
Biological Role of the Intercellular Transfer of Glycosylphosphatidylinositol-Anchored Proteins: Stimulation of Lipid and Glycogen Synthesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
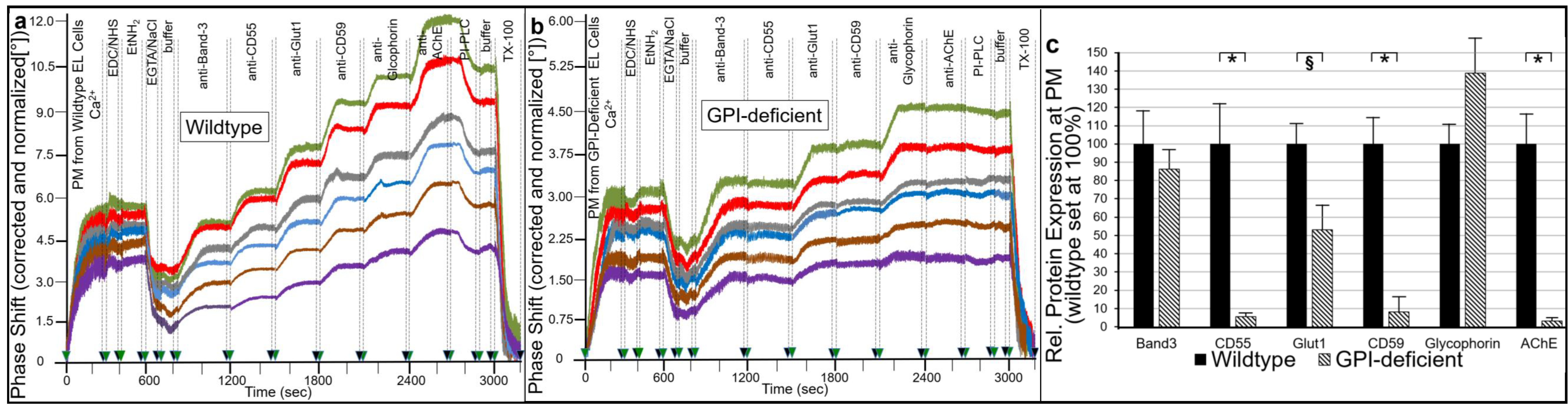
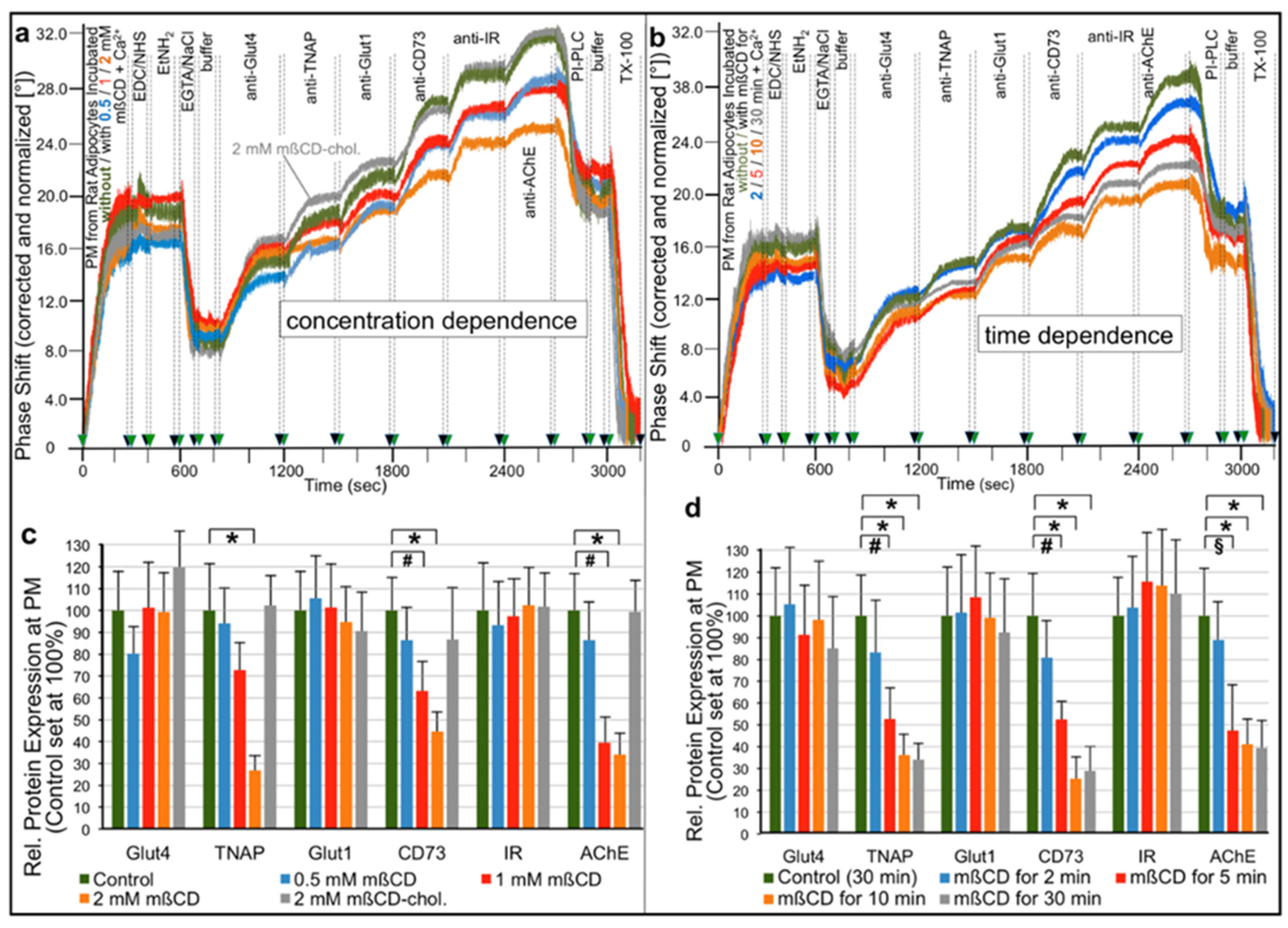
2.1. Different Modes for Depletion of GPI-APs from PM of Different Cell Types
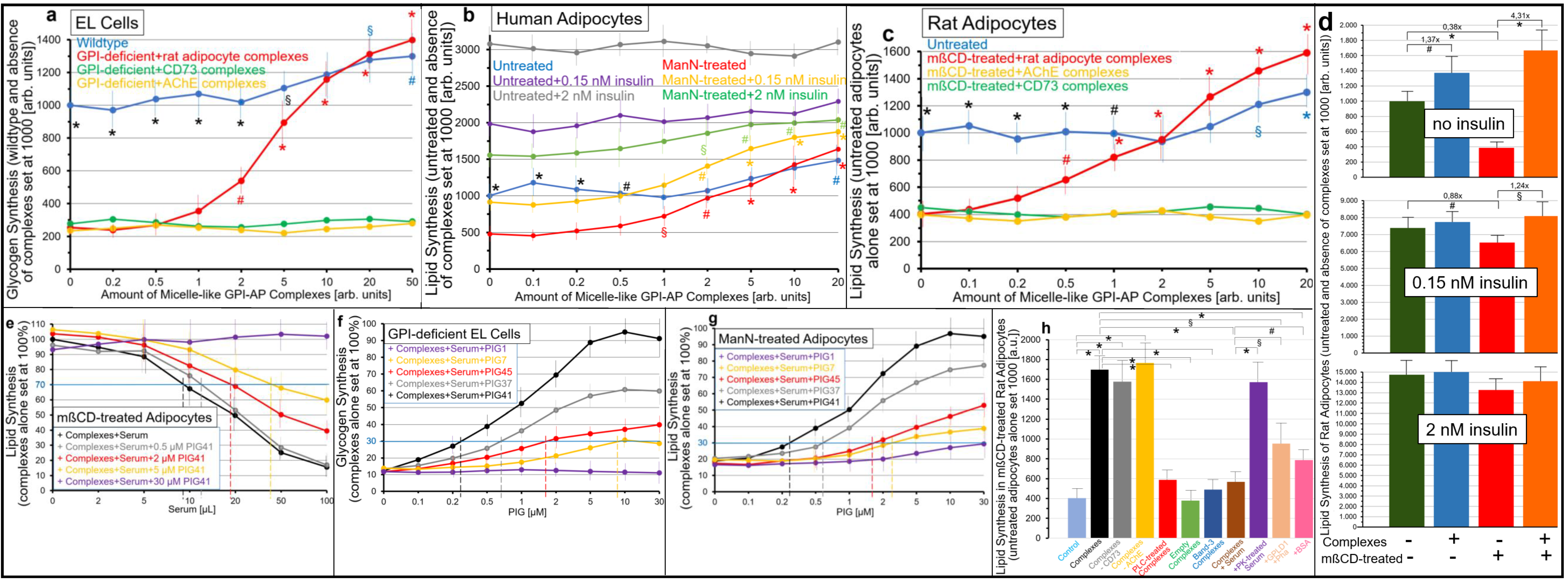
2.2. GPI-APs Are Transferred from Micelle-like GPI-AP Complexes to EL Cells or Adipocytes with Accompanying Stimulation of Glycogen and Lipid Synthesis, Respectively
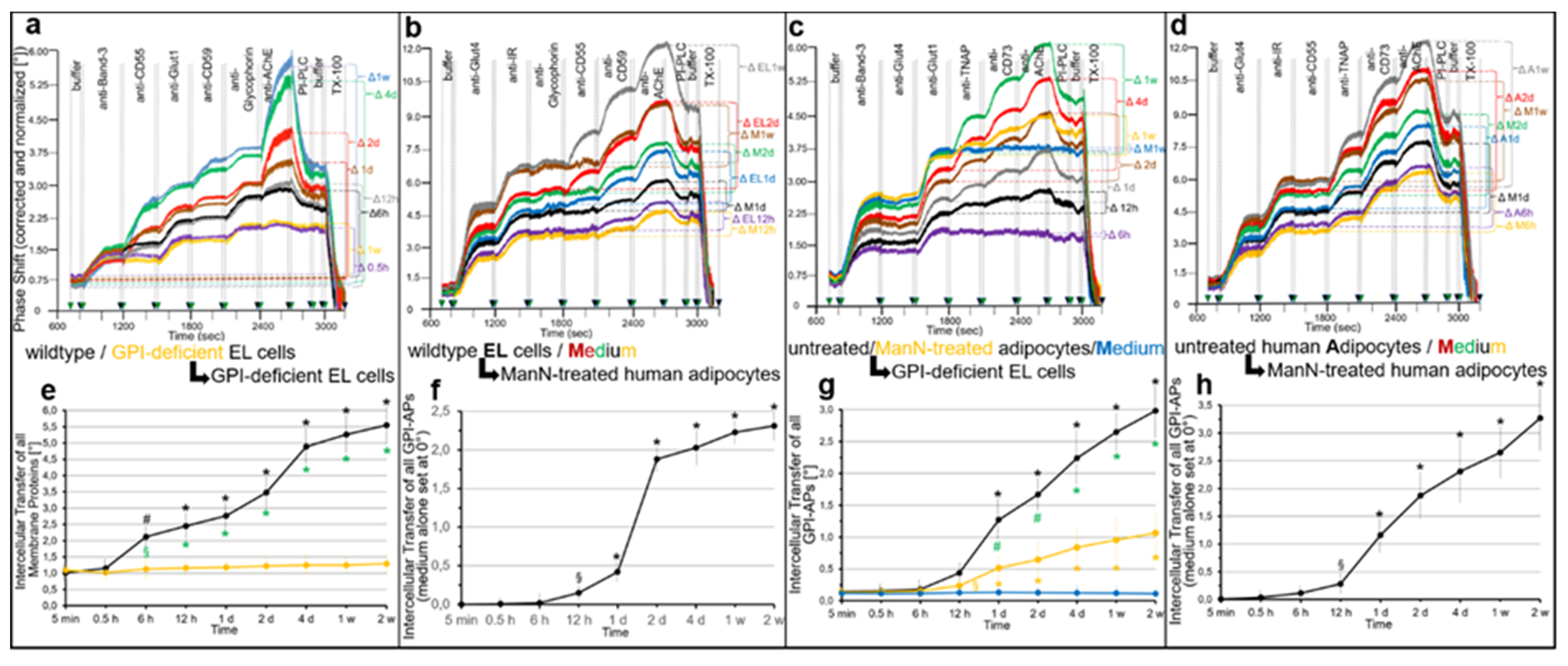
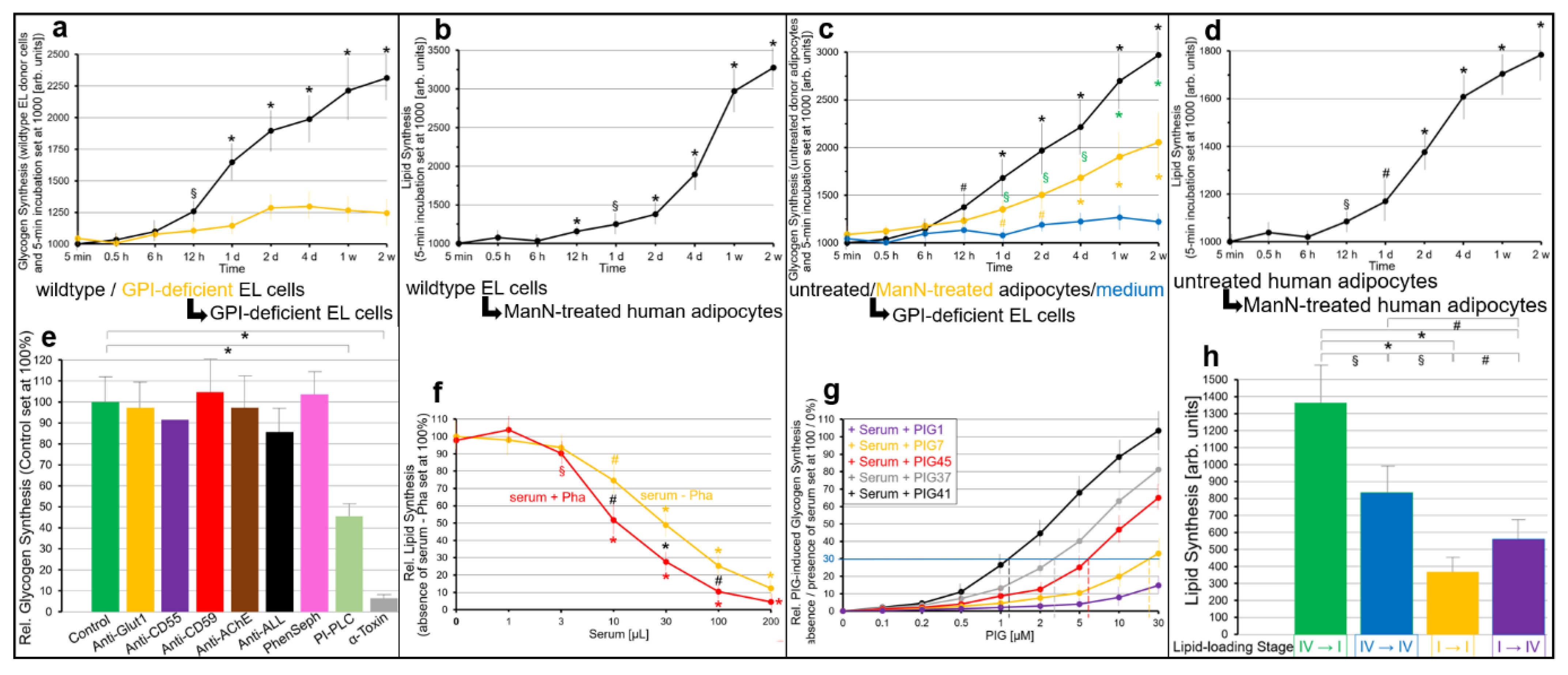
2.3. GPI-APs Are Transferred between EL Cells and Human Adipocytes in Transwell Co-Culture with Accompanying Stimulation of Glycogen and Lipid Synthesis, Respectively
3. Discussion
3.1. (Patho)Physiological Role of Intercellular Transfer of GPI-APs
- (i)
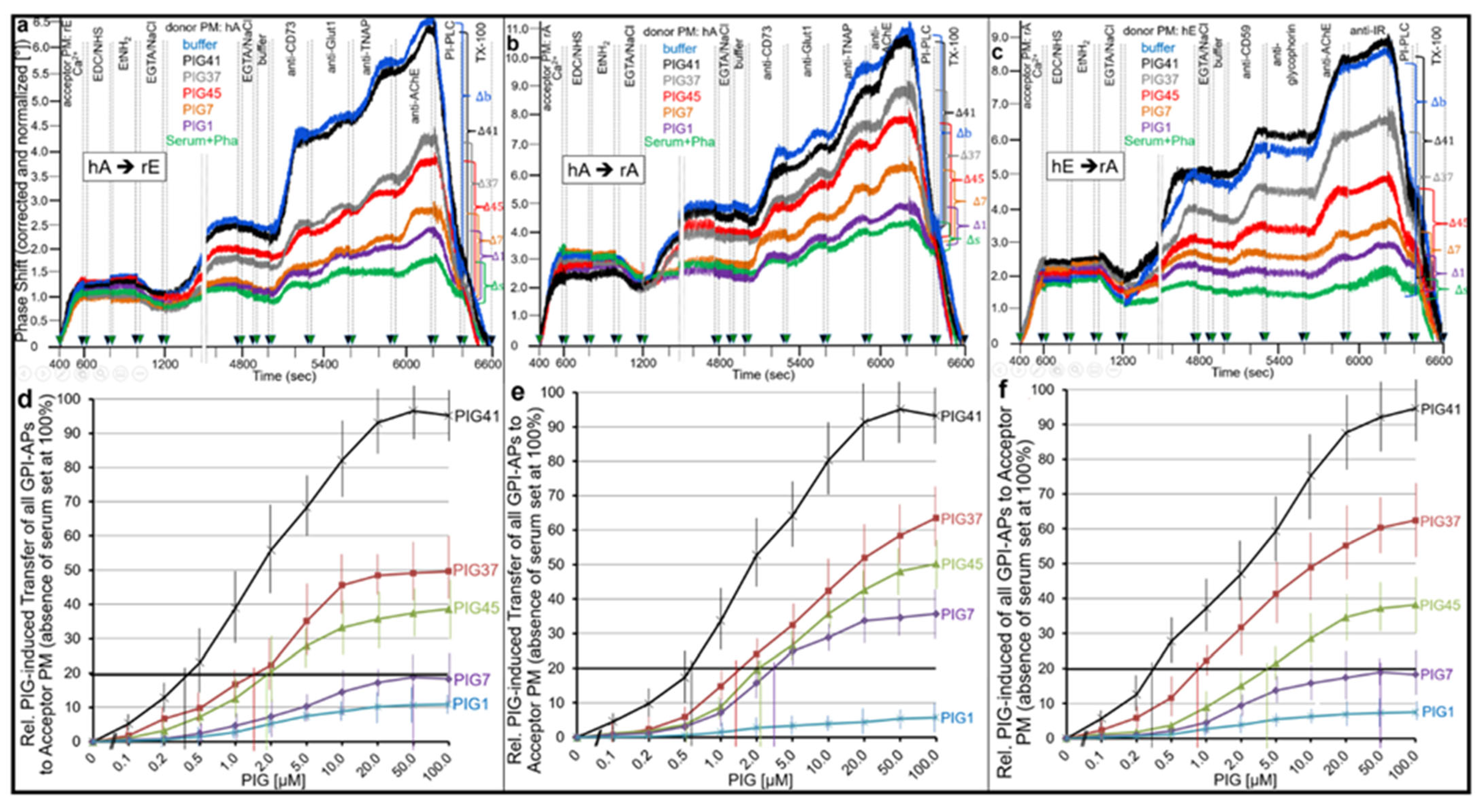
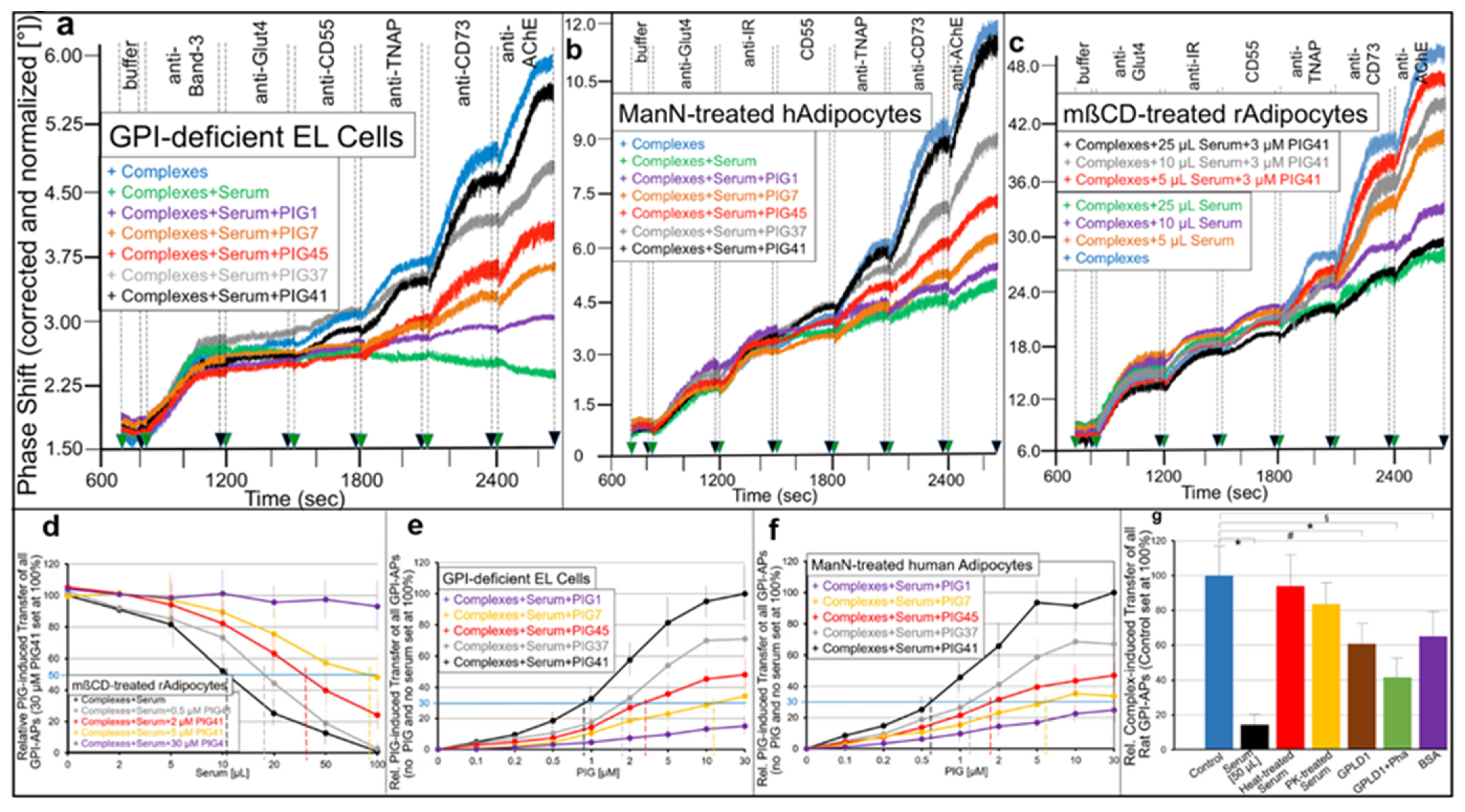
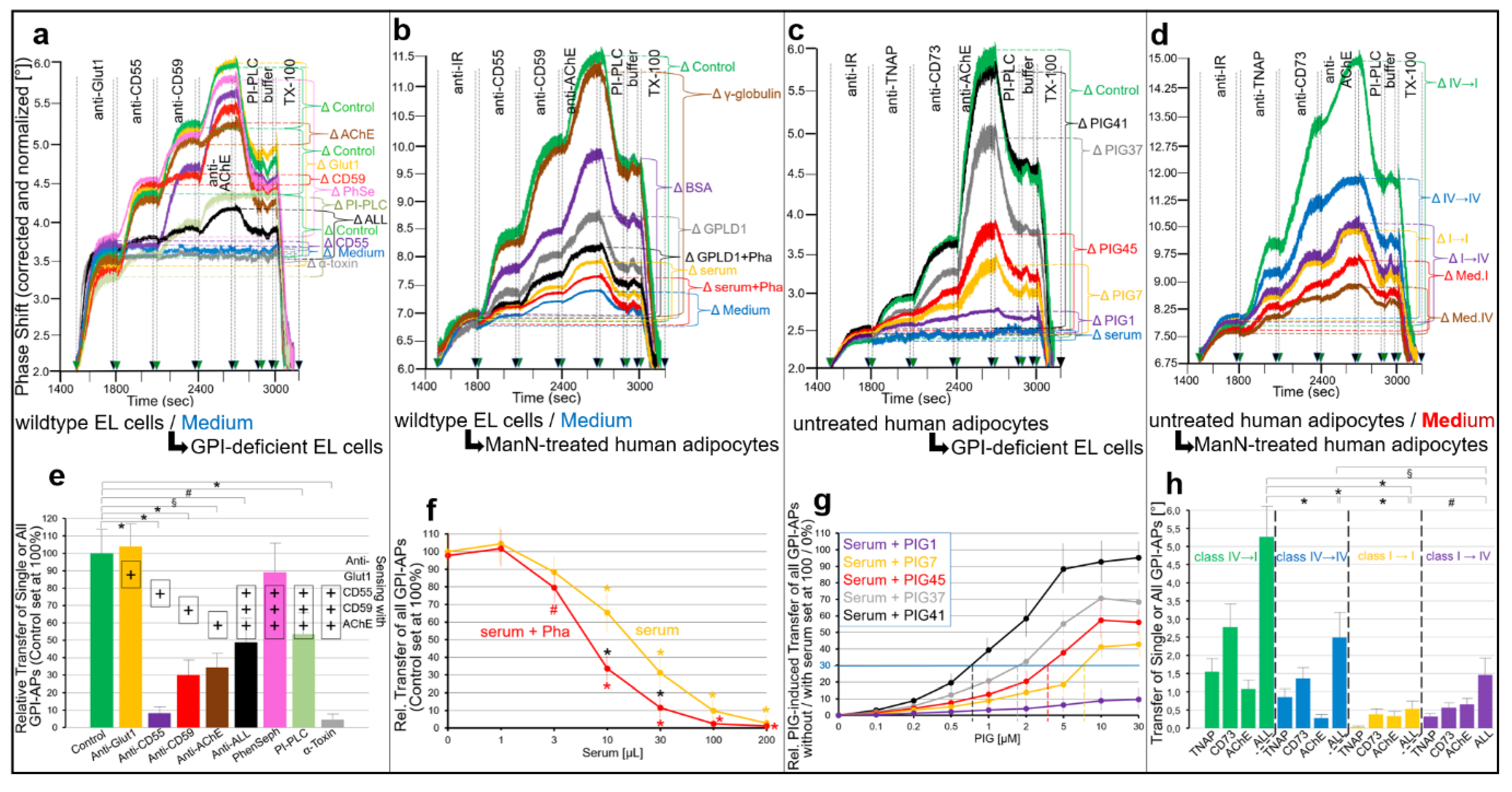
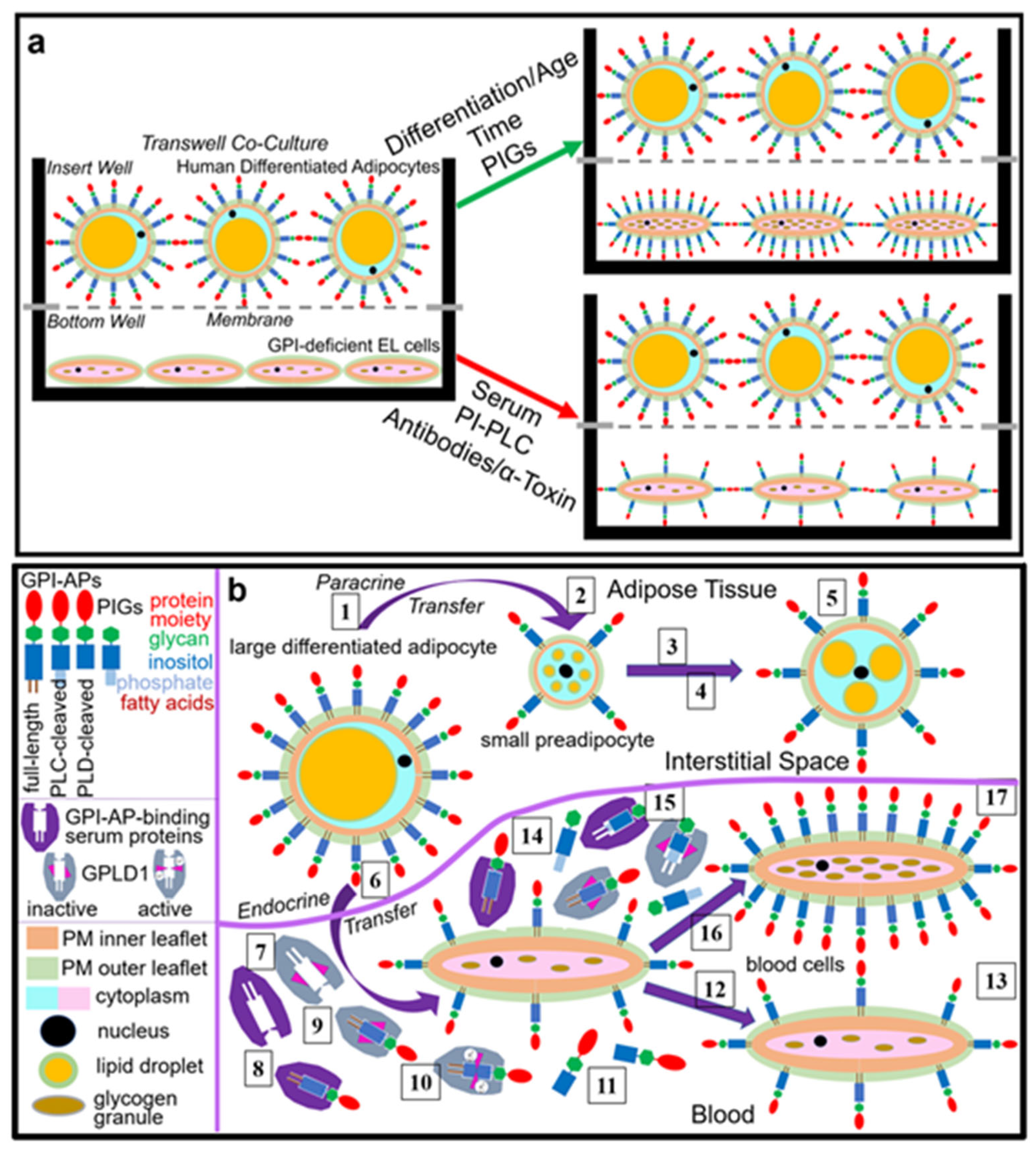
- Transfer of certain full-length GPI-APs, as exemplified by, but actually not involving CD55, CD59, CD73, TNAP and AChE (Figure 4, Figure 8 and Figure 9), to primary rat or cultured human adipocytes and cultured human EL cells with low or missing expression of GPI-APs at PM (Figure 1, Figure 2 and Figure 3) leads to stimulation of lipid and glycogen synthesis, respectively (Figure 7 and Figure 10; Supplementary Materials, Figure S6).
- (ii)
- (iii)
- This blockade is bypassed by the dissociation of GPI-APs from the binding-proteins, as provoked by synthetic PIGs (Figure 6, Figure 7, Figure 9 and Figure 10), which closely resemble the structure of the glycan core of the GPI anchor (see Supplementary Materials, Figure S2) and manage to displace rat and human serum components from micelle-like GPI-AP complexes in vitro (Supplementary Materials, Figure S5).
- (iv)
- Rat and human adipocytes displaying large size or heavy lipid-loading, respectively, are most efficient as donor cells and those of small size or slight lipid-loading as acceptor cells (Supplementary Materials, Figure S6; Figure 10).
3.2. PIGs, GPI-AP Transfer and Matter vs. Information Transfer
3.3. Implications for the Therapy of Metabolic Diseases
4. Materials and Methods
4.1. Materials
4.2. Culture of Wild-Type and GPI-Deficient K562 Erythroleukemia (EL) Cells
4.3. Differentiation and Culture of Human Adipocytes
4.4. Mannosamine (ManN) Treatment of Human Adipocytes
4.5. Preparation of Primary Rat Adipocytes from Epididymal Fat Pads
4.6. Methyl-ß-Cyclodextrin (mßCD) Treatment of Primary Rat Adipocytes
4.7. Transwell Co-Culture of EL Cells and Human Adipocytes
4.8. Preparation of PM
4.9. Preparation of Total GPI-APs from Rat Adipocytes
4.10. Reconstitution of Micelle-like GPI-AP and AChE/CD73 Complexes
4.11. Incubation of EL Cells and Human/Rat Adipocytes with Micelle-like GPI-AP Complexes
4.12. Immune Depletion of Medium or Micelle-like GPI-AP Complexes from AChE/CD73
4.13. Assay of Transfer of GPI-APs from Donor to Acceptor PM In Vitro
4.14. Analysis of PM for the Expression of GPI-APs and Transmembrane Proteins by Chip-Based SAW Sensing
4.15. Immobilization of PM at SAW Chip Surface by Ionic and Subsequent Covalent Capture
4.16. Assay for Glycogen Synthesis with EL Cells
4.17. Assay for Lipid Synthesis with Human and Rat Adipocytes
4.18. Digestion with Phospholipases
4.19. Statistical Analysis
4.20. Miscellaneous
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Eisenhaber, B.; Bork, P.; Eisenhaber, B. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: Analysis of protein sequence data from complete genomes. Protein Eng. 2001, 14, 17–25. [Google Scholar] [CrossRef]
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020, 10, 190290. [Google Scholar] [CrossRef]
- Muniz, M.; Riezman, H. Trafficking of glycosylphosphatidylinositol anchored proteins from the endoplasmic reticulum to the cell surface. J. Lipid Res. 2016, 57, 352–360. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Haldar, K.; Cross, G.A.M. Trypanosoma brucei variant surface glycoprotein has a sn-1,2-dimyristyl glycerol membrane anchor at its COOH terminus. J. Biol. Chem. 1985, 260, 4963–4968. [Google Scholar] [CrossRef]
- Haldar, K.; Ferguson, M.A.J.; Cross, G.A.M. Acylation of a Plasmodium falciparum merozoite surface antigen via sn-1,2-diacyl glycerol. J. Biol. Chem. 1985, 260, 4969–4974. [Google Scholar] [CrossRef]
- Lebreton, S.; Zurzolo, C.; Paladino, S. Organization of GPI-anchored proteins at the cell surface and its physiopathological relevance. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 403–419. [Google Scholar] [CrossRef]
- Paulik, M.G.; Bertozzi, C.R. The glycosylphosphatidylinositol anchor: A complex membrane-anchoring structure for proteins. Biochemistry 2008, 47, 6991–7000. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fujita, M.; Maeda, Y. Biosynthesis, remodeling and functions of mammalian GPI-anchored proteins. Rec. Prog. J. Biochem. 2008, 144, 287–294. [Google Scholar]
- Fujita, M.; Kinoshita, T. GPI anchor remodeling: Potential functions of GPI anchors in intracellular trafficking and membrane dynamics. Biochem. Biophys. Acta 2012, 1821, 1050–1058. [Google Scholar] [CrossRef]
- Maeda, Y.; Kinoshita, T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 2011, 50, 411–424. [Google Scholar] [CrossRef]
- Nakano, M.; Sabido-Bozo, S.; Okazaki, K.; Aguilera-Romero, A.; Rodriguez-Gallardo, S.; Cortes-Gomez, A.; Lopez, S.; Ikeda, A.; Funato, K.; Muniz, M. Structural analysis of the GPI glycan. PLoS ONE 2021, 16, e0257435. [Google Scholar] [CrossRef]
- Yoko-O, T.; Umemura, M.; Komatsuzaki, A.; Ikeda, K.; Ichikawa, D.; Takase, K.; Kanzawa, N.; Saito, K.; Kinoshita, T.; Taguchi, R.; et al. Lipid moiety of glycosylphosphatidylinositol-anchored proteins contributes to the determination of their final destination in yeast. Genes Cells 2018, 23, 880–892. [Google Scholar] [CrossRef]
- Yi-Shi, L.; Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 2020, 48, 1129–1138. [Google Scholar]
- Müller, G.; Jung, C.; Wied, S.; Welte, S.; Jordan, H.; Frick, W. Redistribution of glycolipid raft domain components induces insulin-mimetic signaling in rat adipocytes. Mol. Cell. Biol. 2001, 21, 4553–4567. [Google Scholar] [CrossRef][Green Version]
- Van den Berg, C.W.; Cinek, T.; Hallett, M.B.; Horejsi, V.; Morgan, B.P. Exogenous glycosyl phosphatidylinositol-anchored CD59 associates with kinases in membrane clusters on U937 cells and becomes Ca(2+)-signaling competent. J. Cell Biol. 1995, 131, 669–677. [Google Scholar] [CrossRef]
- Fujihara, Y.; Ikawa, M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2016, 57, 538–545. [Google Scholar] [CrossRef]
- Müller, G.A. The release of glycosylphosphatidylinositol-anchored proteins from the cell surface. Arch. Biochem. Biophys. 2018, 656, 1–18. [Google Scholar] [CrossRef]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI proteins: Sorting, membrane residence and functions. Biochem. Biophys. Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef]
- Müller, G.A. Glycosylphosphatidylinositol-Anchored Proteins and Their Release from Cells—From Phenomenon to Meaning, 1st ed.; Nova Science Publishers Inc.: New York, NY, USA, 2018; pp. 39–91. [Google Scholar]
- Rooney, I.A.; Heuser, J.E.; Atkinson, J.P. GPI-anchored complement regulatory proteins in seminal plasma. An analysis of their physical conditions and the mechanisms of their binding to exogenous cells. J. Clin. Investig. 1996, 97, 1675–1686. [Google Scholar] [CrossRef]
- Rabesandratana, H.; Toutant, J.P.; Reggio, H.; Vidal, M. Decay-accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during in vitro maturation of reticulocytes. Blood 1998, 91, 2573–2580. [Google Scholar] [CrossRef]
- Müller, C.; Jung, C.; Straub, J.; Wied, S.; Kramer, W. Induced release of membrane vesicles and exosomes from rat adipocytes containing lipid droplets, lipid rafts and glycosylphosphatidylinositol-anchored proteins. Cell. Signal. 2009, 21, 324–338. [Google Scholar] [CrossRef]
- Eaton, S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef]
- Eliakim, R.; Alpers, D.H.; Oren, R.; Fich, A.; DeSchryver-Kecskemeti, K. Abundance of surfactant-like particles reflects mucosal integrity in patients with peptic ulcer disease. Gut 1996, 39, 353–359. [Google Scholar] [CrossRef][Green Version]
- Mahmood, A.; Engle, M.J.; Alpers, D.H. Secreted intestinal surfactant-like particles interact with cell membranes and extracellular matrix proteins in rats. J. Physiol. 2002, 542, 237–244. [Google Scholar] [CrossRef]
- Neumann, S.; Harterink, M.; Sprong, H. Hitch-hiking between cells on lipoprotein particles. Traffic 2007, 8, 331–338. [Google Scholar] [CrossRef]
- Panakova, D.; Sprong, H.; Marois, E.; Thiele, C.; Eaton, S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature 2005, 435, 58–65. [Google Scholar] [CrossRef]
- Patton, S.; Huston, G.E. A method for isolation of milk fat globules. Lipids 1986, 21, 170–174. [Google Scholar] [CrossRef]
- Väkevä, A.; Jauhiainen, M.; Ehnholm, C.; Lehto, T.; Meri, S. High density lipoproteins can act as carriers of glycophosphoinositol lipid-anchored CD59 in human plasma. Immunology 1994, 82, 28–33. [Google Scholar]
- Müller, G.A.; Herling, A.W.; Stemmer, K.; Lechner, A.; Tschöp, M.H. Chip-based sensing for release of unprocessed cell surface proteins in vitro and in serum and its (patho)physiological relevance. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E212–E233. [Google Scholar] [CrossRef]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Upregulated phospholipase D activity toward glycosylphosphatidylinositol-anchored proteins in micelle-like serum complexes in metabolically deranged rats and humans. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E462–E479. [Google Scholar] [CrossRef]
- Müller, G.A. Membrane insertion and intercellular transfer of glycosylphosphatidylinositol-anchored proteins: Potential therapeutic applications. Arch. Physiol. Biochem. 2020, 126, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Ahn, J. A common mechanism may be involved in the selective loss of plasma membrane functions during reticulocyte maturation. Biomed. Biochim. Acta 1990, 49, S70–S75. [Google Scholar]
- Geminard, C.; Nault, F.; Johnstone, R.M.; Vidal, M. Characteristics of the interaction between Hsc70 and the transferrin receptor in exosomes released during reticulocyte maturation. J. Biol. Chem. 2001, 276, 9910–9916. [Google Scholar] [CrossRef]
- Johnstone, R.M. Exosomes biological significance: A concise review. Blood Cells Mol. Dis. 2006, 36, 315–321. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Mathew, A.; Mason, A.B.; Teng, K. Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalization of obsolete membrane proteins. J. Cell Physiol. 1991, 147, 27–36. [Google Scholar] [CrossRef]
- Pan, B.T.; Blostein, R.; Johnstone, R.M. Loss of the transferrin receptor during the maturation of sheep reticulocytes in vitro. An immunological approach. Biochem. J. 1983, 210, 37–47. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Bianchini, A.; Teng, K. Reticulocyte maturation and exosome release: Transferrin receptor containing exosomes shows multiple plasma membrane functions. Blood 1989, 74, 1844–1851. [Google Scholar] [CrossRef]
- Kirchhoff, C.; Pera, I.; Derr, P.; Yeung, C.H.; Cooper, T. The molecular biology of the sperm surface. Post-testicular membrane remodelling. Adv. Exp. Med. Biol. 1997, 424, 221–232. [Google Scholar]
- Ilangumaran, S.; Robinson, P.J.; Hoessli, D.C. Transfer of exogenous glycosylphosphatidylinositol (GPI)-linked molecules to plasma membranes. Trends Cell Biol. 1996, 6, 163–167. [Google Scholar] [CrossRef]
- Bütikofer, P.; Kuypers, F.A.; Xu, C.M.; Chiu, D.T.; Lubin, B. Enrichment of two glycosylphosphatidylinositol-anchored proteins, acetylcholinesterase and decay accelerating factor, in vesicles released from human blood cells. Blood 1989, 74, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jones, R.; Martin-DeLeon, P.A. Expression and secretion of rat SPAM1(2B1 or PH-20) in the epididymis: Role of testicular lumicrine factors. Matrix Biol. J. Internat. Soc. Matrix Biol. 2004, 22, 653–661. [Google Scholar] [CrossRef]
- Griffiths, G.S.; Galileo, D.S.; Aravindan, R.G.; Martin-DeLeon, P.A. Clusterin facilitates exchange of glycosyl phosphatidylinositol-linked SPAM1 between reproductive luminal fluids and mouse and human sperm membranes. Biol. Reprod. 2009, 81, 562–570. [Google Scholar] [CrossRef]
- Queiroz, K.C.S.; Tio, R.A.; Zeebregts, C.J.; Bijlsma, M.F.; Zijlstra, F.; Badlou, B.; de Vries, M.; Ferreira, C.V.; Spek, C.A.; Peppelenbosch, M.P.; et al. Human plasma very low density lipoprotein carries Indian Hedgehog. J. Prot. Res. 2010, 9, 6052–6059. [Google Scholar] [CrossRef]
- DeSchryver-Kecskemeti, K.; Eliakim, R.; Carroll, S.; Stenson, W.F.; Moxley, M.A.; Alpers, D.H. Intestinal surfactant-like material. A novel secretory product of the rat enterocyte. J. Clin. Investig. 1989, 84, 1355–1361. [Google Scholar] [CrossRef]
- Eaton, S. Release and trafficking of lipid-linked morphogens. Curr. Opin. Genet. Dev. 2006, 16, 17–22. [Google Scholar] [CrossRef]
- Kirchhoff, C.; Hale, G. Cell-to-cell transfer of glycosylphosphatidylinositol-anchored membrane proteins during sperm maturation. Mol. Hum. Reprod. 1996, 2, 177–184. [Google Scholar] [CrossRef]
- Kooyman, D.L.; Byrne, G.W.; McClellan, S.; Nielsen, D.; Tone, M.; Waldmann, H.; Coffman, T.M.; McCurry, K.R.; Platt, J.L.; Logan, J.S. In vivo transfer of GPI-linked complement restriction factors from erythrocytes to endothelium. Science 1995, 269, 89–92. [Google Scholar] [CrossRef]
- Liu, T.; Li, R.; Pan, T.; Liu, D.; Petersen, R.B.; Wong, B.-S.; Gambetti, P.; Sun Sy, M. Intercellular transfer of the cellular prion protein. J. Biol. Chem. 2002, 277, 47671–47678. [Google Scholar] [CrossRef]
- Watanabe, K.; Salomon, D.S. Intercellular transfer regulation of the paracrine activity of GPI-anchored Cripto-1 as a Nodal co-receptor. Biochem. Biophys. Res. Commun. 2010, 403, 108–113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Chip-based sensing of the intercellular transfer of cell surface proteins: Regulation by the metabolic state. Biomedicines 2021, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Andrä, J.; Böhling, A.; Gronewold, T.M.A.; Schlecht, U.; Perpeet, M.; Gutsmann, T. Surface acoustic wave biosensor as a tool to study the interaction of antimicrobial peptides with phospholipid and lipopolysaccharide model membranes. Langmuir 2008, 24, 9148–9153. [Google Scholar] [CrossRef] [PubMed]
- Gronewold, T.M.A.; Glass, S.; Quandt, E.; Famulok, M. Monitoring complex formation in the blood-coagulation cascade using aptamer-coated SAW sensors. Biosens. Bioelectron. 2005, 20, 2044–2052. [Google Scholar] [CrossRef]
- Frick, W.; Bauer, A.; Bauer, J.; Wied, S.; Müller, G. Structure-activity relationship of synthetic phosphoinositolglycans mimicking metabolic insulin action. Biochemistry 1998, 37, 13421–13436. [Google Scholar] [CrossRef]
- Hirose, S.; Mohney, R.P.; Mutka, S.C.; Ravi, L.; Singleton, D.R.; Perry, G.; Tartakoff, A.M.; Medof, M.E. Derivation and characterization of glycoinositol-phospholipid anchor-defective human K562 cell clones. J. Biol. Chem. 1992, 267, 5272–5278. [Google Scholar] [CrossRef]
- Mohney, R.P.; Knez, J.J.; Ravi, L.; Sevlever, D.; Rosenberry, T.L.; Hirose, S.; Medof, M.E. Glycoinositol phospholipid anchor-defective K562 mutants with biochemical lesions distinct from those in Thy-1- murine lymphoma mutants. J. Biol. Chem. 1994, 269, 6536–6542. [Google Scholar] [CrossRef]
- Kamitani, T.; Chang, H.-M.; Rollins, C.; Waneck, G.L.; Yeh, E.T.H. Correction of the class H defect in glycosylphosphatylinositol anchor biosynthesis in Ltk- cells by a human cDNA clone. J. Biol. Chem. 1993, 268, 20733–20736. [Google Scholar] [CrossRef]
- Stieger, S.; Brodbeck, U. Glycosylphosphatidylinositol anchored acetylcholinesterase as substrate for phosphatidylinositol-specific phospholipase C from Bacillus cereus. Biochimie 1991, 73, 1179–1186. [Google Scholar] [CrossRef]
- Bon, S.; Rosenberry, T.L.; Massoulie, J. Amphiphilic, glycophosphatidylinositol-specific phospholipase C (PI-PLC)-insensitive monomers and dimers of acetylcholinesterase. Cell. Mol. Neurobiol. 1991, 11, 157–172. [Google Scholar] [CrossRef]
- Richier, P.; Arpagaus, M.; Toutant, J.P. Glycolipid-anchored acetylcholinesterases from rabbit lymphocytes and erythrocytes differ in their sensitivity to phosphatidylinositol-specific phospholipase C. Biochim. Biophys. Acta 1992, 1112, 83–88. [Google Scholar] [CrossRef]
- Lazar, D.F.; Knez, J.J.; Medof, M.E.; Cuatrecasas, P.; Saltiel, A.R. Stimulation of glycogen synthesis by insulin in human erythroleukemia cells requires the synthesis of glycosylphosphatidylinositol. Proc. Natl. Acad. Sci. USA 1994, 91, 9665–9669. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Field, M.C.; Caras, I.W.; Menon, A.K.; Rodriguez-Boulan, E. Mannosamine, a novel inhibitor of glycosylphosphatidylinositol incorporation into proteins. EMBO J. 1991, 10, 1969–1977. [Google Scholar] [CrossRef]
- Pan, Y.-T.; Kamitani, T.; Bhuvaneswaran, C.; Hallaq, Y.; Warren, C.D.; Yeh, E.T.H.; Elbein, A.D. Inhibition of glycosylphosphatidylinositol anchor formation by mannosamine. J. Biol. Chem. 1992, 267, 21250–21255. [Google Scholar] [CrossRef]
- Bryson, H.; Buttle, D.J.; Kozaci, L.D.; Johnatty, R.N.; Bunning, R.A.D. Evidence that the inhibition of cartilage proteoglycan breakdown by mannosamine is not mediated via inhibition of glycosylphosphatidylinositol anchor formation. Biochem. J. 2000, 345, 495–501. [Google Scholar] [CrossRef]
- Sandy, J.D.; Thompson, V.; Verscharen, C.; Garnett, D. Chondrocyte-mediated catabolism of aggrecan: Evidence for a glycosyl-phosphatidylinositol-linked protein in the aggrecanase response to interleukin-1 or retinoic acid. Arch. Biochem. Biophys. 1999, 367, 258–264. [Google Scholar] [CrossRef]
- Wei, Y.; Waltz, D.A.; Rao, N.; Drummond, R.J.; Rosenberg, S.; Chapman, H.A. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J. Biol. Chem. 1994, 269, 32380–32388. [Google Scholar] [CrossRef]
- Harasymiak-Krzyzanowska, I.; Niedojadlo, A.; Karwat, J.; Kotula, L.; Gil-Kulik, P.; Sawiuk, M.; Kocki, J. Adipose tissue-derived stem cells show considerable promise for regenerative medicine applications. Cell. Mol. Biol. Lett. 2013, 18, 479–493. [Google Scholar] [CrossRef]
- Bertheuil, N.; Chaput, B.; Menard, C.; Varin, A.; Laloze, J.; Watier, E.; Tarte, K. Adipose mesenchymal stromal cells: Definition, immune modulatory properties, mechanical isolation and interest for plastic surgery. Ann. Chir. Plast. Esthet. 2019, 64, 1–10. [Google Scholar] [CrossRef]
- Müller, G.; Hanekop, N.; Wied, S.; Frick, W. Cholesterol depletion blocks redistribution of lipid raft components and insulin-mimetic signaling by glimepiride and phosphoinositolglycans in rat adipocytes. Mol. Med. 2002, 8, 120–136. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef]
- Goni, F.M. “Rafts”: A nickname for putative transient nanodomains. Chem. Phys. Lipids 2019, 218, 34–39. [Google Scholar] [CrossRef]
- Müller, G. Dynamics of plasma membrane microdomains and cross-talk to the insulin signalling cascade. FEBS Lett. 2002, 531. [Google Scholar] [CrossRef]
- Nishijo, J.; Moriyama, S.; Shiota, S.; Kamigauchi, M.; Sugiura, M. Interaction of heptakis (2,3,6-tri-O-methyl)-beta-cyclodextrin with cholesterol in aqueous solution. Chem. Pharm. Bull. 2004, 52, 1405–1410. [Google Scholar] [CrossRef]
- Loftsson, T.; Magnusdottir, A.; Masson, M.; Sigurjonsdottir, J.F. Self-association and cyclodextrin solubilization of drugs. J. Pharm. Sci. 2002, 91, 2307–2316. [Google Scholar] [CrossRef]
- Cerneus, D.P.; Ueffing, E.; Posthuma, G.; Strous, G.J.; van der Ende, A. Detergent insolubility of alkaline phosphatase during biosynthetic transport and endocytosis. Role of cholesterol. J. Biol. Chem. 1993, 268, 3150–3155. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Crecelius, A.; Kessler, A.; Eckel, J. Phosphoinositolglycan-peptides from yeast potently induce metabolic insulin actions in isolated rat adipocytes, cardiomyocytes and diaphragms. Endocrinology 1997, 138, 3459–3475. [Google Scholar] [CrossRef]
- Kessler, A.; Müller, G.; Wied, S.; Crecelius, A.; Eckel, J. Signalling pathways of an insulin-mimetic phosphoinositolglycan-peptide in muscle and adipose tissue. Biochem. J. 1998, 330, 277–286. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Piossek, C.; Bauer, A.; Bauer, J.; Frick, W. Convergence and divergence of the signaling pathways for insulin and phosphoinositolglycans. Mol. Med. 1998, 4, 299–323. [Google Scholar] [CrossRef]
- Müller, G.A.; Ussar, S.; Tschöp, M.H.; Müller, T.D. Age-dependent membrane release and degradation of full-length glycosylphosphatidylinositol-anchored proteins in rats. Mech. Ageing Dev. 2020, 190, 111307. [Google Scholar] [CrossRef]
- Müller, G.A.; Lechner, A.; Tschöp, M.H.; Müller, T.D. Interaction of full-length glycosylphosphatidylinositol-anchored proteins with serum proteins and their translocation to cells in vitro depend on the (pre-)diabetic state in rats and humans. Biomedicines 2021, 9, 277. [Google Scholar] [CrossRef]
- Bäckdahl, J.; Franzen, L.; Massier, L.; Li, Q.; Jalkanen, J.; Gao, H.; Andersson, A.; Bhalla, N.; Thorell, A.; Ryden, M.; et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. 2021, 33, 1869–1882. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Erlanson-Albertsson, C. Adipose cell size: Importance in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R284–R295. [Google Scholar] [CrossRef]
- Roberts, R.; Hodson, L.; Dennis, A.L.; Neville, M.J.; Humphreys, S.M.; Harnden, K.E.; Micklem, K.J.; Frayn, K.N. Markers of de novo lipogenesis in adipose tissue: Associations with small adipocytes and insulin sensitivity in humans. Diabetologia 2009, 52, 882–890. [Google Scholar] [CrossRef]
- Ryden, M.; Petrus, P.; Andersson, D.P.; Medina-Gomez, G.; Escasany, E.; Corrales Cordon, P.; Dahlman, I.; Kulyte, A.; Arner, P. Insulin action is severely impaired in adipocytes of apparently healthy overweight and obese subjects. J. Intern. Med. 2019, 285, 578–588. [Google Scholar] [CrossRef]
- Medof, M.E.; Walter, E.I.; Roberts, W.L.; Haas, R.; Rosenberry, T.L. Decay accelerating factor of complement is anchored to cells by a C-terminal glycolipid. Biochemistry 1986, 25, 6740–6747. [Google Scholar] [CrossRef]
- Davitz, M.A.; Hereld, D.; Shak, S.; Krakow, J.; Englund, P.T.; Nussenzweig, V. A glycan-phosphatidylinositol-specific phospholipase D in human serum. Science 1987, 238, 81–84. [Google Scholar] [CrossRef]
- Davitz, M.A.; Low, M.G.; Nussenzweig, V. Release of decay-accelerating factor (DAF) from the cell membrane by phosphatidylinositol-specific phospholipase C (PIPLC). Selective modification of a complement regulatory protein. J. Exp. Med. 1986, 163, 1150–1161. [Google Scholar] [CrossRef]
- Mishra, V.; Heath, R.J. Structural and biochemical features of human albumin essential for eukaryotic cell culture. Int. J. Mol. Sci. 2021, 22, 8411. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Ducharme, N.A.; Bickel, P.E. Lipid droplets in lipogenesis and lipolysis. Endocrinology 2008, 149, 942–949. [Google Scholar] [CrossRef]
- Jung, H.N.; Jung, C.H. The role of anti-inflammatory adipokines in cardiometabolic disorders: Moving beyond adiponectin. Int. J. Mol. Sci. 2021, 22, 13529. [Google Scholar] [CrossRef]
- Romero, A.; Eckel, J. Organ crosstalk and the modulation of insulin signaling. Cells 2021, 10, 2082. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Low, M.G.; Cross, G.A.M. Glycosyl-sn-1,2-dimyristylphosphatidylinositol is covalently linked to Trypanosoma brucei variant surface glycoprotein. J. Biol. Chem. 1985, 260, 14547–14555. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Homans, S.W.; Dwek, R.A.; Rademacher, T.W. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science 1988, 239, 753–759. [Google Scholar] [CrossRef]
- Medof, M.E.; Kinoshita, T.; Silber, R.; Nussenzweig, V. Amelioration of lytic abnormalities of paroxysmale nocturnal hemoglobinuria with decay-accelerating factor. Proc. Natl. Acad. Sci. USA 1985, 82, 2980–2984. [Google Scholar] [CrossRef]
- Zalman, L.S.; Wood, L.M.; Frank, M.M.; Müller-Eberhard, H.J. Deficiency of the homologous restriction factor in paroxysmal nocturnal hemoglobinuria. J. Exp. Med. 1987, 165, 572–577. [Google Scholar] [CrossRef]
- Wilcox, L.A.; Ezzel, J.L.; Bernshaw, N.J.; Parker, C.J. Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serum. Blood 1991, 78, 820–829. [Google Scholar] [CrossRef]
- Cross, B.; Ronzon, F.; Roux, B.; Rieu, J.-P. Measurement of the anchorage force between GPI-anchored alkaline phosphatase and supported membranes by AFM force spectroscopy. Langmuir 2005, 21, 5149–5153. [Google Scholar] [CrossRef]
- Caseli, L.; Masui, D.C.; Furriel, R.P.M.; Leone, F.A.; Zaniquelli, M.E.D.; Orbulescu, J.; Leblanc, R.M. Rat osseous plate alkaline phosphatase as Langmuir monolayer—An infrared study at the air-water interface. J. Colloid Interface Sci. 2008, 320, 476–482. [Google Scholar] [CrossRef]
- Ronzon, F.; Rieu, J.-P.; Chauvet, J.-P.; Roux, B. A thermodynamic study of GPI-anchored and soluble form of alkaline phosphatase films at the air-water interface. J. Colloid Interface Sci. 2006, 301, 493–502. [Google Scholar] [CrossRef]
- Suzuki, K.; Okumura, Y. Mechanism of selective release of membrane proteins from human erythrocytes in the presence of liposomes. Arch. Biochem. Biophys. 2000, 379, 344–352. [Google Scholar] [CrossRef]
- Nakamura, M.; Tsujii, K.; Katsuragi, Y.; Kurihara, K.; Sunamoto, J. Taste receptor proteins directly extracted by liposome from intact epithelium of bullfrog tongue. Biochem. Biophys. Res. Commun. 1994, 201, 415–422. [Google Scholar] [CrossRef]
- Okumura, Y.; Ishitobi, M.; Sobel, M.; Akiyoshi, K.; Sunamoto, J. Transfer of membrane proteins from human platelet to liposomal fraction by interaction with liposomes containing an artificial boundary lipid. Biochem. Biophys. Acta 1994, 1194, 335–340. [Google Scholar] [CrossRef]
- Kogure, K.; Nakamura, C.; Okuda, O.; Hayashi, K.; Ueno, M. Effect of dicetylphosphate or stearic acid on spontaneous transfer of protein from influenza virus-infected cells to dimyristoylphosphatidylcholine liposomes. Biochim. Biophys. Acta 1997, 1329, 174–182. [Google Scholar] [CrossRef]
- Medof, M.E.; Kinoshita, T.; Nussenzweig, V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J. Exp. Med. 1984, 160, 1558–1578. [Google Scholar] [CrossRef]
- Nagarajan, S.; Anderson, M.; Ahmed, S.N.; Sell, K.W.; Selvaraj, P. Purification and optimization of functional reconstitution on the surface of leukemic cell lines of GPI-anchored Fc gamma receptor III. J. Immunol. Methods 1995, 184, 241–251. [Google Scholar] [CrossRef]
- Zhang, F.; Schmidt, W.G.; Hou, Y.; Williams, A.F.; Jacobson, K. Spontaneous incorporation of the glycosyl-phosphatidylinositol-linked protein Thy-1 into cell membranes. Proc. Natl. Acad. Sci. USA 1992, 89, 5231–5235. [Google Scholar] [CrossRef]
- Rieu, J.-P.; Ronzon, F.; Place, C.; Dekkiche, F.; Cross, B.; Roux, B. Insertion of GPI-anchored alkaline phosphatase into supported membranes: A combined AFM and fluorescence microscopy study. Acta Biochim. Pol. 2004, 51, 189–197. [Google Scholar] [CrossRef]
- Kouzayha, A.; Besson, F. GPI-alkaline phosphatase insertion into phosphatidylcholine monolayers: Phase behavior and morphology changes. Biochem. Biophys. Res. Commun. 2005, 333, 1315–1321. [Google Scholar] [CrossRef]
- Morandat, S.; Bortolato, M.; Roux, B. Cholesterol-dependent insertion of glycosylphosphatidylinositol-anchored enzyme. Biochim. Biophys. Acta 2002, 1564, 473–478. [Google Scholar] [CrossRef]
- Premkumar, D.R.D.; Fukuoka, Y.; Sevlever, D.; Brunschwig, E.; Rosenberry, T.L.; Tykocinski, M.L.; Medof, M.E. Properties of exogenously added GPI-anchored proteins following their incorporation into cells. J. Cell. Biochem. 2001, 82, 234–245. [Google Scholar] [CrossRef]
- Müller, G.; Jung, C.; Frick, W.; Bandlow, W.; Kramer, W. Interaction of phosphatidylinositolglycan(-peptides) with plasma membrane lipid rafts triggers insulin-mimetic signaling in rat adipocytes. Arch. Biochem. Biophys. 2002, 408, 7–16. [Google Scholar] [CrossRef]
- Müller, G.; Hanekop, N.; Kramer, W.; Bandlow, W.; Frick, W. Interaction of phosphoinositolglycan(-peptides) with plasma membrane lipid rafts of rat adipocytes. Arch. Biochem. Biophys. 2002, 408, 17–32. [Google Scholar] [CrossRef]
- Kiechle, F.L.; Jarett, L.; Kotagal, N.; Popp, D.A. Partial purification from rat adipocyte plasma membranes of a chemical mediator which stimulates the action of insulin on pyruvate dehydrogenase. J. Biol. Chem. 1981, 256, 2945–2951. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Cuatrecasas, P. Insulin stimulates the generation from hepatic plasma membranes of modulators derived from an inositol glycolipid. Proc. Natl. Sci. Acad. USA 1986, 83, 5793–5797. [Google Scholar] [CrossRef]
- Romero, G.; Luttrell, L.; Rogol, A.; Zeller, K.; Hewlett, E.; Larner, J. Phosphatidylinositol-glycan anchors of membrane proteins: Potential precursors of insulin mediators. Science 1988, 240, 509–511. [Google Scholar] [CrossRef]
- Saltiel, A.R. Second messengers of insulin action. Diabetes Care 1990, 13, 244–256. [Google Scholar] [CrossRef]
- Stralfors, P. Insulin second messengers. Bioessays 1997, 19, 327–335. [Google Scholar] [CrossRef]
- Larner, J.; Brautigan, D.L.; Thorner, M.O. D-Chiro-inositol glycans in insulin signaling and insulin resistance. Mol. Med. 2010, 16, 543–551. [Google Scholar] [CrossRef]
- Misek, D.E.; Saltiel, A.R. An inositol phosphate glycan derived from a Trypanosoma brucei glycosyl-phosphatidylinositol mimics some of the metabolic actions of insulin. J. Biol. Chem. 1992, 267, 16266–16273. [Google Scholar] [CrossRef]
- Hecht, M.-L.; Tsai, Y.-H.; Liu, X.; Wolfrum, C.; Seeberger, P.H. Synthetic inositol phosphoglycans related to GPI lack insulin-mimetic activity. ACS Chem. Biol. 2010, 5, 1075–1086. [Google Scholar] [CrossRef]
- Asplin, I.; Galasko, G.; Larner, J. Chiro-inositol deficiency and insulin resistance: A comparison of the chiro-inositol- and the myo-inositol-containing insulin mediators isolated from urine, hemodialysate, and muscle of control and type II diabetic subjects. Proc. Natl. Acad. Sci. USA 1993, 90, 5924–5928. [Google Scholar] [CrossRef]
- Chakraborty, N.; d’Alarcao, M. An anionic inositol phosphate glycan pseudotetrasaccharide exhibits high insulin-mimetic activity in rat adipocytes. Bioorg. Med. Chem. 2005, 13, 6732–6741. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Rome, S.; Blandin, A.; Le Lay, S. Adipocyte-derived extracellular vesicles: State of the art. Int. J. Mol. Sci. 2021, 22, 1788. [Google Scholar] [CrossRef]
- Müller, G. Microvesicles/exosomes as potential novel biomarkers of metabolic diseases. Diabetes Metab. Syndr. Obes. 2012, 5, 247–282. [Google Scholar] [CrossRef]
- Lakhter, A.J.; Sims, E.K. Emerging roles for extracellular vesicles in diabetes and related metabolic disorders. Mol. Endocrinol. 2015, 29, 1535–1548. [Google Scholar] [CrossRef]
- Medof, M.E.; Nagarajan, S.; Tykocinski, M.L. Cell-surface engineering with GPI-anchored proteins. FASEB J. 1996, 10, 574–586. [Google Scholar] [CrossRef]
- Bouwens, E.A.; Stavenuiter, F.; Mosnier, L.O. Cell painting with an EPCR to augment the protein C system. Thromb. Haemost. 2015, 114, 1144–1155. [Google Scholar] [CrossRef]
- Müller, G.A. Oral delivery of protein drugs: Driver for personalized medicine. Curr. Issues Mol. Biol. 2011, 13, 13–24. [Google Scholar]
- Müller, G.; Wetekam, E.; Jung, C.; Bandlow, W. Membrane association of lipoprotein lipase and a cAMP-binding ectoprotein in rat adipocytes. Endocrinology 1997, 33, 12149–12159. [Google Scholar] [CrossRef][Green Version]
- Huang, K.; Park, S. Affinity purification of glycosylphosphatidylinositol-anchored proteins by alpha-toxin. In Glycosaminoglycans; Methods in Molecular Biology; Balagurunathan, K., Nakato, H., Desai, U., Saijoh, Y., Eds.; Humana Press: New York, NY, USA, 2003; Volume 2303. [Google Scholar]
- Müller, G. Control of lipid storage and cell size between adipocytes by vesicle-associated glycosylphosphatidylinositol-anchored proteins. Arch. Physiol. Biochem. 2010, 117, 23–43. [Google Scholar] [CrossRef]
- Lawrence, J.C., Jr.; Guinovart, J.J.; Larner, J. Activation of rat adipocyte glycogen synthase by insulin. J. Biol. Chem. 1977, 252, 444–450. [Google Scholar] [CrossRef]
- Müller, G.; Jordan, H.; Petry, S.; Wetekam, E.-M.; Schindler, P. Analysis of lipid metabolism in adipocytes using a fluorescent fatty acid derivative. I. Insulin stimulation of lipogenesis. Biochim. Biophys. Acta 1997, 1347, 23–39. [Google Scholar] [CrossRef]
- Müller, G.; Schubert, K.; Fiedler, F.; Bandlow, W. The cAMP-binding ectoprotein from Saccharomyces cerevisiae is membrane-anchored by glycosylphosphatdylinositol. J. Biol. Chem. 1992, 267, 25337–25346. [Google Scholar] [CrossRef]
- Stieger, S.; Diem, S.; Jakob, A.; Brodbeck, U. Enzymatic properties of phosphatidylinositol-glycan-specific phospholipase C from rat liver and phosphatidylinositol-glycan-specific phospholipase D from rat serum. Eur. J. Biochem. 1991, 197, 67–73. [Google Scholar] [CrossRef]
- Li, J.-Y.; Hollfelder, K.; Huang, K.-S.; Low, M.G. Structural features of GPI-specific phospholipase D revealed by proteolytic fragmentation and Ca2+ binding studies. J. Biol. Chem. 1994, 269, 28963–28971. [Google Scholar] [CrossRef]
- Gnagey, A.L.; Forte, M.; Rosenberry, T.L. Isolation and characterization of acetylcholinesterase from Drosophila. J. Biol. Chem. 1987, 262, 13920–13928. [Google Scholar] [CrossRef]
- Kaya, A.L.; Özcan, B.; Sisecioglu, M.; Özdemir, H. Purification of acetylcholinesterase by 9-amino-1,2,3,4-tetrahydroacridine from human erythrocytes. Appl. Biochem. Biotechnol. 2013, 170, 198–209. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, G.A.; Müller, T.D. Biological Role of the Intercellular Transfer of Glycosylphosphatidylinositol-Anchored Proteins: Stimulation of Lipid and Glycogen Synthesis. Int. J. Mol. Sci. 2022, 23, 7418. https://doi.org/10.3390/ijms23137418
Müller GA, Müller TD. Biological Role of the Intercellular Transfer of Glycosylphosphatidylinositol-Anchored Proteins: Stimulation of Lipid and Glycogen Synthesis. International Journal of Molecular Sciences. 2022; 23(13):7418. https://doi.org/10.3390/ijms23137418
Chicago/Turabian StyleMüller, Günter A., and Timo D. Müller. 2022. "Biological Role of the Intercellular Transfer of Glycosylphosphatidylinositol-Anchored Proteins: Stimulation of Lipid and Glycogen Synthesis" International Journal of Molecular Sciences 23, no. 13: 7418. https://doi.org/10.3390/ijms23137418
APA StyleMüller, G. A., & Müller, T. D. (2022). Biological Role of the Intercellular Transfer of Glycosylphosphatidylinositol-Anchored Proteins: Stimulation of Lipid and Glycogen Synthesis. International Journal of Molecular Sciences, 23(13), 7418. https://doi.org/10.3390/ijms23137418