
New Frontiers in Neurodegeneration and Regeneration Associated with Brain-Derived Neurotrophic Factor and the rs6265 Single Nucleotide Polymorphism


Abstract
1. Introduction
2. Brain-Derived Neurotrophic Factor (BDNF)
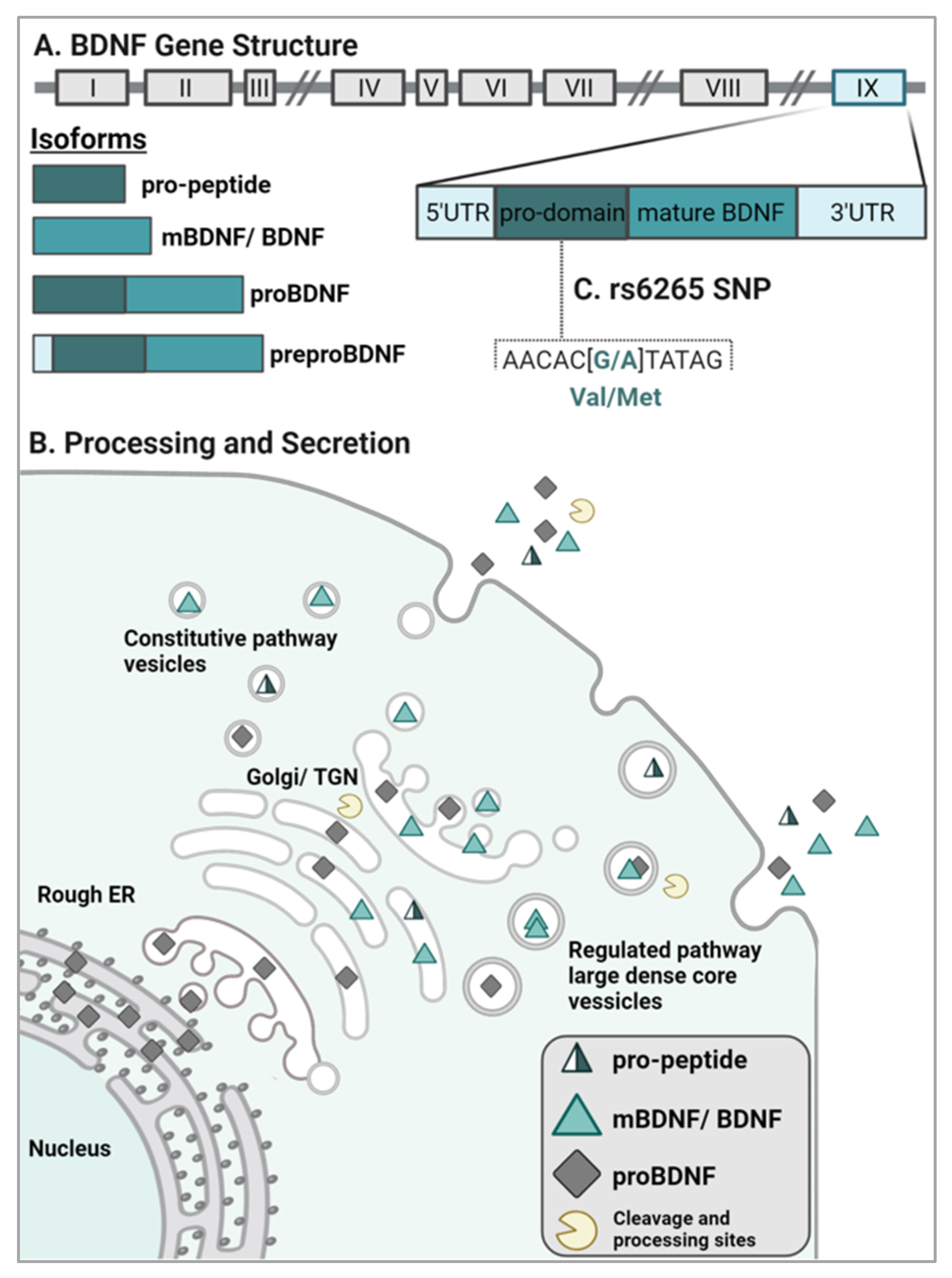
2.1. BDNF Gene Structure and Isoform Processing
2.2. BDNF Sorting and Release
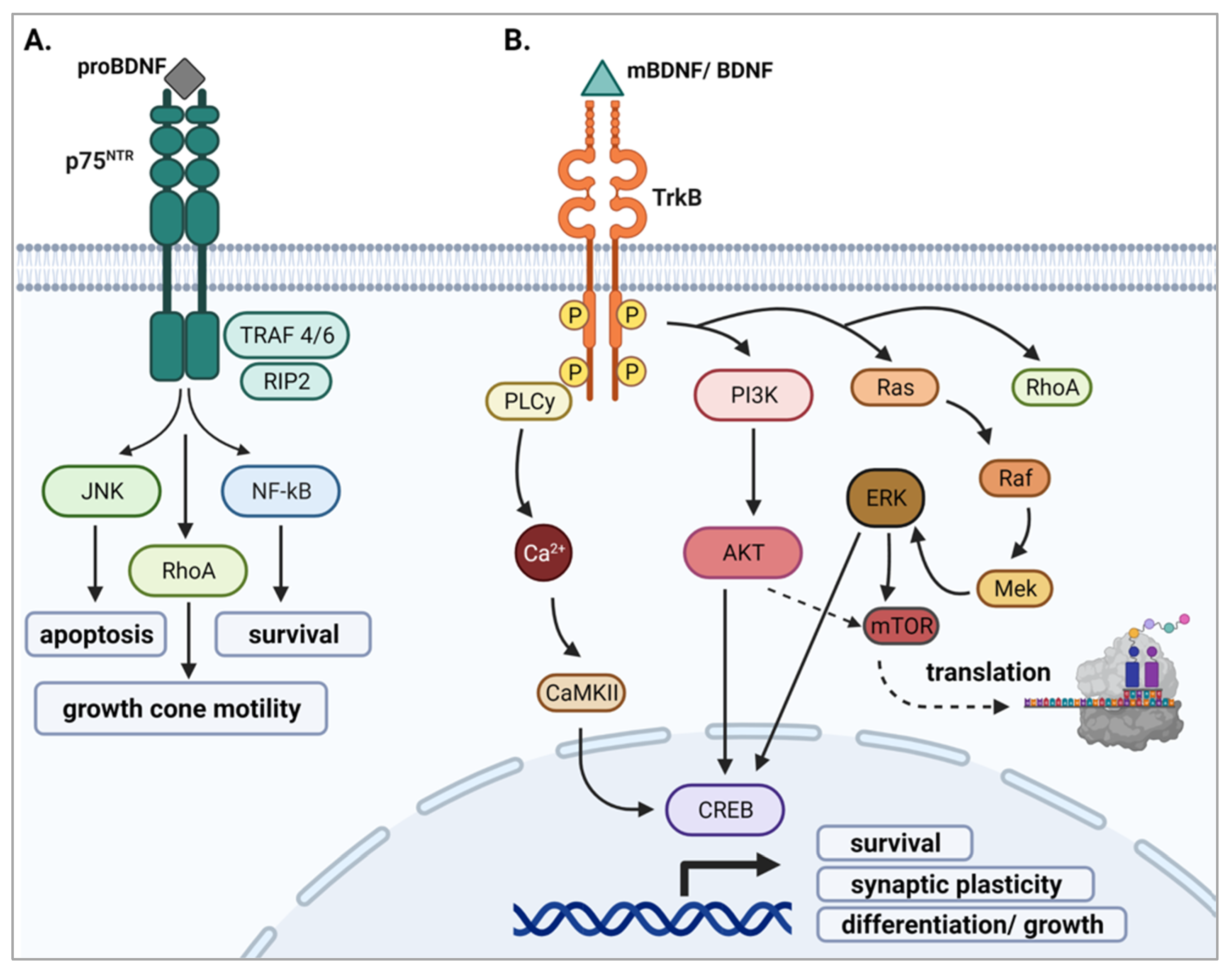
2.3. BDNF Signaling
2.3.1. proBDNF and p75NTR
2.3.2. mBDNF and TrkB

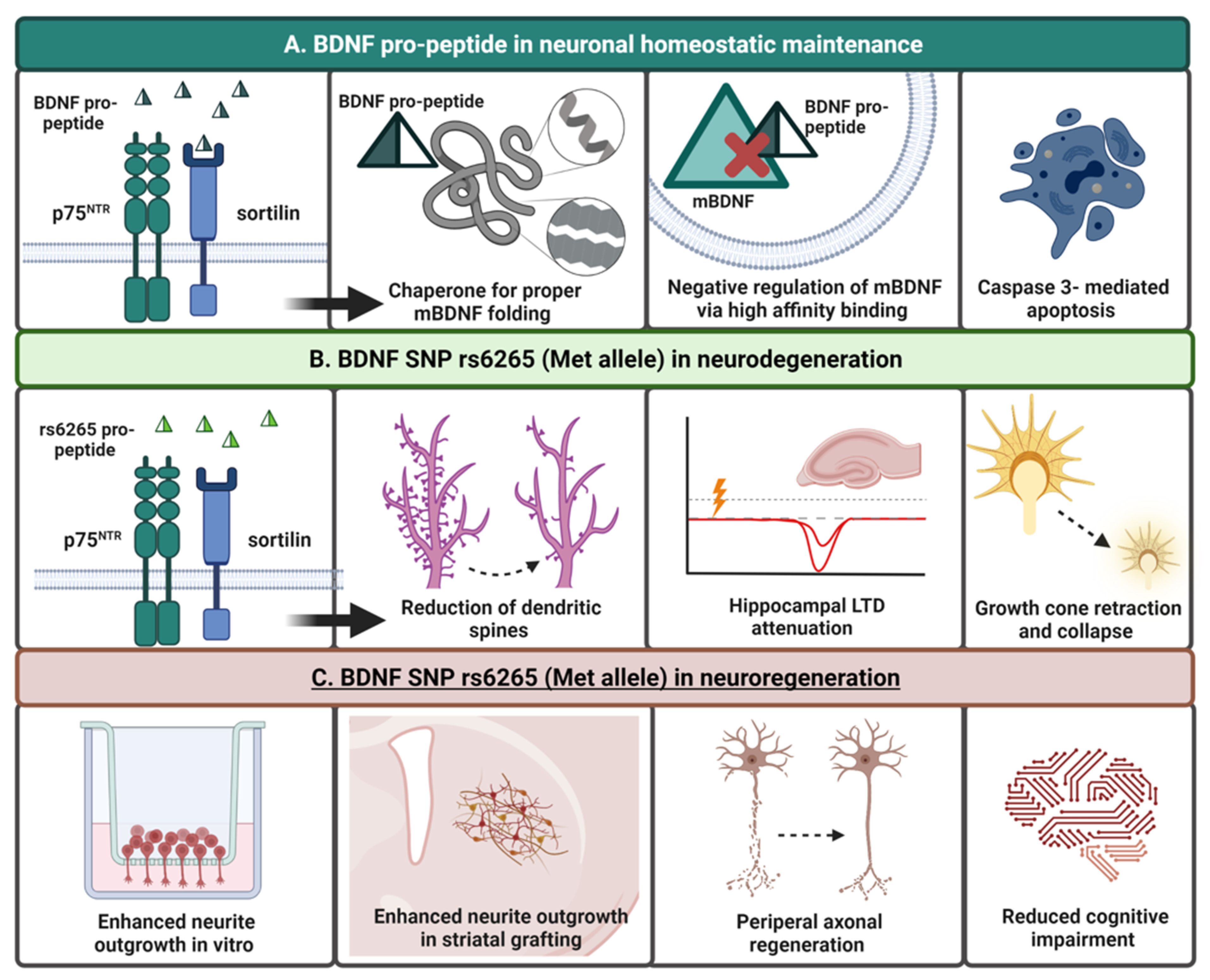
2.3.3. BDNF Pro-Peptide and Sortilin
3. Genetic Polymorphisms of BDNF
rs6265 (Val66Met)
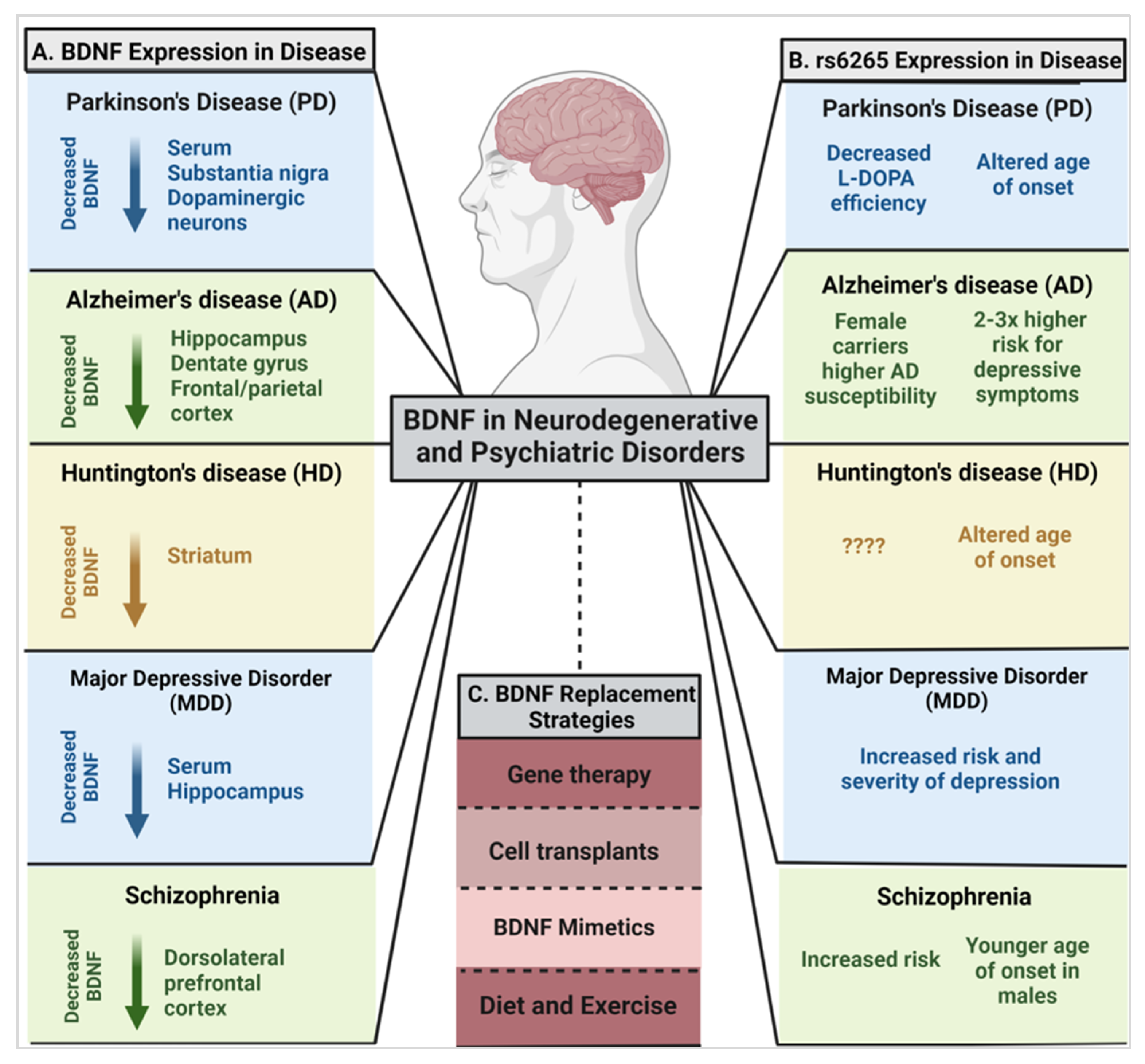
4. BDNF and the rs6265 SNP in Neurodegeneration
4.1. Parkinson’s Disease (PD) and BDNF
rs6265 in PD
4.2. Alzheimer’s Disease (AD) and BDNF
rs6265 in AD
4.3. Huntington’s Disease (HD) and BDNF
rs6265 in HD
5. BDNF and rs6265 in Psychiatric Disorders
5.1. Major Depressive Disorder (MDD)
rs6265 in MDD
5.2. Schizophrenia
rs6265 in Schizophrenia
6. Targeting BDNF in the Brain: Therapeutic Challenges and Potential
6.1. Gene- and Cell-Based Therapy
6.2. BDNF Mimetics
6.3. Diet and Exercise
7. The BDNF Pro-Peptide: A Functional “Third Ligand”
Harnessing the Neurogenerative Benefits of the BDNF Pro-Peptide and rs6265 SNP
8. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Subjects/Model | Region/Source | Effect |
|---|---|---|---|
| Dieni et al. (2012) [43] | C57BL/6, Bdnf-Myc, cbdnf ko, Bsn mutant mice all 8 weeks of age. | Hippocampus | Mature BDNF and the BDNF pro-peptide are stored at equimolar ratios in large dense core vesicles in presynaptic terminals of excitatory neurons. |
| Anastasia et al. (2013) [27] | Cultures prepared from E18 BDNFVal/Val and BDNFMet/Met knock-in mice Primary neurons isolated from E15 C57BL/6 mouse embryos. | Hippocampal-cortical neurons Hippocampus | In hippocampal-cortical neurons, secreted levels of Met prodomain was significantly lower compared to Val prodomain secretion. In hippocampal neurons, growth cone retraction was induced by Met prodomain application in p75+ cells; Val prodomain was inactive. Met prodomain only interacted with SorCS2 receptor. |
| Lim et al. (2015) [243] | SH-SY5Y neuroblastoma cells Extracts from post-mortem tissue (AD patients) | Hippocampus | In culture, application of the Met prodomain negatively affected cell viability only in the presence of Aβ; Val prodomain had no effect. Levels of pro-peptide were 16-fold higher in AD patients and correlated with Aβ accumulation. |
| Mizui et al. (2015) [31] | Slices prepared from 3–4-week-old C57BL/6 and Bdnf KO mice. DIV21 cultures prepared from E18 Wistar rats. | Hippocampal tissue slices Hippocampus | Application of the Val pro-peptide facilitated LTD in hippocampal slices and required the activation of GluN2B-containing NMDA receptors. In cultured neurons, Val pro-peptide also induced endocytosis of AMPA receptors. In cultured neurons, the presence of the Val66Met SNP in the pro-peptide inhibited LTD. |
| Guo et al. (2016) [235] | DIV16 rat neuronal cultures electroporated with plasmid-expressing eGFP. | Hippocampus | Val prodomain application reduced spine density and increased spine length. Val prodomain increased caspase-3 activity and mitochondria elongation. *Met prodomain was not studied. |
| Yang et al. (2016) [244] | 7-week-old male Sprague Dawley rats of learned helplessness (LH) model of depression (WT and Bdnf KO). | Medial prefrontal cortex (mPFC), CA3 and dentate gyrus of hippocampus, nucleus accumbens. | Significantly higher expression of BDNF pro-peptide in mPFC and CA3 regions of LH rats compared to controls. Significantly lower expression of BDNF pro-peptide in nucleus accumbens and dentate gyrus compared to controls. |
| Uegaki et al. (2017) [67] | BIAcore sensor chip and recombinant human BDNF protein Slices prepared from male C57BL/6J mice (3-4-weeks-old) | Hippocampus | Using BIAcore chip, the BDNF pro-peptide binds to mature BDNF with high affinity. Using BIAcore chip, The Met pro-peptide is more stable in acidic and neutral pH environments compared to Val pro-peptide. In hippocampal slices, pre-incubation of the Val pro-peptide reduced the ability of mBDNF to inhibit LTD. |
| Yang et al. (2017) [245] | Patients with MDD, SCZ, and bipolar disorder (BD) | Postmortem samples of cerebellum, parietal cortex, liver, and spleen | BDNF pro-peptide levels were significantly lower in the cerebellum and the spleen of MDD, SCZ, and BD patients compared to control groups. BDNF pro-peptide levels were significantly higher in the parietal cortex of MDD, SCZ, and BD patients compared to control groups. |
| Yang et al. (2017) [234] | Cell culture | C6 glioma cells | Application of the WT BDNF pro-peptide promoted C6 glioma cell apoptosis and decreased cell growth through caspase-3 activation. |
| Giza et al. (2018) [68] | DIV21 primary neurons prepared from C57BL/6 mice. BDNFVal/Val, BDNFVal/Met, BDNFMet/Met P23-P60 male mice. | Hippocampus (ventral CA1 neurons) Hippocampus | In culture, the Met prodomain decreased mushroom spines and reduced PSD95 density in p75+ and SorCS2+ cells; Val prodomain had no effect. Increased freezing behavior/decreased fear extinction was demonstrated in Met-prodomain injected mice. Fewer spines were also found in Met-prodomain treated mice compared with the Val-prodomain injected mice. |
| Mizui et al. (2019) [242] | Japanese patients with Major depressive disorder (MDD) or Schizophrenia (SCZ) | Cerebral spinal fluid | The ratio of BDNF pro-peptide to total protein in MDD patients was lower in males and not females compared to controls. The ratio of BDNF pro-peptide to total protein was lower in SCZ patients, but it was not statistically significant. |
References
- Al-Qudah, M.A.; Al-Dwairi, A. Mechanisms and regulation of neurotrophin synthesis and secretion. Neurosciences 2016, 21, 306–313. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Hamburger, V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 1951, 116, 321–362. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Hamburger, V. A diffusible agent of mouse sarcoma, producing hyperplasia of sympathetic ganglia and hyperneurotization of viscera in the chick embryo. J. Exp. Zool. 1953, 123, 233–287. [Google Scholar] [CrossRef]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoenen, H.; Barde, Y.A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature 1989, 341, 149–152. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Baydyuk, M.; Xu, B. BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 2014, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Brigadski, T.; Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res. 2020, 382, 15–45. [Google Scholar] [CrossRef] [PubMed]
- Zagrebelsky, M.; Tacke, C.; Korte, M. BDNF signaling during the lifetime of dendritic spines. Cell Tissue Res. 2020, 382, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Moya-Alvarado, G.; Gonzalez-Billaut, C.; Bronfman, F.C. Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton 2016, 73, 612–628. [Google Scholar] [CrossRef]
- Sasi, M.; Vignoli, B.; Canossa, M.; Blum, R. Neurobiology of local and intercellular BDNF signaling. Pflugers Arch. 2017, 469, 593–610. [Google Scholar] [CrossRef]
- Liu, Q.; Lei, L.; Yu, T.; Jiang, T.; Kang, Y. Effect of Brain-Derived Neurotrophic Factor on the Neurogenesis and Osteogenesis in Bone Engineering. Tissue Eng. Part A 2018, 24, 1283–1292. [Google Scholar] [CrossRef]
- Urbina-Varela, R.; Soto-Espinoza, M.I.; Vargas, R.; Quiñones, L.; del Campo, A. Influence of BDNF genetic polymorphisms in the pathophysiology of aging-related diseases. Aging Dis. 2020, 11, 1513. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Begni, V.; Pariante, C.M.; Riva, M.A. The human BDNF gene: Peripheral gene expression and protein levels as biomarkers for psychiatric disorders. Transl. Psychiatry 2016, 6, e958. [Google Scholar] [CrossRef]
- Vaghi, V.; Polacchini, A.; Baj, G.; Pinheiro, V.L.M.; Vicario, A.; Tongiorgi, E. Pharmacological profile of brain-derived neurotrophic factor (BDNF) splice variant translation using a novel drug screening assay b. J. Biol. Chem. 2014, 289, 27702–27713. [Google Scholar] [CrossRef]
- Notaras, M.; van den Buuse, M. Brain-Derived Neurotrophic Factor (BDNF): Novel Insights into Regulation and Genetic Variation. Neuroscientist 2019, 25, 434–454. [Google Scholar] [CrossRef]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef]
- Chiaruttini, C.; Vicario, A.; Li, Z.; Baj, G.; Braiuca, P.; Wu, Y.; Lee, F.S.; Gardossi, L.; Baraban, J.M.; Tongiorgi, E. Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation. Proc. Natl. Acad. Sci. USA 2009, 106, 16481–16486. [Google Scholar] [CrossRef]
- Pruunsild, P.; Kazantseval, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef]
- Pang, P.T.; Nagappan, G.; Guo, W.; Lu, B. Extracellular and intracellular cleavages of proBDNF required at two distinct stages of late-phase LTP. NPJ Sci. Learn. 2016, 1, 16003. [Google Scholar] [CrossRef]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Nakade, J.; Tachibana, M.; Ibi, D.; Someya, E.; Koike, H.; Kamei, H.; Nabeshima, T.; Itohara, S.; Takuma, K.; et al. Matrix metalloproteinase-9 contributes to kindled seizure development in pentylenetetrazole-treated mice by converting pro-BDNF to mature BDNF in the hippocampus. J. Neurosci. 2011, 31, 12963–12971. [Google Scholar] [CrossRef]
- McGregor, C.E.; English, A.W. The role of BDNF in peripheral nerve regeneration: Activity-dependent treatments and Val66Met. Front. Cell. Neurosci. 2019, 12, 522. [Google Scholar] [CrossRef]
- Anastasia, A.; Deinhardt, K.; Chao, M.V.; Will, N.E.; Irmady, K.; Lee, F.S.; Hempstead, B.L.; Bracken, C. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat. Commun. 2013, 4, 2490. [Google Scholar] [CrossRef]
- Leßmann, V.; Brigadski, T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: An update. Neurosci. Res. 2009, 65, 11–22. [Google Scholar] [CrossRef]
- Wong, Y.H.; Lee, C.M.; Xie, W.; Cui, B.; Poo, M.M. Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc. Natl. Acad. Sci. USA 2015, 112, E4475–E4484. [Google Scholar] [CrossRef]
- Cunha, C.; Brambilla, R.; Thomas, K.L. A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 2010, 3, 1. [Google Scholar] [CrossRef]
- Mizui, T.; Ishikawa, Y.; Kumanogoh, H.; Lume, M.; Matsumoto, T.; Hara, T.; Yamawaki, S.; Takahashi, M.; Shiosaka, S.; Itami, C.; et al. BDNF pro-peptide actions facilitate hippocampal LTD and are altered by the common BDNF polymorphism Val66Met. Proc. Natl. Acad. Sci. USA 2015, 112, E3067–E3074. [Google Scholar] [CrossRef] [PubMed]
- Frodl, T.; Schaub, A.; Banac, S.; Charypar, M.; Jäger, M.; Kümmler, P.; Bottlender, R.; Zetzsche, T.; Born, C.; Leinsinger, G.; et al. Reduced hippocampal volume correlates with executive dysfunctioning in major depression. J. Psychiatry Neurosci. 2006, 31, 316–323. [Google Scholar] [PubMed]
- Mercado, N.M.; Stancati, J.A.; Sortwell, C.E.; Mueller, R.L.; Boezwinkle, S.A.; Duffy, M.F.; Fischer, D.L.; Sandoval, I.M.; Manfredsson, F.P.; Collier, T.J.; et al. The BDNF Val66Met polymorphism (rs6265) enhances dopamine neuron graft efficacy and side-effect liability in rs6265 knock-in rats. Neurobiol. Dis. 2021, 148, 105175. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Jing, D.; Bath, K.G.; Ieraci, A.; Khan, T.; Siao, C.J.; Herrera, D.G.; Toth, M.; Yang, C.; McEwen, B.S.; et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 2006, 314, eaay8477. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Ieraci, A.; Teng, H.; Dall, H.; Meng, C.X.; Herrera, D.G.; Nykjaer, A.; Hempstead, B.L.; Lee, F.S. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 2005, 25, 6156–6166. [Google Scholar] [CrossRef]
- Lou, H.; Kim, S.K.; Zaitsev, E.; Snell, C.R.; Lu, B.; Loh, Y.P. Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase E. Neuron 2005, 45, 585–595. [Google Scholar] [CrossRef]
- Evans, S.F.; Irmady, K.; Ostrow, K.; Kim, T.; Nykjaer, A.; Saftig, P.; Blobel, C.; Hempstead, B.L. Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J. Biol. Chem. 2011, 286, 29556–29567. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Caldeira, M.V.; Santos, S.D.; Duarte, C.B. Role of the brain-derived neurotrophic factor at glutamatergic synapses. Br. J. Pharmacol. 2008, 153, S310–S324. [Google Scholar] [CrossRef]
- Skaper, S.D. Neurotrophic factors: An overview. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1727. [Google Scholar]
- Lu, J.J.; Yang, M.; Sun, Y.; Zhou, X.F. Synthesis, trafficking and release of BDNF. In Handbook of Neurotoxicity; Springer: New York, NY, USA, 2014; Volume 3. [Google Scholar]
- Adachi, N.; Kohara, K.; Tsumoto, T. Difference in trafficking of brain-derived neurotrophic factor between axons and dendrites of cortical neurons, revealed by live-cell imaging. BMC Neurosci. 2005, 6, 42. [Google Scholar] [CrossRef]
- Dieni, S.; Matsumoto, T.; Dekkers, M.; Rauskolb, S.; Ionescu, M.S.; Deogracias, R.; Gundelfinger, E.D.; Kojima, M.; Nestel, S.; Frotscher, M.; et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol. 2012, 196, 775–788. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef]
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721. [Google Scholar] [CrossRef]
- Binder, D.K.; Scharfman, H.E. Brain-derived neurotrophic factor. Growth Factors 2004, 22, 123. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Friedman, W.J. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J. Neurosci. 2000, 20, 6340–6346. [Google Scholar] [CrossRef]
- Meeker, R.; Williams, K. Dynamic Nature of the p75 Neurotrophin Receptor in Response to Injury and Disease. J. Neuroimmune Pharmacol. 2014, 9, 615–628. [Google Scholar] [CrossRef]
- Hempstead, B. Dissecting the Diverse Actions of Pro- and Mature Neurotrophins. Curr. Alzheimer Res. 2006, 3, 19–24. [Google Scholar] [CrossRef]
- Zanin, J.P.; Montroull, L.E.; Volosin, M.; Friedman, W.J. The p75 Neurotrophin Receptor Facilitates TrkB Signaling and Function in Rat Hippocampal Neurons. Front. Cell. Neurosci. 2019, 13, 485. [Google Scholar] [CrossRef]
- Diniz, C.R.A.F.; Casarotto, P.C.; Resstel, L.; Joca, S.R.L. Beyond good and evil: A putative continuum-sorting hypothesis for the functional role of proBDNF/BDNF-propeptide/mBDNF in antidepressant treatment. Neurosci. Biobehav. Rev. 2018, 90, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Segal, R.A. Selectivity in neurotrophin signaling: Theme and variations. Annu. Rev. Neurosci. 2003, 26, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of dendritic arborization by the phosphoinositide-3′-kinase- Akt-mammalian target of rapamycin pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK Pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- Sakuragi, S.; Tominaga-Yoshino, K.; Ogura, A. Involvement of TrkB- and p75MNTR -signaling pathways in two contrasting forms of long-lasting synaptic plasticity. Sci. Rep. 2013, 3, 3185. [Google Scholar] [CrossRef]
- Woo, N.H.; Teng, H.K.; Siao, C.J.; Chiaruttini, C.; Pang, P.T.; Milner, T.A.; Hempstead, B.L.; Lu, B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat. Neurosci. 2005, 8, 1069–1077. [Google Scholar] [CrossRef]
- Deinhardt, K.; Kim, T.; Spellman, D.S.; Mains, R.E.; Eipper, B.A.; Neubert, T.A.; Chao, M.V.; Hempstead, B.L. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through rac. Sci. Signal. 2011, 4, ra82. [Google Scholar] [CrossRef]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; LaFrancois, J.J.; Bath, K.G.; Mark, W.; et al. ProBDNF Negatively Regulates Neuronal Remodeling, Synaptic Transmission, and Synaptic Plasticity in Hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef]
- Suelves, N.; Miguez, A.; López-Benito, S.; Barriga, G.G.D.; Giralt, A.; Alvarez-Periel, E.; Arévalo, J.C.; Alberch, J.; Ginés, S.; Brito, V. Early Downregulation of p75 NTR by Genetic and Pharmacological Approaches Delays the Onset of Motor Deficits and Striatal Dysfunction in Huntington’s Disease Mice. Mol. Neurobiol. 2019, 56, 935–953. [Google Scholar] [CrossRef]
- Brito, V.; Puigdellívol, M.; Giralt, A.; Del Toro, D.; Alberch, J.; Ginés, S. Imbalance of p75NTR/TrkB protein expression in Huntington’s disease: Implication for neuroprotective therapies. Cell Death Dis. 2013, 4, e595. [Google Scholar] [CrossRef]
- Yi, X.; Yang, Y.; Zhao, Z.; Xu, M.; Zhang, Y.; Sheng, Y.; Tian, J.; Xu, Z. Serum mBDNF and ProBDNF Expression Levels as Diagnosis Clue for Early Stage Parkinson’s Disease. Front. Neurol. 2021, 12, 1200. [Google Scholar] [CrossRef]
- Mizui, T.; Ohira, K.; Kojima, M. BDNF pro-peptide: A novel synaptic modulator generated as an N-terminal fragment from the BDNF precursor by proteolytic processing. Neural Regen. Res. 2017, 12, 1024–1027. [Google Scholar] [CrossRef]
- Mizui, T.; Ishikawa, Y.; Kumanogoh, H.; Kojima, M. Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: Multi-ligand model of growth factor signaling. Pharmacol. Res. 2016, 105, 93–98. [Google Scholar] [CrossRef]
- Uegaki, K.; Kumanogoh, H.; Mizui, T.; Hirokawa, T.; Ishikawa, Y.; Kojima, M. BDNF binds its pro-peptide with high affinity and the common val66met polymorphism attenuates the interaction. Int. J. Mol. Sci. 2017, 18, 1042. [Google Scholar] [CrossRef]
- Giza, J.I.; Kim, J.; Meyer, H.C.; Anastasia, A.; Dincheva, I.; Zheng, C.I.; Lopez, K.; Bains, H.; Yang, J.; Bracken, C.; et al. The BDNF Val66Met Prodomain Disassembles Dendritic Spines Altering Fear Extinction Circuitry and Behavior. Neuron 2018, 99, 163–178. [Google Scholar] [CrossRef]
- Tudor, L.; Konjevod, M.; Perkovic, M.N.; Strac, D.S.; Erjavec, G.N.; Uzun, S.; Kozumplik, O.; Sagud, M.; Petrovic, Z.K.; Pivac, N. Genetic variants of the brain-derived neurotrophic factor and metabolic indices in veterans with posttraumatic stress disorder. Front. Psychiatry 2018, 9, 637. [Google Scholar] [CrossRef]
- Huang, R.; Huang, J.; Cathcart, H.; Smith, S.; Poduslo, S.E. Genetic variants in brain-derived neurotrophic factor associated with Alzheimer’s disease. J. Med. Genet. 2007, 44, e66. [Google Scholar] [CrossRef]
- Shen, T.; You, Y.; Joseph, C.; Mirzaei, M.; Klistorner, A.; Graham, S.L.; Gupta, V. BDNF polymorphism: A review of its diagnostic and clinical relevance in neurodegenerative disorders. Aging Dis. 2018, 9, 523. [Google Scholar] [CrossRef]
- Petryshen, T.L.; Sabeti, P.C.; Aldinger, K.A.; Fry, B.; Fan, J.B.; Schaffner, S.F.; Waggoner, S.G.; Tahl, A.R.; Sklar, P. Population genetic study of the brain-derived neurotrophic factor (BDNF) gene. Mol. Psychiatry 2010, 15, 810–815. [Google Scholar] [CrossRef]
- Tsai, S.J. Critical issues in BDNF Val66met genetic studies of neuropsychiatric disorders. Front. Mol. Neurosci. 2018, 11, 156. [Google Scholar] [CrossRef]
- Lee, Y.; Lim, S.W.; Kim, S.Y.; Chung, J.W.; Kim, J.; Myung, W.; Song, J.; Kim, S.; Carroll, B.J.; Kim, D.K. Association between the BDNF Val66Met Polymorphism and Chronicity of Depression. Psychiatry Investig. 2013, 10, 56. [Google Scholar] [CrossRef]
- Mei, S.; Chen, W.; Chen, S.; Hu, Y.; Dai, X.; Liu, X. Evaluation of the Relationship Between BDNF Val66Met Gene Polymorphism and Attention Deficit Hyperactivity Disorder: A Meta-Analysis. Front. Psychiatry 2022, 13, 827658. [Google Scholar] [CrossRef]
- Di Carlo, P.; Punzi, G.; Ursini, G. Brain-derived neurotrophic factor and schizophrenia. Psychiatr. Genet. 2019, 29, 200–210. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, N.; Zhang, E.; Liu, Q.; Li, H.; Zhao, X. The Brain-Derived Neurotrophic Factor Val66Met Polymorphism Is Associated With Female Obsessive-Compulsive Disorder: An Updated Meta-Analysis of 2765 Obsessive-Compulsive Disorder Cases and 5558 Controls. Front. Psychiatry 2022, 12, 685041. [Google Scholar] [CrossRef]
- Cai, X.; Shi, X.; Zhang, X.; Zhang, A.; Zheng, M.; Fang, Y. The association between brain-derived neurotrophic factor gene polymorphism and migraine: A meta-analysis. J. Headache Pain 2017, 18, 13. [Google Scholar] [CrossRef]
- Siokas, V.; Kardaras, D.; Aloizou, A.M.; Asproudis, I.; Boboridis, K.G.; Papageorgiou, E.; Hadjigeorgiou, G.M.; Tsironi, E.E.; Dardiotis, E. BDNF rs6265 (Val66Met) Polymorphism as a Risk Factor for Blepharospasm. NeuroMol. Med. 2019, 21, 68–74. [Google Scholar] [CrossRef]
- Huang, Y.; Yun, W.; Zhang, M.; Luo, W.; Zhou, X. Serum concentration and clinical significance of brain-derived neurotrophic factor in patients with Parkinson’s disease or essential tremor. J. Int. Med. Res. 2018, 46, 1477–1485. [Google Scholar] [CrossRef]
- Scalzo, P.; Kümmer, A.; Bretas, T.L.; Cardoso, F.; Teixeira, A.L. Serum levels of brain-derived neurotrophic factor correlate with motor impairment in Parkinson’s disease. J. Neurol. 2010, 257, 540–545. [Google Scholar] [CrossRef]
- Razgado-Hernandez, L.F.; Espadas-Alvarez, A.J.; Reyna-Velazquez, P.; Sierra-Sanchez, A.; Anaya-Martinez, V.; Jimenez-Estrada, I.; Bannon, M.J.; Martinez-Fong, D.; Aceves-Ruiz, J. The transfection of BDNF to dopamine neurons potentiates the effect of dopamine D3 receptor agonist recovering the striatal innervation, dendritic spines and motor behavior in an aged rat model of Parkinson’s disease. PLoS ONE 2015, 10, e0117391. [Google Scholar] [CrossRef]
- Baquet, Z.C.; Gorski, J.A.; Jones, K.R. Early Striatal Dendrite Deficits followed by Neuron Loss with Advanced Age in the Absence of Anterograde Cortical Brain-Derived Neurotrophic Factor. J. Neurosci. 2004, 24, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.F.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-Specific Neurotrophin Imbalances in Alzheimer Disease. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 1996, 7, 2925–2928. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Ferrer, I.; Goutan, E.; Marín, C.; Rey, M.J.; Ribalta, T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000, 866, 257–261. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Kingsbury, A.; Welcher, A.A.; Wilkin, G.P. Elevated glial brain-derived neurotrophic factor in Parkinson’s diseased nigra. Park. Relat. Disord. 2002, 8, 329–341. [Google Scholar] [CrossRef]
- Molendijk, M.L.; Spinhoven, P.; Polak, M.; Bus, B.A.A.; Penninx, B.W.J.H.; Elzinga, B.M. Serum BDNF concentrations as peripheral manifestations of depression: Evidence from a systematic review and meta-analyses on 179 associations (N = 9484). Mol. Psychiatry 2014, 19, 791–800. [Google Scholar] [CrossRef]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.J.O.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef]
- Shimizu, E.; Hashimoto, K.; Okamura, N.; Koike, K.; Komatsu, N.; Kumakiri, C.; Nakazato, M.; Watanabe, H.; Shinoda, N.; Okada, S.I.; et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol. Psychiatry 2003, 54, 70–75. [Google Scholar] [CrossRef]
- Januar, V.; Ancelin, M.L.; Ritchie, K.; Saffery, R.; Ryan, J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef]
- Pandey, G.N.; Ren, X.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Dwivedi, Y. Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int. J. Neuropsychopharmacol. 2008, 11, 1047–1061. [Google Scholar] [CrossRef]
- Dwivedi, Y.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Tamminga, C.A.; Pandey, G.N. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch. Gen. Psychiatry 2003, 60, 804–815. [Google Scholar] [CrossRef]
- Weickert, C.S.; Hyde, T.M.; Lipska, B.K.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2003, 8, 592–610. [Google Scholar] [CrossRef]
- Weickert, C.S.; Ligons, D.L.; Romanczyk, T.; Ungaro, G.; Hyde, T.M.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2005, 10, 637–650. [Google Scholar] [CrossRef]
- Reinhart, V.; Bove, S.E.; Volfson, D.; Lewis, D.A.; Kleiman, R.J.; Lanz, T.A. Evaluation of TrkB and BDNF transcripts in prefrontal cortex, hippocampus, and striatum from subjects with schizophrenia, bipolar disorder, and major depressive disorder. Neurobiol. Dis. 2015, 77, 220–227. [Google Scholar] [CrossRef]
- Hashimoto, T.; Bergen, S.E.; Nguyen, Q.L.; Xu, B.; Monteggia, L.M.; Pierri, J.N.; Sun, Z.; Sampson, A.R.; Lewis, D.A. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 2005, 25, 372–383. [Google Scholar] [CrossRef]
- Xiu, M.H.; Hui, L.; Dang, Y.F.; De Hou, T.; Zhang, C.X.; Zheng, Y.L.; Chen, D.C.; Kosten, T.R.; Zhang, X.Y. Decreased serum BDNF levels in chronic institutionalized schizophrenia on long-term treatment with typical and atypical antipsychotics. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 1508–1512. [Google Scholar] [CrossRef]
- Sortwell, C.E.; Hacker, M.L.; Fischer, D.L.; Konrad, P.E.; Davis, T.L.; Neimat, J.S.; Wang, L.; Song, Y.; Mattingly, Z.R.; Cole-Strauss, A.; et al. BDNF rs6265 Genotype Influences Outcomes of Pharmacotherapy and Subthalamic Nucleus Deep Brain Stimulation in Early-Stage Parkinson’s Disease. Neuromodulation 2021, in press. [CrossRef]
- Fischer, D.L.; Auinger, P.; Goudreau, J.L.; Paumier, K.L.; Cole-Strauss, A.; Kemp, C.J.; Lipton, J.W.; Sortwell, C.E. Bdnf variant is associated with milder motor symptom severity in early-stage Parkinson’s disease. Park. Relat. Disord. 2018, 53, 70–75. [Google Scholar] [CrossRef]
- Drozdzik, M.; Bialecka, M.; Kurzawski, M. Pharmacogenetics of Parkinson’s Disease—Through Mechanisms of Drug Actions. Curr. Genomics 2014, 14, 568–577. [Google Scholar] [CrossRef]
- Karamohamed, S.; Latourelle, J.C.; Racette, B.A.; Perlmutter, J.S.; Wooten, G.F.; Lew, M.; Klein, C.; Shill, H.; Golbe, L.I.; Mark, M.H.; et al. BDNF genetic variants are associated with onset age of familial Parkinson disease: GenePD Study. Neurology 2005, 65, 1823–1825. [Google Scholar] [CrossRef]
- Fukumoto, N.; Fujii, T.; Combarros, O.; Kamboh, M.I.; Tsai, S.J.; Matsushita, S.; Nacmias, B.; Comings, D.E.; Arboleda, H.; Ingelsson, M.; et al. Sexually dimorphic effect of the Val66Met oolymorphism of BDNF on susceptibility to Alzheimer’s disease: New data and meta-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153, 235–242. [Google Scholar] [CrossRef]
- Laing, K.R.; Mitchell, D.; Wersching, H.; Czira, M.E.; Berger, K.; Baune, B.T. Brain-derived neurotrophic factor (BDNF) gene: A gender-specific role in cognitive function during normal cognitive aging of the MEMO-Study? Age 2012, 34, 1011–1022. [Google Scholar] [CrossRef]
- Borroni, B.; Grassi, M.; Archetti, S.; Costanzi, C.; Bianchi, M.; Caimi, L.; Caltagirone, C.; Di Luca, M.; Padovani, A. BDNF genetic variations increase the risk of Alzheimer’s disease-related depression. J. Alzheimer’s Dis. 2009, 18, 867–875. [Google Scholar] [CrossRef]
- Alberch, J.; López, M.; Badenas, C.; Carrasco, J.L.; Milà, M.; Muñoz, E.; Canals, J.M. Association between BDNF Val66Met polymorphism and age at onset in Huntington disease. Neurology 2005, 65, 964–965. [Google Scholar] [CrossRef]
- Losenkov, I.S.; Mulder, N.J.V.; Levchuk, L.A.; Vyalova, N.M.; Loonen, A.J.M.; Bosker, F.J.; Simutkin, G.G.; Boiko, A.S.; Bokhan, N.A.; Wilffert, B.; et al. Association Between BDNF Gene Variant Rs6265 and the Severity of Depression in Antidepressant Treatment-Free Depressed Patients. Front. Psychiatry 2020, 11, 38. [Google Scholar] [CrossRef]
- Pei, Y.; Smith, A.K.; Wang, Y.; Pan, Y.; Yang, J.; Chen, Q.; Pan, W.; Bao, F.; Zhao, L.; Tie, C.; et al. The brain-derived neurotrophic-factor (BDNF) val66met polymorphism is associated with geriatric depression: A meta-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159B, 560–566. [Google Scholar] [CrossRef]
- Hosang, G.M.; Shiles, C.; Tansey, K.E.; McGuffin, P.; Uher, R. Interaction between stress and the BDNF Val66Met polymorphism in depression: A systematic review and meta-analysis. BMC Med. 2014, 12, 7. [Google Scholar] [CrossRef]
- Kheirollahi, M.; Kazemi, E.; Ashouri, S. Brain-Derived Neurotrophic Factor Gene Val66Met Polymorphism and Risk of Schizophrenia: A Meta-analysis of Case–Control Studies. Cell. Mol. Neurobiol. 2016, 36, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Gratacòs, M.; González, J.R.; Mercader, J.M.; de Cid, R.; Urretavizcaya, M.; Estivill, X. Brain-Derived Neurotrophic Factor Val66Met and Psychiatric Disorders: Meta-Analysis of Case-Control Studies Confirm Association to Substance-Related Disorders, Eating Disorders, and Schizophrenia. Biol. Psychiatry 2007, 61, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Suchanek, R.; Owczarek, A.; Paul-Samojedny, M.; Kowalczyk, M.; Kucia, K.; Kowalski, J. BDNF val66met polymorphism is associated with age at onset and intensity of symptoms of paranoid schizophrenia in a Polish population. J. Neuropsychiatry Clin. Neurosci. 2013, 25, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Zhang, C.; Wu, Z.; Hong, W.; Li, Z.; Fang, Y.; Yu, S. Lack of effect of brain derived neurotrophic factor (BDNF) Val66Met polymorphism on early onset schizophrenia in Chinese Han population. Brain Res. 2011, 1417, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.M.; Kao, H.T.; Porton, B. BDNF Val66Met variant and age of onset in schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 505–506. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Yurek, D.M.; Fletcher-Turner, A. Differential expression of GDNF, BDNF, and NT-3 in the aging nigrostriatal system following a neurotoxic lesion. Brain Res. 2001, 891, 228–235. [Google Scholar] [CrossRef]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef]
- Mercado, N.M.; Collier, T.J.; Sortwell, C.E.; Steece-Collier, K. BDNF in the Aged Brain: Translational Implications for Parkinson’s Disease. Austin Neurol. Neurosci. 2017, 2, 1021. [Google Scholar]
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Ventriglia, M.; Zanardini, R.; Bonomini, C.; Zanetti, O.; Volpe, D.; Pasqualetti, P.; Gennarelli, M.; Bocchio-Chiavetto, L. Serum brain-derived neurotrophic factor levels in different neurological diseases. Biomed Res. Int. 2013, 2013, 901082. [Google Scholar] [CrossRef]
- Jin, W. Regulation of bdnf-trkb signaling and potential therapeutic strategies for parkinson’s disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef]
- Fenner, M.E.; Achim, C.L.; Fenner, B.M. Expression of full-length and truncated trkB in human striatum and substantia nigra neurons: Implications for Parkinson’s disease. J. Mol. Histol. 2014, 45, 349–361. [Google Scholar] [CrossRef]
- Mitre, M.; Mariga, A.; Chao, M.V. Neurotrophin signalling: Novel insights into mechanisms and pathophysiology. Clin. Sci. 2017, 131, 13–23. [Google Scholar] [CrossRef]
- Porritt, M.J.; Batchelor, P.E.; Howells, D.W. Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp. Neurol. 2005, 192, 226–234. [Google Scholar] [CrossRef]
- Fedosova, A.; Titova, N.; Kokaeva, Z.; Shipilova, N.; Katunina, E.; Klimov, E. Genetic markers as risk factors for the development of impulsive-compulsive behaviors in patients with parkinson’s disease receiving dopaminergic therapy. J. Pers. Med. 2021, 11, 1321. [Google Scholar] [CrossRef]
- Gorzkowska, A.; Cholewa, J.; Cholewa, J.; Wilk, A.; Klimkowicz-Mrowiec, A. Risk factors for apathy in Polish patients with parkinson’s disease. Int. J. Environ. Res. Public Health 2021, 18, 10196. [Google Scholar] [CrossRef]
- Fischer, D.L.; Auinger, P.; Goudreau, J.L.; Cole-Strauss, A.; Kieburtz, K.; Elm, J.J.; Hacker, M.L.; Charles, P.D.; Lipton, J.W.; Pickut, B.A.; et al. BDNF rs6265 Variant Alters Outcomes with Levodopa in Early-Stage Parkinson’s Disease. Neurotherapeutics 2020, 17, 1785–1795. [Google Scholar] [CrossRef]
- Foltynie, T.; Cheeran, B.; Williams-Gray, C.H.; Edwards, M.J.; Schneider, S.A.; Weinberger, D.; Rothwell, J.C.; Barker, R.A.; Bhatia, K.P. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 141–144. [Google Scholar] [CrossRef]
- Białecka, M.; Kurzawski, M.; Roszmann, A.; Robowski, P.; Sitek, E.J.; Honczarenko, K.; Mak, M.; Deptuła-Jarosz, M.; Gołab-Janowska, M.; Droździk, M.; et al. BDNF G196A (Val66Met) polymorphism associated with cognitive impairment in Parkinson’s disease. Neurosci. Lett. 2014, 561, 86–90. [Google Scholar] [CrossRef]
- Svetel, M.; Pekmezovic, T.; Markovic, V.; Novaković, I.; Dobričić, V.; Djuric, G.; Stefanova, E.; Kostić, V. No association between brain-derived neurotrophic factor g196a polymorphism and clinical features of parkinson’s disease. Eur. Neurol. 2013, 70, 257–262. [Google Scholar] [CrossRef]
- Hauser, R.A.; Auinger, P.; Oakes, D. Levodopa response in early Parkinson’s disease. Mov. Disord. 2009, 24, 2328–2336. [Google Scholar] [CrossRef]
- Parkinson’s Disease 2014: Advancing Research, Improving Lives, U.S. Dep. Heal. Hum. Serv. (n.d.). Available online: https://www.ninds.nih.gov/parkinsons-disease-2014-advancing-research-improving-lives (accessed on 6 June 2022).
- Schroeder, S.K.; Joly-Amado, A.; Gordon, M.N.; Morgan, D. Tau-Directed Immunotherapy: A Promising Strategy for Treating Alzheimer’s Disease and Other Tauopathies. J. Neuroimmune Pharmacol. 2016, 11, 9–25. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ikonomovic, M.D.; Counts, S.E.; Perez, S.E.; Malek-Ahmadi, M.; Scheff, S.W.; Ginsberg, S.D. Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav. Brain Res. 2016, 311, 54–69. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical Implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Malek-Ahmadi, M.H.; Alldred, M.J.; Chen, Y.; Chen, K.; Chao, M.V.; Counts, S.E.; Mufson, E.J. Brain-derived neurotrophic factor (BDNF) and TrkB hippocampal gene expression are putative predictors of neuritic plaque and neurofibrillary tangle pathology. Neurobiol. Dis. 2019, 132, 104540. [Google Scholar] [CrossRef] [PubMed]
- Murer, M.G.; Boissiere, F.; Yan, Q.; Hunot, S.; Villares, J.; Faucheux, B.; Agid, Y.; Hirsch, E.; Raisman-Vozari, R. An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience 1999, 88, 1015–1032. [Google Scholar] [CrossRef]
- Ferrer, I.; Marín, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Martí, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Maetzler, W.; Wittorf, A.; Soekadar, S.; Richartz, E.; Koehler, N.; Bartels, M.; et al. BDNF serum and CSF concentrations in Alzheimer’s disease, normal pressure hydrocephalus and healthy controls. J. Psychiatr. Res. 2007, 41, 387–394. [Google Scholar] [CrossRef]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol. Aging 2018, 70, 308–324. [Google Scholar] [CrossRef]
- Li, G.D.; Bi, R.; Zhang, D.F.; Xu, M.; Luo, R.; Wang, D.; Fang, Y.; Li, T.; Zhang, C.; Yao, Y.G. Female-specific effect of the BDNF gene on Alzheimer’s disease. Neurobiol. Aging 2017, 53, 192-e11. [Google Scholar] [CrossRef]
- Bagnoli, S.; Nacmias, B.; Tedde, A.; Guarnieri, B.M.; Cellini, E.; Petruzzi, C.; Bartoli, A.; Ortenzi, L.; Sorbi, S. Brain-Derived Neurotrophic Factor Genetic Variants Are Not Susceptibility Factors to Alzheimer’s Disease in Italy. Ann. Neurol. 2004, 55, 447–448. [Google Scholar] [CrossRef]
- Chuu, J.Y.J.; Taylor, J.L.; Tinklenberg, J.; Noda, A.; Yesavage, J.; Murphy, G.M. The brain-derived neurotrophic factor Val66Met polymorphism and rate of decline in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 43–49. [Google Scholar] [CrossRef]
- He, X.M.; Zhang, Z.X.; Zhang, J.W.; Zhou, Y.T.; Tang, M.N.; Wu, C.B.; Hong, Z. Lack of association between the BDNF gene Val66Met polymorphism and Alzheimer disease in a Chinese Han population. Neuropsychobiology 2007, 55, 151–155. [Google Scholar] [CrossRef]
- Saarela, M.S.; Lehtimäki, T.; Rinne, J.O.; Huhtala, H.; Rontu, R.; Hervonen, A.; Röyttä, M.; Ahonen, J.P.; Mattila, K.M. No association between the brain-derived neurotrophic factor 196G>A or 270C>T polymorphisms and Alzheimer’s or Parkinson’s disease. Folia Neuropathol. 2006, 44, 12–16. [Google Scholar]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008, 18, 399–411. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef]
- Mai, M.; Akkad, A.D.; Wieczorek, S.; Saft, C.; Andrich, J.; Kraus, P.H.; Epplen, J.T.; Arning, L. No association between polymorphisms in the BDNF gene and age at onset in Huntington disease. BMC Med. Genet. 2006, 7, 10. [Google Scholar] [CrossRef]
- Di Maria, E.; Marasco, A.; Tartari, M.; Ciotti, P.; Abbruzzese, G.; Novelli, G.; Bellone, E.; Cattaneo, E.; Mandich, P. No evidence of association between BDNF gene variants and age-at-onset of Huntington’s disease. Neurobiol. Dis. 2006, 24, 274–279. [Google Scholar] [CrossRef]
- Kishikawa, S.; Li, J.L.; Gillis, T.; Hakky, M.M.; Warby, S.; Hayden, M.; MacDonald, M.E.; Myers, R.H.; Gusella, J.F. Brain-derived neurotrophic factor does not influence age at neurologic onset of Huntington’s disease. Neurobiol. Dis. 2006, 24, 280–285. [Google Scholar] [CrossRef]
- Metzger, S.; Bauer, P.; Tomiuk, J.; Laccone, F.; DiDonato, S.; Gellera, C.; Mariotti, C.; Lange, H.W.; Weirich-Schwaiger, H.; Wenning, G.K.; et al. Genetic analysis of candidate genes modifying the age-at-onset in Huntington’s disease. Hum. Genet. 2006, 120, 285–292. [Google Scholar] [CrossRef]
- Levy, M.J.F.; Boulle, F.; Steinbusch, H.W.; van den Hove, D.L.A.; Kenis, G.; Lanfumey, L. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology 2018, 235, 2195–2220. [Google Scholar] [CrossRef]
- Elfving, B.; Buttenschøn, H.N.; Foldager, L.; Poulsen, P.H.P.; Andersen, J.H.; Grynderup, M.B.; Hansen, Å.M.; Kolstad, H.A.; Kaerlev, L.; Mikkelsen, S.; et al. Depression, the Val66Met polymorphism, age, and gender influence the serum BDNF level. J. Psychiatr. Res. 2012, 46, 1118–1125. [Google Scholar] [CrossRef]
- Ray, M.T.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 2011, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Paumier, K.L.; Sortwell, C.E.; Madhavan, L.; Terpstra, B.; Celano, S.L.; Green, J.J.; Imus, N.M.; Marckini, N.; Daley, B.; Steece-Collier, K.; et al. Chronic amitriptyline treatment attenuates nigrostriatal degeneration and significantly alters trophic support in a rat model of parkinsonism. Neuropsychopharmacology 2015, 40, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Youssef, M.M.; Underwood, M.D.; Huang, Y.Y.; Hsiung, S.C.; Liu, Y.; Simpson, N.R.; Bakalian, M.J.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; et al. Association of BDNF Val66MET polymorphism and brain BDNF levels with major depression and suicide. Int. J. Neuropsychopharmacol. 2018, 21, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Chagnon, Y.C.; Potvin, O.; Hudon, C.; Préville, M. DNA methylation and single nucleotide variants in the brain-derived neurotrophic factor (BDNF) and oxytocin receptor (OXTR) genes are associated with anxiety/depression in older women. Front. Genet. 2015, 6, 230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.K.; Mitra, P.; Ghosh, R.; Sharma, S.; Nebhinani, N.; Sharma, P. Association of circulating BDNF levels with BDNF rs6265 polymorphism in schizophrenia. Behav. Brain Res. 2020, 394, 112832. [Google Scholar] [CrossRef]
- Lee, M.T.; Mouri, A.; Kubota, H.; Lee, H.-J.; Chang, M.-H.; Wu, C.-Y.; Knutson, D.E.; Mihovilovic, M.; Cook, J.; Sieghart, W.; et al. Targeting α6GABAA receptors as a novel therapy for schizophrenia: A proof-of-concept preclinical study using various animal models. Biomed. Pharmacother. 2022, 150, 113022. [Google Scholar] [CrossRef]
- Miyazawa, A.; Kanahara, N.; Shiko, Y.; Ozawa, Y.; Kawasaki, Y.; Komatsu, H.; Masumo, Y.; Nakata, Y.; Iyo, M. The cortical silent period in schizophrenia: A systematic review and meta-analysis focusing on disease stage and antipsychotic medication. J. Psychopharmacol. 2022, 36, 479–488. [Google Scholar] [CrossRef]
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.G.; Steiner, J.; Bogerts, B.; Braun, K.; Kumaratilake, J.; Henneberg, M.; et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef]
- Nieto, R.; Kukuljan, M.; Silva, H. BDNF and schizophrenia: From neurodevelopment to neuronal plasticity, learning, and memory. Front. Psychiatry 2013, 4, 45. [Google Scholar] [CrossRef]
- Green, M.J.; Matheson, S.L.; Shepherd, A.; Weickert, C.S.; Carr, V.J. Brain-derived neurotrophic factor levels in schizophrenia: A systematic review with meta-analysis. Mol. Psychiatry 2011, 16, 960–972. [Google Scholar] [CrossRef]
- Toyooka, K.; Asama, K.; Watanabe, Y.; Muratake, T.; Takahashi, M.; Someya, T.; Nawa, H. Decreased levels of brain-derived neurotrophic factor in serum of chronic schizophrenic patients. Psychiatry Res. 2002, 110, 249–257. [Google Scholar] [CrossRef]
- Dong, E.; Dzitoyeva, S.G.; Matrisciano, F.; Tueting, P.; Grayson, D.R.; Guidotti, A. Brain-derived neurotrophic factor epigenetic modifications associated with schizophrenia-like phenotype induced by prenatal stress in mice. Biol. Psychiatry 2015, 77, 589–596. [Google Scholar] [CrossRef]
- Fillman, S.G.; Cloonan, N.; Catts, V.S.; Miller, L.C.; Wong, J.; Mccrossin, T.; Cairns, M.; Weickert, C.S. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2013, 18, 206–214. [Google Scholar] [CrossRef]
- Takahashi, M.; Shirakawa, O.; Toyooka, K.; Kitamura, N.; Hashimoto, T.; Maeda, K.; Koizumi, S.; Wakabayashi, K.; Takahashi, H.; Someya, T.; et al. Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol. Psychiatry 2000, 5, 293–300. [Google Scholar] [CrossRef]
- Xia, H.; Zhang, G.; Du, X.; Zhang, Y.; Yin, G.; Dai, J.; He, M.X.; Soares, J.C.; Li, X.; Zhang, X.Y. Suicide attempt, clinical correlates, and BDNF Val66Met polymorphism in chronic patients with schizophrenia. Neuropsychology 2018, 32, 199. [Google Scholar] [CrossRef]
- Skibinska, M.; Groszewska, A.; Kapelski, P.; Rajewska-Rager, A.; Pawlak, J.; Dmitrzak-Weglarz, M.; Szczepankiewicz, A.; Twarowska-Hauser, J. Val66Met functional polymorphism and serum protein level of brain-derived neurotrophic factor (BDNF) in acute episode of schizophrenia and depression. Pharmacol. Rep. 2018, 70, 55–59. [Google Scholar] [CrossRef]
- Zakharyan, R.; Boyajyan, A.; Arakelyan, A.; Gevorgyan, A.; Mrazek, F.; Petrek, M. Functional variants of the genes involved in neurodevelopment and susceptibility to schizophrenia in an Armenian population. Hum. Immunol. 2011, 72, 746–748. [Google Scholar] [CrossRef]
- Numata, S.; Ueno, S.I.; Iga, J.I.; Yamauchi, K.; Hongwei, S.; Ohta, K.; Kinouchi, S.; Shibuya-Tayoshi, S.; Tayoshi, S.; Aono, M.; et al. Brain-derived neurotrophic factor (BDNF) Val66Met polymorphism in schizophrenia is associated with age at onset and symptoms. Neurosci. Lett. 2006, 401, 1–5. [Google Scholar] [CrossRef]
- Hung, H.C.; Lee, E.H.Y. The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: Role of BDNF gene expression. Mol. Brain Res. 1996, 41, 16–26. [Google Scholar] [CrossRef]
- Arancibia, S.; Silhol, M.; Moulière, F.; Meffre, J.; Höllinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef]
- Deng, P.; Anderson, J.D.; Yu, A.S.; Annett, G.; Fink, K.D.; Nolta, J.A. Engineered BDNF producing cells as a potential treatment for neurologic disease. Expert Opin. Biol. Ther. 2016, 16, 1025–1033. [Google Scholar] [CrossRef]
- Altar, C.A.; Boylan, C.B.; Fritsche, M.; Jones, B.E.; Jackson, C.; Wiegand, S.J.; Lindsay, R.M.; Hyman, C. Efficacy of Brain-Derived Neurotrophic Factor and Neurotrophin-3 on Neurochemical and Behavioral Deficits Associated with Partial Nigrostriatal Dopamine Lesions. J. Neurochem. 1994, 63, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.G. A controlled trial of recombinant methionyl human BDNF in ALS. Neurology 1999, 52, 1427–1433. [Google Scholar] [CrossRef]
- Ochs, G.; Penn, R.D.; York, M.; Giess, R.; Beck, M.; Tonn, J.; Haigh, J.; Malta, E.; Traub, M.; Sendtner, M.; et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2000, 1, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Genge, A.; Arnold, D.L. A prospective, randomized, placebo-controlled evaluation of corticoneuronal response to intrathecal BDNF therapy in ALS using magnetic resonance spectroscopy: Feasibility and results. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. The brain-derived neurotrophic factor in neuronal plasticity and neuroregeneration: New pharmacological concepts for old and new drugs. Neural Regen. Res. 2018, 13, 983. [Google Scholar] [CrossRef]
- Yeom, C.W.; Park, Y.J.; Choi, S.W.; Bhang, S.Y. Association of peripheral BDNF level with cognition, attention and behavior in preschool children. Child Adolesc. Psychiatry Ment. Health 2016, 10, 10. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef]
- Chen, C.; Dong, Y.; Liu, F.; Gao, C.; Ji, C.; Dang, Y.; Ma, X.; Liu, Y. A study of antidepressant effect and mechanism on intranasal delivery of BDNF-HA2TAT/AAV to rats with post-stroke depression. Neuropsychiatr. Dis. Treat. 2020, 16, 637. [Google Scholar] [CrossRef]
- Kells, A.P.; Fong, D.M.; Dragunow, M.; During, M.J.; Young, D.; Connor, B. AAV-mediated gene delivery of BDNF or GDNF is neuroprotective in a model of Huntington disease. Mol. Ther. 2004, 9, 682–688. [Google Scholar] [CrossRef]
- Manfredsson, F.P.; Polinski, N.K.; Subramanian, T.; Boulis, N.; Wakeman, D.R.; Mandel, R.J. The Future of GDNF in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 593572. [Google Scholar] [CrossRef]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Merola, A.; Van Laar, A.; Lonser, R.; Bankiewicz, K. Gene therapy for Parkinson’s disease: Contemporary practice and emerging concepts. Expert Rev. Neurother. 2020, 20, 577–590. [Google Scholar] [CrossRef]
- Levivier, M.; Przedborski, S.; Bencsics, C.; Kang, U.J. Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 1995, 15, 7810–7820. [Google Scholar] [CrossRef]
- Crane, A.T.; Rossignol, J.; Dunbar, G.L. Use of genetically altered stem cells for the treatment of Huntington’s disease. Brain Sci. 2014, 4, 202–219. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Sang, U.H.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef]
- Baum, C.; Düllmann, J.; Li, Z.; Fehse, B.; Meyer, J.; Williams, D.A.; Von Kalle, C. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood 2003, 101, 2099–2113. [Google Scholar] [CrossRef]
- Baum, C.; von Kalle, C.; Staal, F.J.T.; Li, Z.; Fehse, B.; Schmidt, M.; Weerkamp, F.; Karlsson, S.; Wagemaker, G.; Williams, D.A. Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol. Ther. 2004, 9, 5–13. [Google Scholar] [CrossRef]
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat. Commun. 2020, 11, 5820. [Google Scholar] [CrossRef]
- Du, X.; Hill, R.A. 7,8-Dihydroxyflavone as a pro-neurotrophic treatment for neurodevelopmental disorders. Neurochem. Int. 2015, 89, 170–180. [Google Scholar] [CrossRef]
- Kazim, S.F.; Iqbal, K. Neurotrophic factor small-molecule mimetics mediated neuroregeneration and synaptic repair: Emerging therapeutic modality for Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 50. [Google Scholar] [CrossRef]
- Cardenas-Aguayo, M.d.C.; Kazim, S.F.; Grundke-Iqbal, I.; Iqbal, K. Neurogenic and Neurotrophic Effects of BDNF Peptides in Mouse Hippocampal Primary Neuronal Cell Cultures. PLoS ONE 2013, 8, e53596. [Google Scholar] [CrossRef]
- Zainullina, L.F.; Vakhitova, Y.V.; Lusta, A.Y.; Gudasheva, T.A.; Seredenin, S.B. Dimeric mimetic of BDNF loop 4 promotes survival of serum-deprived cell through TrkB-dependent apoptosis suppression. Sci. Rep. 2021, 11, 14336. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef]
- Bollen, E.; Vanmierlo, T.; Akkerman, S.; Wouters, C.; Steinbusch, H.M.W.; Prickaerts, J. 7,8-Dihydroxyflavone improves memory consolidation processes in rats and mice. Behav. Brain Res. 2013, 257, 8–12. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of alzheimer’s disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef]
- Parrini, M.; Ghezzi, D.; Deidda, G.; Medrihan, L.; Castroflorio, E.; Alberti, M.; Baldelli, P.; Cancedda, L.; Contestabile, A. Aerobic exercise and a BDNF-mimetic therapy rescue learning and memory in a mouse model of Down syndrome. Sci. Rep. 2017, 7, 16825. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Povarnina, P.Y.; Tarasiuk, A.V.; Seredenin, S.B. Low-molecular mimetics of nerve growth factor and brain-derived neurotrophic factor: Design and pharmacological properties. Med. Res. Rev. 2021, 41, 2746–2774. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Tallerova, A.V.; Mezhlumyan, A.G.; Antipova, T.A.; Logvinov, I.O.; Firsova, Y.N.; Povarnina, P.Y.; Seredenin, S.B. Low-molecular weight bdnf mimetic, dimeric dipeptide GSB-106, reverses depressive symptoms in mouse chronic social defeat stress. Biomolecules 2021, 11, 252. [Google Scholar] [CrossRef]
- Maswood, N.; Young, J.; Tilmont, E.; Zhang, Z.; Gash, D.M.; Gerhardt, G.A.; Grondin, R.; Roth, G.S.; Mattison, J.; Lane, M.A.; et al. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 18171–18176. [Google Scholar] [CrossRef]
- Duan, W.; Guo, Z.H.; Mattson, M.P. Brain-derived neurotrophic factor mediates an excitoprotective effect of dietary restriction in mice. J. Neurochem. 2001, 76, 619–626. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L.; Duan, W. Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol. Rev. 2002, 82, 637–672. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Marchese, M.; Head, E.; Pop, V.; Michalski, B.; Milgram, W.N.; Cotman, C.W. BDNF increases with behavioral enrichment and an antioxidant diet in the aged dog. Neurobiol. Aging 2012, 33, 553-e1. [Google Scholar] [CrossRef] [PubMed]
- Tuon, T.; Valvassori, S.S.; Lopes-Borges, J.; Luciano, T.; Trom, C.B.; Silva, L.A.; Quevedo, J.; Souza, C.T.; Lira, F.S.; Pinho, R.A. Physical training exerts neuroprotective effects in the regulation of neurochemical factors in an animal model of Parkinson’s disease. Neuroscience 2012, 227, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Wang, T.F.; Yu, L.; Jen, C.J.; Chuang, J.I.; Wu, F.S.; Wu, C.W.; Kuo, Y.M. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain. Behav. Immun. 2011, 25, 135–146. [Google Scholar] [CrossRef]
- Fredriksson, A.; Stigsdotter, I.M.; Hurtig, A.; Ewalds-Kvist, B.; Archer, T. Running wheel activity restores MPTP-induced functional deficits. J. Neural Transm. 2011, 118, 407–420. [Google Scholar] [CrossRef]
- Tajiri, N.; Yasuhara, T.; Shingo, T.; Kondo, A.; Yuan, W.; Kadota, T.; Wang, F.; Baba, T.; Tayra, J.T.; Morimoto, T.; et al. Exercise exerts neuroprotective effects on Parkinson’s disease model of rats. Brain Res. 2010, 1310, 200–207. [Google Scholar] [CrossRef]
- Lau, Y.S.; Patki, G.; Das-Panja, K.; Le, W.D.; Ahmad, S.O. Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson’s disease with moderate neurodegeneration. Eur. J. Neurosci. 2011, 33, 1264–1274. [Google Scholar] [CrossRef]
- Toy, W.A.; Petzinger, G.M.; Leyshon, B.J.; Akopian, G.K.; Walsh, J.P.; Hoffman, M.V.; Vučković, M.G.; Jakowec, M.W. Treadmill exercise reverses dendritic spine loss in direct and indirect striatal medium spiny neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. Neurobiol. Dis. 2014, 63, 201–209. [Google Scholar] [CrossRef]
- Petzinger, G.M.; Walsh, J.P.; Akopian, G.; Hogg, E.; Abernathy, A.; Arevalo, P.; Turnquist, P.; Vučković, M.; Fisher, B.E.; Togasaki, D.M.; et al. Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. J. Neurosci. 2007, 27, 5291–5300. [Google Scholar] [CrossRef]
- Herman, T.; Giladi, N.; Gruendlinger, L.; Hausdorff, J.M. Six Weeks of Intensive Treadmill Training Improves Gait and Quality of Life in Patients With Parkinson’s Disease: A Pilot Study. Arch. Phys. Med. Rehabil. 2007, 88, R713–R715. [Google Scholar] [CrossRef]
- Stuckenschneider, T.; Helmich, I.; Raabe-Oetker, A.; Feodoroff, B.; Froböse, I.; Schneider, S. Parkinson’s Disease Patients Show Long-term Gait Improvements After Active Assistive Forced Exercise Training. Med. Sci. Sport. Exerc. 2016, 48, 983. [Google Scholar] [CrossRef]
- Fisher, B.E.; Wu, A.D.; Salem, G.J.; Song, J.; Lin, C.H. (Janice); Yip, J.; Cen, S.; Gordon, J.; Jakowec, M.; Petzinger, G. The Effect of Exercise Training in Improving Motor Performance and Corticomotor Excitability in People with Early Parkinson’s Disease. Arch. Phys. Med. Rehabil. 2008, 89, 1221–1229. [Google Scholar] [CrossRef]
- Gerecke, K.M.; Jiao, Y.; Pagala, V.; Smeyne, R.J. Exercise does not protect against MPTP-induced neurotoxicity in BDNF happloinsufficent mice. PLoS ONE 2012, 7, e43250. [Google Scholar] [CrossRef]
- Real, C.C.; Ferreira, A.F.B.; Chaves-Kirsten, G.P.; Torrão, A.S.; Pires, R.S.; Britto, L.R.G. BDNF receptor blockade hinders the beneficial effects of exercise in a rat model of Parkinson’s disease. Neuroscience 2013, 237, 118–129. [Google Scholar] [CrossRef]
- Szuhany, K.L.; Bugatti, M.; Otto, M.W. A meta-analytic review of the effects of exercise on brain-derived neurotrophic factor. J. Psychiatr. Res. 2015, 60, 56–64. [Google Scholar] [CrossRef]
- Reycraft, J.T.; Islam, H.; Townsend, L.K.; Hayward, G.C.; Hazell, T.O.M.J.; MacPherson, R.E.K. Exercise Intensity and Recovery on Circulating Brain-derived Neurotrophic Factor. Med. Sci. Sports Exerc. 2020, 52, 1210–1217. [Google Scholar] [CrossRef]
- Voisey, J.; Lawford, B.; Bruenig, D.; Harvey, W.; Morris, C.P.; Young, R.M.D.; Mehta, D. Differential BDNF methylation in combat exposed veterans and the association with exercise. Gene 2019, 698, 107–112. [Google Scholar] [CrossRef]
- Zanin, J.P.; Unsain, N.; Anastasia, A. Growth factors and hormones pro-peptides: The unexpected adventures of the BDNF prodomain. J. Neurochem. 2017, 141, 330–340. [Google Scholar] [CrossRef]
- Kojima, M.; Mizui, T. BDNF Propeptide: A Novel Modulator of Synaptic Plasticity. Vitam. Horm. 2017, 104, 19–28. [Google Scholar]
- Yang, J.; Siao, C.J.; Nagappan, G.; Marinic, T.; Jing, D.; McGrath, K.; Chen, Z.Y.; Mark, W.; Tessarollo, L.; Lee, F.S.; et al. Neuronal release of proBDNF. Nat. Neurosci. 2009, 12, 113–115. [Google Scholar] [CrossRef]
- Yang, B.; Qin, J.; Nie, Y.; Li, Y.; Chen, Q. Brain-derived neurotrophic factor propeptide inhibits proliferation and induces apoptosis in C6 glioma cells. Neuroreport 2017, 28, 726. [Google Scholar] [CrossRef]
- Guo, J.; Ji, Y.; Ding, Y.; Jiang, W.; Sun, Y.; Lu, B.; Nagappan, G. BDNF pro-peptide regulates dendritic spines via caspase-3. Cell Death Dis. 2016, 7, e2264. [Google Scholar] [CrossRef]
- McGregor, C.E.; Irwin, A.M.; English, A.W. The Val66Met BDNF Polymorphism and Peripheral Nerve Injury: Enhanced Regeneration in Mouse Met-Carriers Is Not Further Improved With Activity-Dependent Treatment. Neurorehabil. Neural Repair 2019, 33, 407–418. [Google Scholar] [CrossRef]
- Barbey, A.K.; Colom, R.; Paul, E.; Forbes, C.; Krueger, F.; Goldman, D.; Grafman, J. Preservation of general intelligence following traumatic brain injury: Contributions of the Met66 brain-derived neurotrophic factor. PLoS ONE 2014, 9, e88733. [Google Scholar] [CrossRef]
- Krueger, F.; Pardini, M.; Huey, E.D.; Raymont, V.; Solomon, J.; Lipsky, R.H.; Hodgkinson, C.A.; Goldman, D.; Grafman, J. The role of the met66 brain-derived neurotrophic factor allele in the recovery of executive functioning after combat-related traumatic brain injury. J. Neurosci. 2011, 31, 598–606. [Google Scholar] [CrossRef]
- Finan, J.D.; Udani, S.V.; Patel, V.; Bailes, J.E. The Influence of the Val66Met Polymorphism of Brain-Derived Neurotrophic Factor on Neurological Function after Traumatic Brain Injury. J. Alzheimer’s Dis. 2018, 65, 1055–1064. [Google Scholar] [CrossRef]
- Zivadinov, R.; Weinstock-Guttman, B.; Benedict, R.; Tamaño-Blanco, M.; Hussein, S.; Abdelrahman, N.; Durfee, J.; Ramanathan, M. Preservation of gray matter volume in multiple sclerosis patients with the Met allele of the rs6265 (Val66Met) SNP of brain-derived neurotrophic factor. Hum. Mol. Genet. 2007, 16, 2659–2668. [Google Scholar] [CrossRef]
- Kojima, M.; Matsui, K.; Mizui, T. BDNF pro-peptide: Physiological mechanisms and implications for depression. Cell Tissue Res. 2019, 377, 73–79. [Google Scholar] [CrossRef]
- Mizui, T.; Hattori, K.; Ishiwata, S.; Hidese, S.; Yoshida, S.; Kunugi, H.; Kojima, M. Cerebrospinal fluid BDNF pro-peptide levels in major depressive disorder and schizophrenia. J. Psychiatr. Res. 2019, 113, 190–198. [Google Scholar] [CrossRef]
- Lim, J.Y.; Reighard, C.P.; Crowther, D.C. The pro-domains of neurotrophins, including BDNF, are linked to Alzheimer’s disease through a toxic synergy with Aβ. Hum. Mol. Genet. 2015, 24, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yang, C.; Ren, Q.; Zhang, J.C.; Chen, Q.X.; Shirayama, Y.; Hashimoto, K. Regional differences in the expression of brain-derived neurotrophic factor (BDNF) pro-peptide, proBDNF and preproBDNF in the brain confer stress resilience. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ren, Q.; Zhang, J.C.; Chen, Q.X.; Hashimoto, K. Altered expression of BDNF, BDNF pro-peptide and their precursor proBDNF in brain and liver tissues from psychiatric disorders: Rethinking the brain-liver axis. Transl. Psychiatry 2017, 7, e1128. [Google Scholar] [CrossRef] [PubMed]
- Bellani, M.; Ferro, A.; Brambilla, P. The potential role of the parietal lobe in schizophrenia. Epidemiol. Psichiatr. Soc. 2010, 19, 118–119. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, M.; Borgwardt, S.J.; Berger, G.E. Parietal Lobes in Schizophrenia: Do They Matter? Schizophr. Res. Treatment 2011, 2011, 581686. [Google Scholar] [CrossRef]
- Bittar, T.P.; Labonté, B. Functional Contribution of the Medial Prefrontal Circuitry in Major Depressive Disorder and Stress-Induced Depressive-Like Behaviors. Front. Behav. Neurosci. 2021, 15, 128. [Google Scholar] [CrossRef]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef]
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 2012, 18, 1413–1417. [Google Scholar] [CrossRef]
- Mavridis, I. The role of the nucleus accumbens in psychiatric disorders. Psychiatrike 2015, 25, 282–294. [Google Scholar]
- Hempstead, B.L. Brain-Derived Neurotrophic Factor: Three Ligands, Many Actions. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 9. [Google Scholar]
- Hall, A.; Lalli, G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb. Perspect. Biol. 2010, 2, a001818. [Google Scholar] [CrossRef]
- Kailainathan, S.; Piers, T.M.; Yi, J.H.; Choi, S.; Fahey, M.S.; Borger, E.; Gunn-Moore, F.J.; O’Neill, L.; Lever, M.; Whitcomb, D.J.; et al. Activation of a synapse weakening pathway by human Val66 but not Met66 pro-brain-derived neurotrophic factor (proBDNF). Pharmacol. Res. 2016, 104, 97–107. [Google Scholar] [CrossRef]
- Voineskos, A.N.; Lerch, J.P.; Felsky, D.; Shaikh, S.; Rajji, T.K.; Miranda, D.; Lobaugh, N.J.; Mulsant, B.H.; Pollock, B.G.; Kennedy, J.L. The brain-derived neurotrophic factor Val66Met polymorphism and prediction of neural risk for alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 198–206. [Google Scholar] [CrossRef]
- Qin, L.; Jing, D.; Parauda, S.; Carmel, J.; Ratan, R.R.; Lee, F.S.; Cho, S. An adaptive role for BDNF Val66Met polymorphism in motor recovery in chronic stroke. J. Neurosci. 2014, 34, 2493–2502. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szarowicz, C.A.; Steece-Collier, K.; Caulfield, M.E. New Frontiers in Neurodegeneration and Regeneration Associated with Brain-Derived Neurotrophic Factor and the rs6265 Single Nucleotide Polymorphism. Int. J. Mol. Sci. 2022, 23, 8011. https://doi.org/10.3390/ijms23148011
Szarowicz CA, Steece-Collier K, Caulfield ME. New Frontiers in Neurodegeneration and Regeneration Associated with Brain-Derived Neurotrophic Factor and the rs6265 Single Nucleotide Polymorphism. International Journal of Molecular Sciences. 2022; 23(14):8011. https://doi.org/10.3390/ijms23148011
Chicago/Turabian StyleSzarowicz, Carlye A., Kathy Steece-Collier, and Margaret E. Caulfield. 2022. "New Frontiers in Neurodegeneration and Regeneration Associated with Brain-Derived Neurotrophic Factor and the rs6265 Single Nucleotide Polymorphism" International Journal of Molecular Sciences 23, no. 14: 8011. https://doi.org/10.3390/ijms23148011
APA StyleSzarowicz, C. A., Steece-Collier, K., & Caulfield, M. E. (2022). New Frontiers in Neurodegeneration and Regeneration Associated with Brain-Derived Neurotrophic Factor and the rs6265 Single Nucleotide Polymorphism. International Journal of Molecular Sciences, 23(14), 8011. https://doi.org/10.3390/ijms23148011