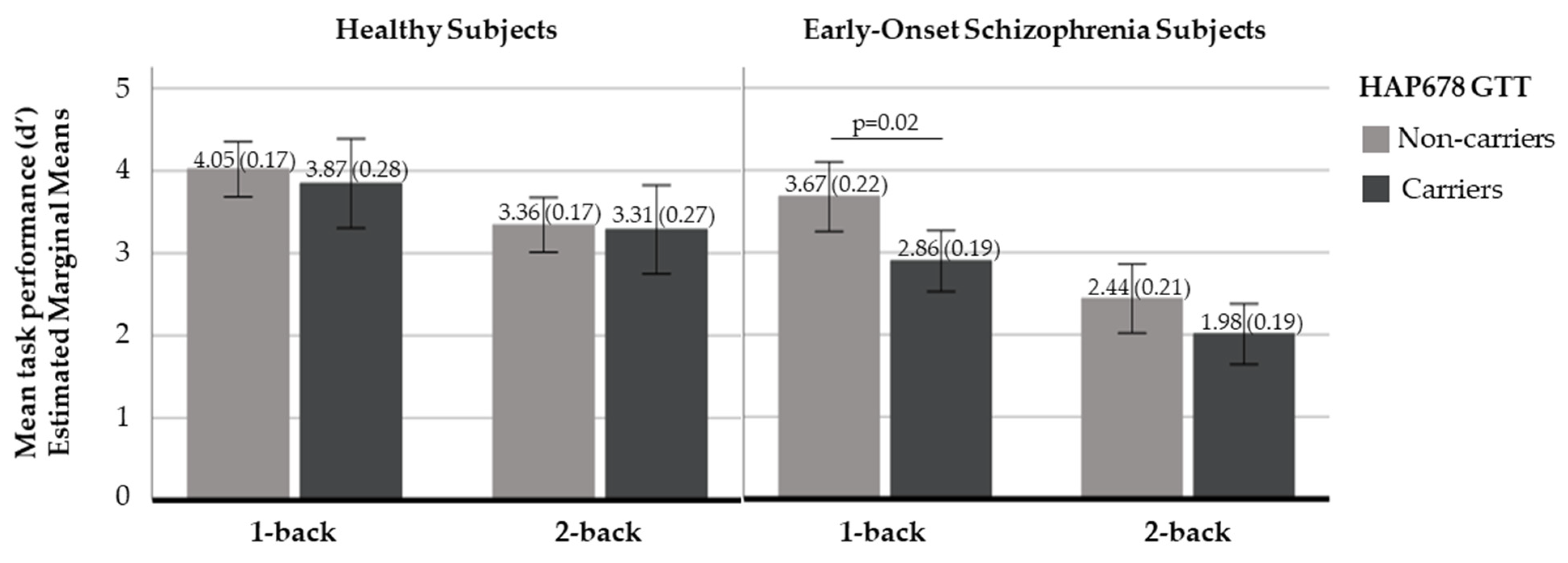{kind=link}
| SNPs | Haplotype | Transmitted EO SSD | Not Transmitted EO SSD | OR (CI 95%) | TDT; pperm |
| HAP678 | GCT | 13 | 27 | 0.48 (0.25–0.93) | 4.90; 0.03 |
| SNPs | Genotypes Haplotypes | Frequency EO SSD | Frequency HS | OR (CI 95%) | Wald; pperm |
| SNP6 | TT/TG/GG | 11 (0.13)/40 (0.48)/33 (0.39) | 31 (0.26)/58 (0.49)/30 (0.25) | 1.68 (1.01–2.57) | 2.39; 0.02 a,b |
| SNP7 | CC/CT/TT | 43 (0.50)/35 (0.41)/8 (0.09) | 78 (0.66)/35 (0.29)/6 (0.05) | 1.69 (1.06–2.71) | 2.19; 0.03 b |
| SNP8 | CC/CT/TT | 14 (0.17)/41 (0.49)/29 (0.35) | 33 (0.29)/58 (0.51)/23 (0.20) | 1.66 (1.08–2.56) | 2.31; 0.02 a,c |
| HAP678 | TCC | 0.37 | 0.50 | 0.59 (0.39–0.89) | 6.44; 0.01 |
| HAP678 | GTT | 0.30 | 0.20 | 1.70 (1.08–2.67) | 5.28; 0.02 |
| Sample 1: Family-Based a (n = 453) | AO Offspring (n = 71) | EO Offspring (n = 80) | AO Parents (n = 142) | EO Parents (n = 160) | ||
| Male | 58 (81.70) | 54 (67.50) | n.s. | 71 (50.00) | 80 (50.00) | n.s. |
| Age at interview | 27.45 (5.03) | 18.04 (4.94) | t = −11.51, p < 0.001 | 50.04 (7.97) | 58.34 (8.44) | t = −7.27, p < 0.001 |
| Age at onset | 23.24 (4.24) | 15.41 (2.12) d | t = −13.35, p < 0.001 | – | – | – |
| Sample 2: Case-control b (n = 345) | AO Subjects (n = 138) | EO Subjects (n = 87) | Healthy Subjects (n = 120) | |||
| Male | 93 (67.40) | 67 (77.00) | n.s. | 60 (50.00) | χ2 = 17.13, p < 0.001 | |
| Age at interview | 41.97 (10.03) | 39.79 (10.87) | n.s. | 38.24 (11.21) | F = 3.45; p = 0.033 e | |
| Age at onset | 25.12 (5.86) | 16.38 (2.00) | t = 16.11, p < 0.001 | – | – | |
| Sample 3: Neuroimaging c (n = 117) | AO Subjects (n = 39) | EO Subjects (n = 39) | Healthy Subjects (n = 39) | |||
| Male | 37 (94.87) | 37 (94.87) | n.s. | 37 (94.87) | n.s. | |
| Age at interview | 39.49 (1.90) | 39.30 (1.87) | n.s. | 38.43 (1.78) | n.s. | |
| Age at onset | 24.56 (0.80) | 16.85 (0.26) | t = 9.17; p < 0.001 | - | - | |
| Illness duration | 14.92 (11.01) | 22.46 (11.31) | t = −2.98; p = 0.004 | - | - | |
| PANSS total | 68.72 (20.46) | 80.05 (21.11) | t = −2.60; p = 0.011 | - | - | |
| CPZE | 367.01 (188.83) | 633.66 (304.39) | t = −4.28; p < 0.001 | - | - | |
| SNPs | Chromosome Position | Gene Position | Alleles (Minor/Major) | MAF (All/Eur) | Family-Based MAF | Case-Control MAF | RegulomeDB Score a | |
|---|---|---|---|---|---|---|---|---|
| SNP1 | rs2208870 | 5,992,257 | intergenic | G/A | 0.33/0.34 | 0.33 | 0.33 | 0.61 |
| SNP2 | rs12333117 | 5,994,759 | intergenic | T/C | 0.35/0.40 | 0.43 | 0.38 | 0.61 |
| SNP3 | rs582186 | 6,001,148 | downstream | A/G | 0.45/0.62 | 0.61 | 0.40 | 0.61 |
| SNP4 | rs645649 | 6,004,726 | intronic | C/G | 0.45/0.64 | 0.64 | 0.38 | 0.61 |
| SNP5 | rs582262 | 6,007,758 | intronic | C/G | 0.30/0.48 | 0.28 | 0.27 | 0.70 |
| SNP6 | rs3763180 | 6,009,615 | upstream | T/G | 0.40/0.46 | 0.45 | 0.43 | 0.63 |
| SNP7 | rs10484320 | 6,010,204 | upstream | T/C | 0.15/0.22 | 0.26 | 0.24 | 0.16 |
| SNP8 | rs4960155 | 6,010,306 | upstream | T/C | 0.43/0.49 | 0.50 | 0.49 | 0.13 |
| SNP9 | rs9379002 | 6,012,158 | intergenic | G/T | 0.29/0.42 | 0.24 | 0.26 | 0.13 |
| SNP10 | rs9405890 | 6,012,488 | intergenic | C/T | 0.31/0.38 | 0.28 | 0.33 | 0.18 |
| SNP11 | rs1475157 | 6,016,936 | intergenic | G/A | 0.16/0.17 | 0.16 | 0.16 | 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almodóvar-Payá, C.; Guardiola-Ripoll, M.; Giralt-López, M.; Gallego, C.; Salgado-Pineda, P.; Miret, S.; Salvador, R.; Muñoz, M.J.; Lázaro, L.; Guerrero-Pedraza, A.; et al. NRN1 Gene as a Potential Marker of Early-Onset Schizophrenia: Evidence from Genetic and Neuroimaging Approaches. Int. J. Mol. Sci. 2022, 23, 7456. https://doi.org/10.3390/ijms23137456
Almodóvar-Payá C, Guardiola-Ripoll M, Giralt-López M, Gallego C, Salgado-Pineda P, Miret S, Salvador R, Muñoz MJ, Lázaro L, Guerrero-Pedraza A, et al. NRN1 Gene as a Potential Marker of Early-Onset Schizophrenia: Evidence from Genetic and Neuroimaging Approaches. International Journal of Molecular Sciences. 2022; 23(13):7456. https://doi.org/10.3390/ijms23137456
Chicago/Turabian StyleAlmodóvar-Payá, Carmen, Maria Guardiola-Ripoll, Maria Giralt-López, Carme Gallego, Pilar Salgado-Pineda, Salvador Miret, Raymond Salvador, María J. Muñoz, Luisa Lázaro, Amalia Guerrero-Pedraza, and et al. 2022. "NRN1 Gene as a Potential Marker of Early-Onset Schizophrenia: Evidence from Genetic and Neuroimaging Approaches" International Journal of Molecular Sciences 23, no. 13: 7456. https://doi.org/10.3390/ijms23137456
APA StyleAlmodóvar-Payá, C., Guardiola-Ripoll, M., Giralt-López, M., Gallego, C., Salgado-Pineda, P., Miret, S., Salvador, R., Muñoz, M. J., Lázaro, L., Guerrero-Pedraza, A., Parellada, M., Carrión, M. I., Cuesta, M. J., Maristany, T., Sarró, S., Fañanás, L., Callado, L. F., Arias, B., Pomarol-Clotet, E., & Fatjó-Vilas, M. (2022). NRN1 Gene as a Potential Marker of Early-Onset Schizophrenia: Evidence from Genetic and Neuroimaging Approaches. International Journal of Molecular Sciences, 23(13), 7456. https://doi.org/10.3390/ijms23137456