
The Transcription Coregulator RIP140 Inhibits Cancer Cell Proliferation by Targeting the Pentose Phosphate Pathway



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
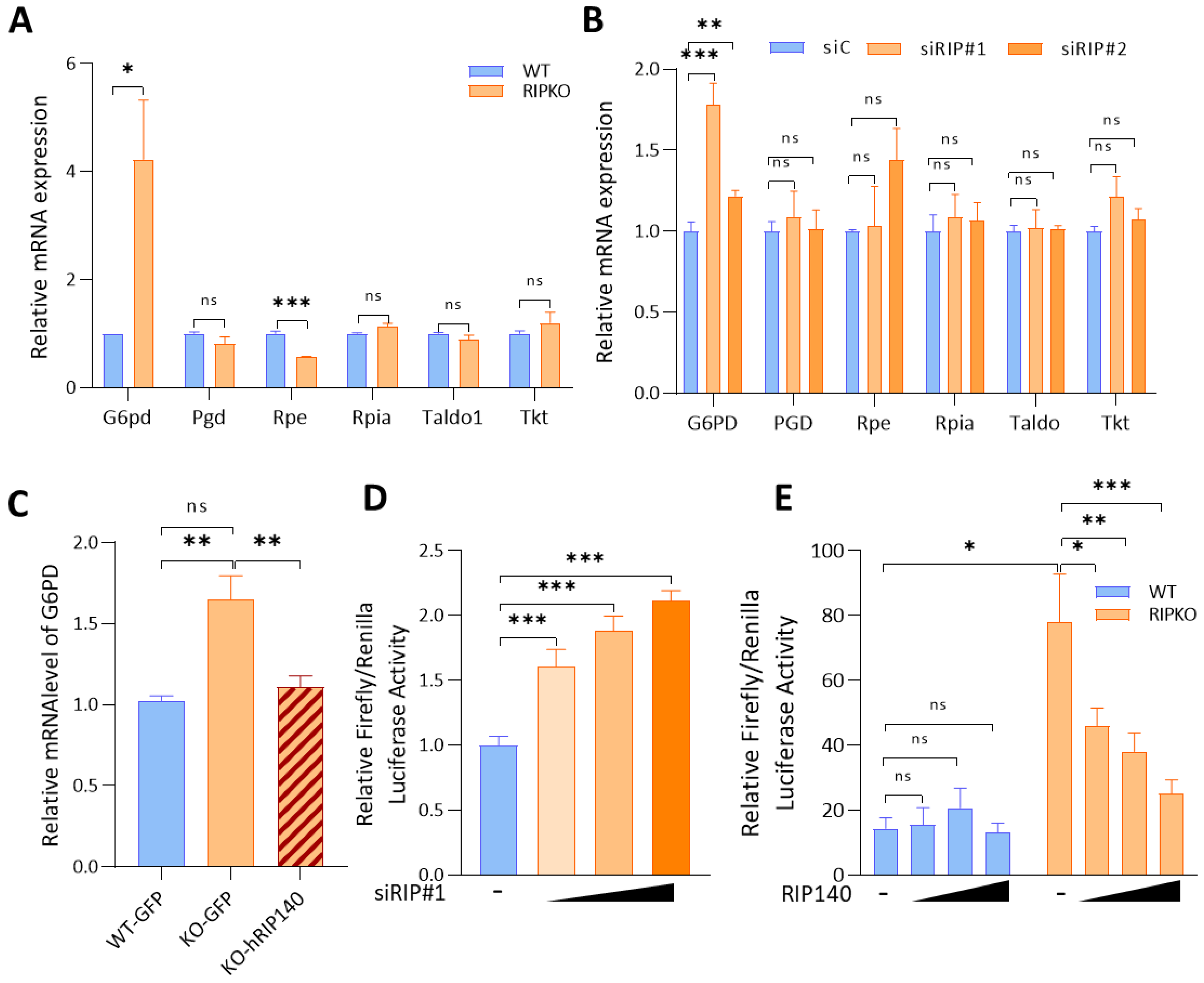
2.1. RIP140 Inhibits G6PD Gene Expression at the Transcriptional Level
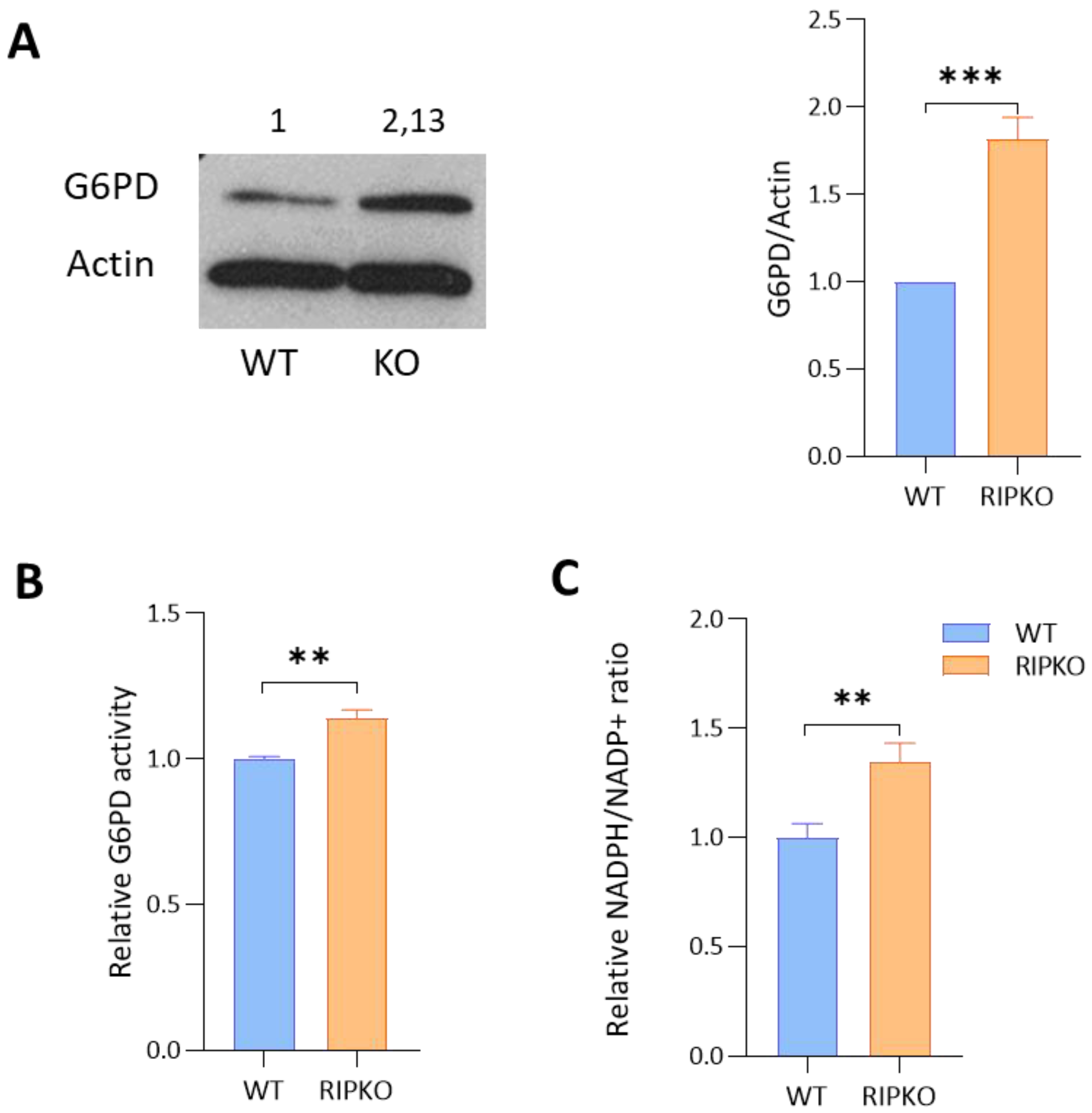
2.2. RIP140 Deficiency Increases G6PD Activity
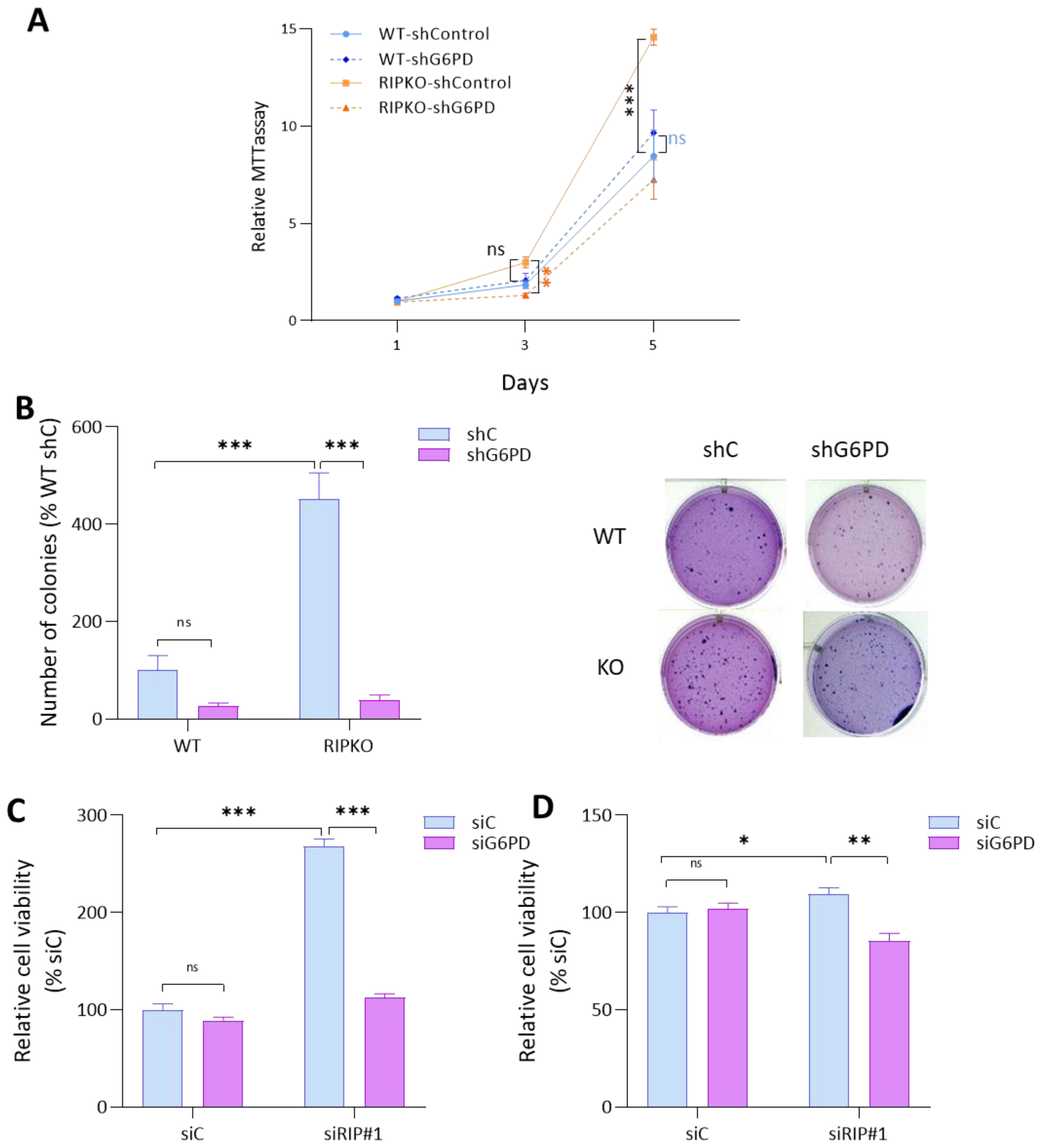
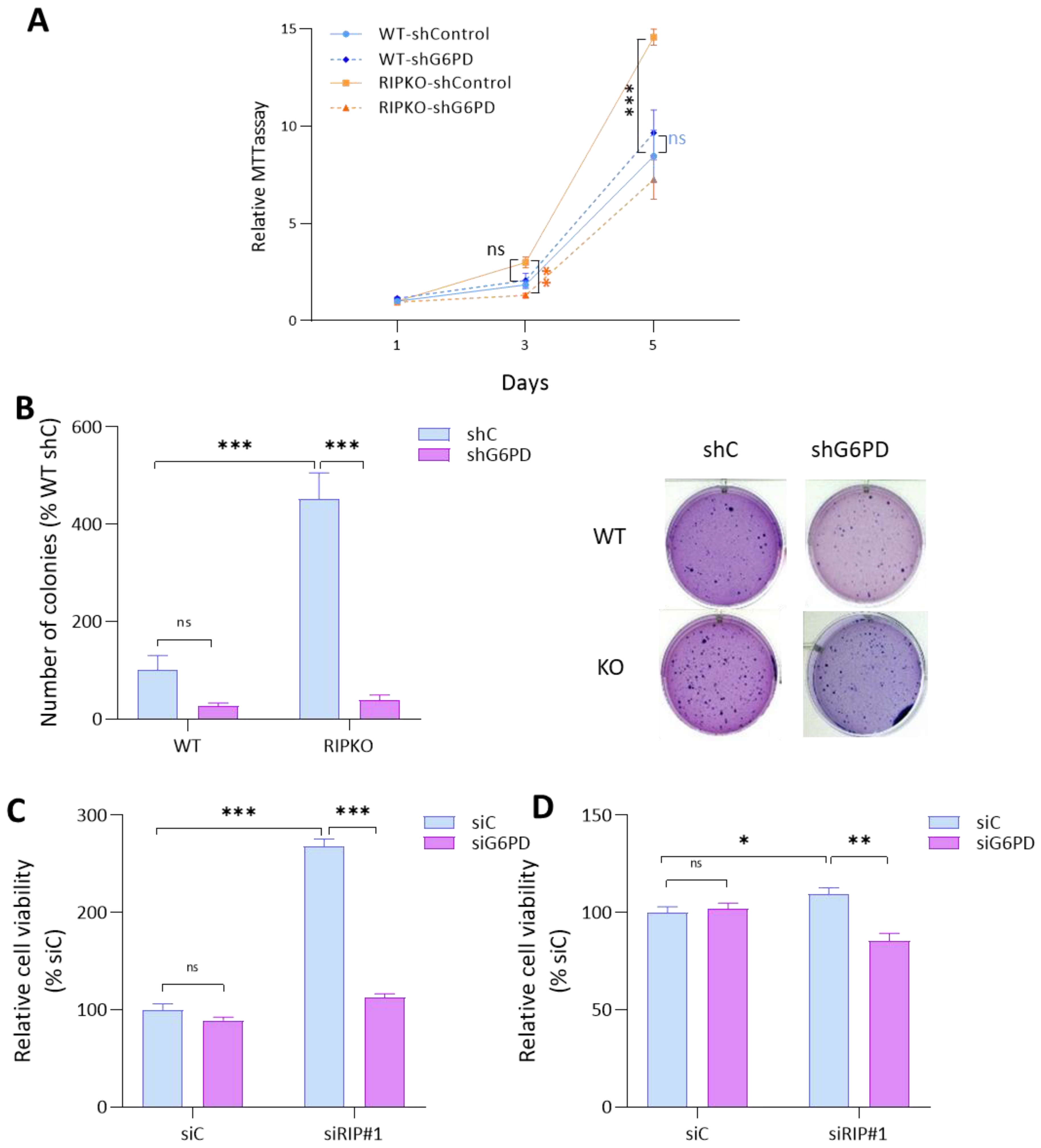
2.3. G6PD Is Required for the Proliferative Advantage of RIP140-Deficient Cells
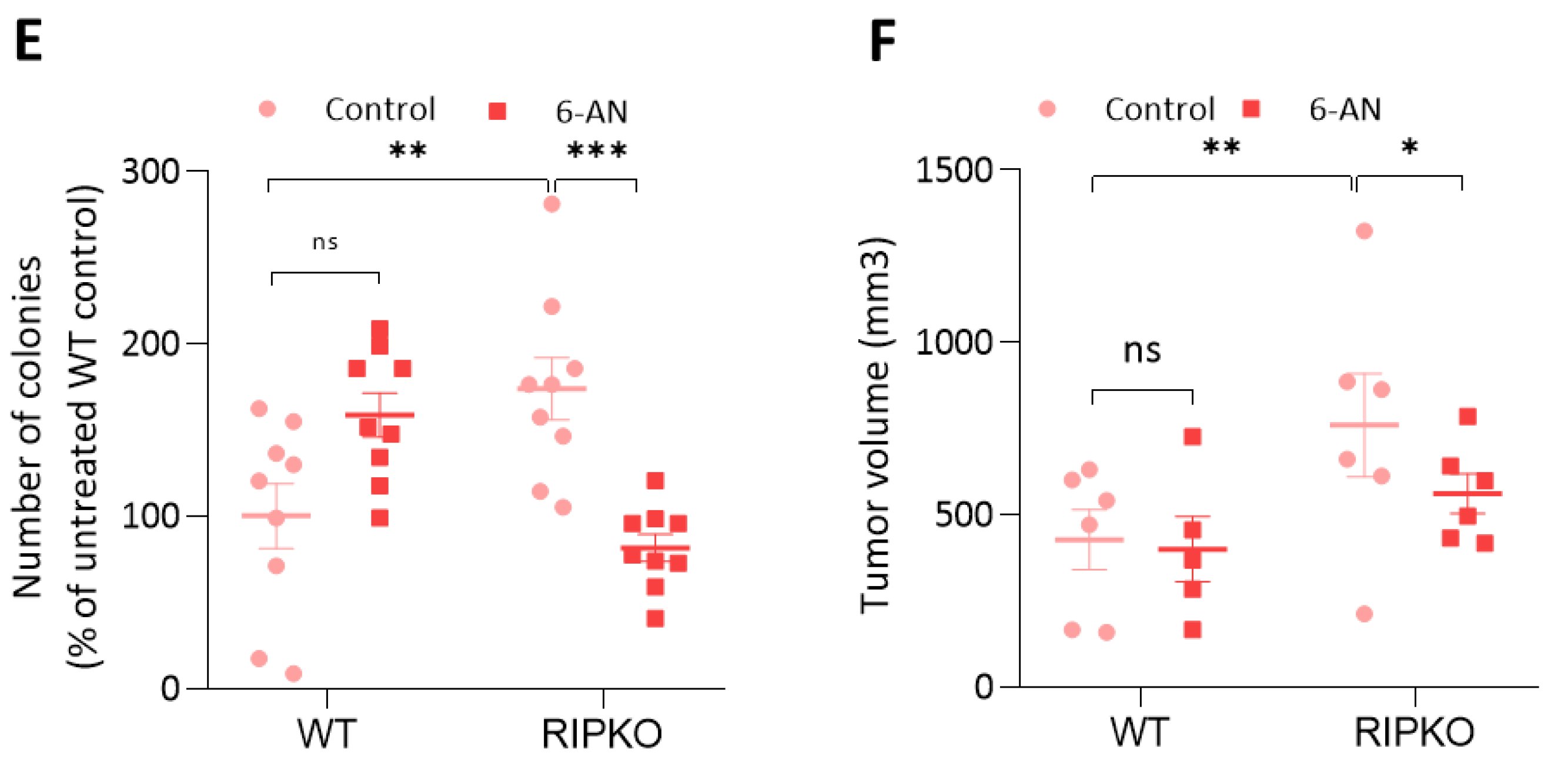
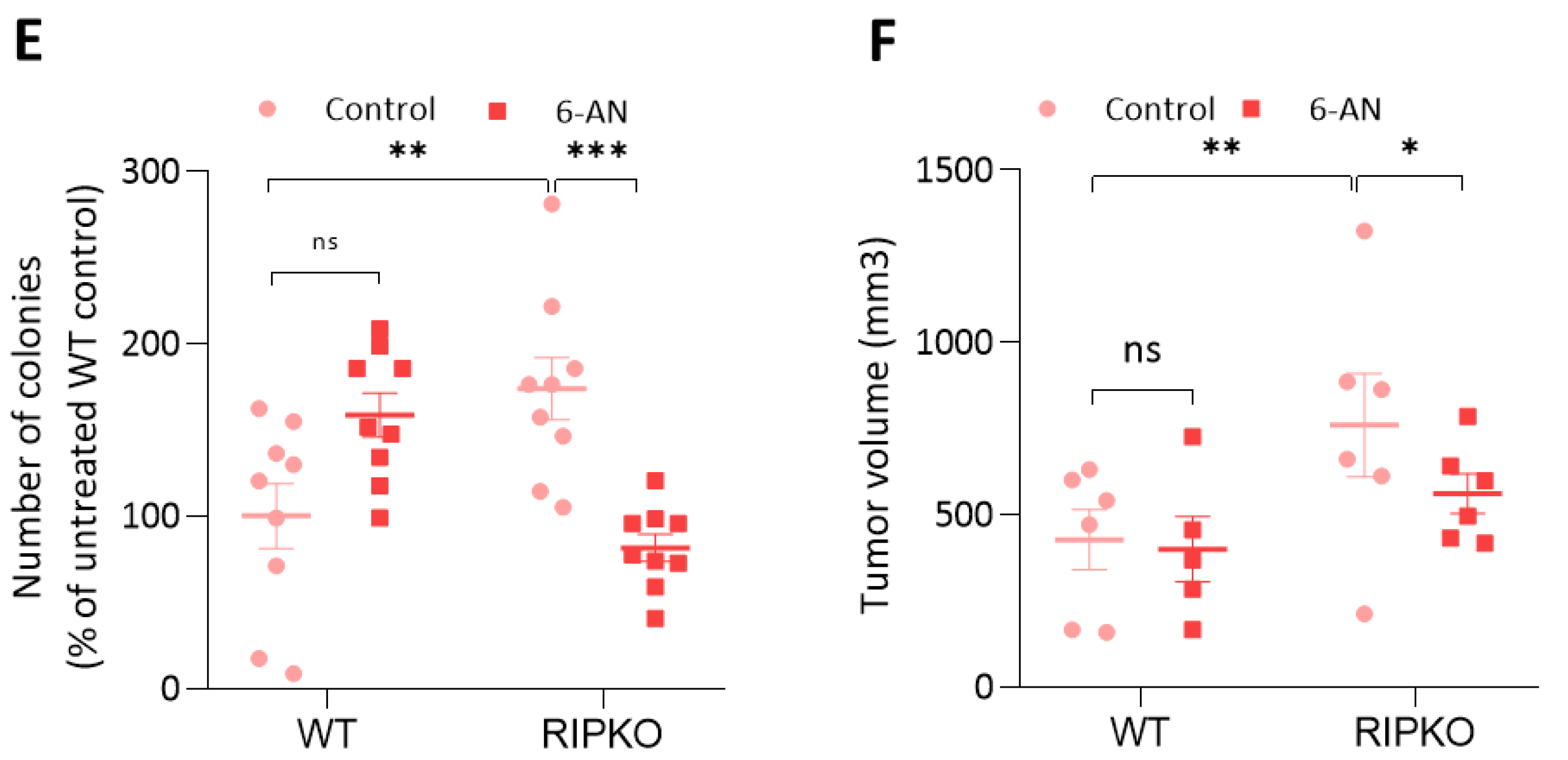
2.4. RIP140 Deficiency Sensitizes Cells to G6PD Inhibition
3. Discussion
4. Materials and Methods
4.1. Plasmids, siRNA and Reagents
4.2. Cell Culture
4.3. Real-Time qPCR
4.4. Protein Detection
4.5. Luciferase Reporter Assay
4.6. Cell Proliferation Analysis
4.7. Soft-Agar Colony Assay
4.8. G6PD Activity Assay
4.9. NADP/NADPH Assays
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, L.; Zhou, Y. Crucial Role of the Pentose Phosphate Pathway in Malignant Tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stern, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 Regulates Biosynthesis through Direct Inactivation of Glucose-6-Phosphate Dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augereau, P.; Badia, E.; Balaguer, P.; Carascossa, S.; Castet, A.; Jalaguier, S.; Cavaillès, V. Negative Regulation of Hormone Signaling by RIP140. J. Steroid Biochem. Mol. Biol. 2006, 102, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, M.; Docquier, A.; Castet-Nicolas, A.; Gitenay, D.; Jalaguier, S.; Teyssier, C.; Cavaillès, V. The Emerging Role of the Transcriptional Coregulator RIP140 in Solid Tumors. Biochim. Biophys. Acta 2015, 1856, 144–150. [Google Scholar] [CrossRef]
- Lapierre, M.; Bonnet, S.; Bascoul-Mollevi, C.; Ait-Arsa, I.; Jalaguier, S.; Del Rio, M.; Plateroti, M.; Roepman, P.; Ychou, M.; Pannequin, J.; et al. RIP140 Increases APC Expression and Controls Intestinal Homeostasis and Tumorigenesis. J. Clin. Investig. 2014, 124, 1899–1913. [Google Scholar] [CrossRef] [Green Version]
- Triki, M.; Lapierre, M.; Cavailles, V.; Mokdad-Gargouri, R. Expression and Role of Nuclear Receptor Coregulators in Colorectal Cancer. World J. Gastroenterol. 2017, 23, 4480–4490. [Google Scholar] [CrossRef]
- Docquier, A.; Harmand, P.-O.; Fritsch, S.; Chanrion, M.; Darbon, J.-M.; Cavaillès, V. The Transcriptional Coregulator RIP140 Represses E2F1 Activity and Discriminates Breast Cancer Subtypes. Clin. Cancer Res. 2010, 16, 2959–2970. [Google Scholar] [CrossRef] [Green Version]
- Aziz, M.H.; Chen, X.; Zhang, Q.; DeFrain, C.; Osland, J.; Luo, Y.; Shi, X.; Yuan, R. Suppressing NRIP1 Inhibits Growth of Breast Cancer Cells in Vitro and in Vivo. Oncotarget 2015, 6, 39714–39724. [Google Scholar] [CrossRef] [Green Version]
- Rosell, M.; Jones, M.C.; Parker, M.G. Role of Nuclear Receptor Corepressor RIP140 in Metabolic Syndrome. Biochim. Biophys. Acta 2011, 1812, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.-H.; Xue, X.; Zhu, X.; Li, X. Downregulation of RIP140 in Triple-Negative Breast Cancer Inhibits the Growth and Proliferation of Cancer Cells. Oncol. Lett. 2018, 15, 8784–8788. [Google Scholar] [CrossRef] [Green Version]
- Jalaguier, S.; Teyssier, C.; Nait Achour, T.; Lucas, A.; Bonnet, S.; Rodriguez, C.; Elarouci, N.; Lapierre, M.; Cavaillès, V. Complex Regulation of LCoR Signaling in Breast Cancer Cells. Oncogene 2017, 36, 4790–4801. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, V.; Gitenay, D.; Fritsch, S.; Bonnet, S.; Győrffy, B.; Jalaguier, S.; Linares, L.K.; Cavaillès, V.; Teyssier, C. RIP140 Inhibits Glycolysis-Dependent Proliferation of Breast Cancer Cells by Regulating GLUT3 Expression through Transcriptional Crosstalk between Hypoxia Induced Factor and P53. Cell. Mol. Life Sci. 2022, 79, 270. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, J. Transcriptional Coregulator RIP140: An Essential Regulator of Physiology. J. Mol. Endocrinol. 2017, 58, R147–R158. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The Pentose Phosphate Pathway and Cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, W.; Yang, Y.; Gu, C. Exploring the Role of Glucose-6-phosphate Dehydrogenase in Cancer (Review). Oncol. Rep. 2020, 44, 2325–2336. [Google Scholar] [CrossRef]
- Pu, H.; Zhang, Q.; Zhao, C.; Shi, L.; Wang, Y.; Wang, J.; Zhang, M. Overexpression of G6PD Is Associated with High Risks of Recurrent Metastasis and Poor Progression-Free Survival in Primary Breast Carcinoma. World J. Surg. Oncol. 2015, 13, 323. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Sun, H.; Zhang, S.; Shan, C. The Multiple Roles of Glucose-6-Phosphate Dehydrogenase in Tumorigenesis and Cancer Chemoresistance. Life 2022, 12, 271. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Z.; Ni, Y.; Bai, H.; Han, Q.; Yi, Z.; Yi, X.; Agbana, Y.L.; Kuang, Y.; Zhu, Y. NF-ΚB and PSTAT3 Synergistically Drive G6PD Overexpression and Facilitate Sensitivity to G6PD Inhibition in CcRCC. Cancer Cell Int. 2020, 20, 483. [Google Scholar] [CrossRef]
- Wu, S.; Wang, H.; Li, Y.; Xie, Y.; Huang, C.; Zhao, H.; Miyagishi, M.; Kasim, V. Transcription Factor YY1 Promotes Cell Proliferation by Directly Activating the Pentose Phosphate Pathway. Cancer Res. 2018, 78, 4549–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Gu, X.; Zhang, E.; Park, M.-A.; Pereira, A.M.; Wang, S.; Morrison, T.; Li, C.; Blenis, J.; Gerbaudo, V.H.; et al. Estradiol Promotes Pentose Phosphate Pathway Addiction and Cell Survival via Reactivation of Akt in MTORC1 Hyperactive Cells. Cell Death Dis. 2014, 5, e1231. [Google Scholar] [CrossRef] [PubMed]
- Cavaillès, V.; Dauvois, S.; L’Horset, F.; Lopez, G.; Hoare, S.; Kushner, P.J.; Parker, M.G. Nuclear Factor RIP140 Modulates Transcriptional Activation by the Estrogen Receptor. EMBO J. 1995, 14, 3741–3751. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-C.; Stern, A.; Chiu, D.T.-Y. G6PD: A Hub for Metabolic Reprogramming and Redox Signaling in Cancer. Biomed. J. 2021, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, Z.; Zhu, Z.; Chen, A.; Fu, G.; Wang, Y.; Pan, H.; Jin, B. Modulation of G6PD Affects Bladder Cancer via ROS Accumulation and the AKT Pathway in Vitro. Int. J. Oncol. 2018, 53, 1703–1712. [Google Scholar] [CrossRef] [Green Version]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High MTORC1 Activity Drives Glycolysis Addiction and Sensitivity to G6PD Inhibition in Acute Myeloid Leukemia Cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef]
- Zilliacus, J.; Holter, E.; Wakui, H.; Tazawa, H.; Treuter, E.; Gustafsson, J.A. Regulation of Glucocorticoid Receptor Activity by 14--3-3-Dependent Intracellular Relocalization of the Corepressor RIP140. Mol. Endocrinol. 2001, 15, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Makarona, K.; Caputo, V.S.; Costa, J.R.; Liu, B.; O’Connor, D.; Iskander, D.; Roper, D.; Robertson, L.; Bhatnagar, N.; Terpos, E.; et al. Transcriptional and Epigenetic Basis for Restoration of G6PD Enzymatic Activity in Human G6PD-Deficient Cells. Blood 2014, 124, 134–141. [Google Scholar] [CrossRef]
- Carascossa, S.; Gobinet, J.; Georget, V.; Lucas, A.; Badia, E.; Castet, A.; White, R.; Nicolas, J.-C.; Cavaillès, V.; Jalaguier, S. Receptor-Interacting Protein 140 Is a Repressor of the Androgen Receptor Activity. Mol. Endocrinol. 2006, 20, 1506–1518. [Google Scholar] [CrossRef]
- White, R.; Leonardsson, G.; Rosewell, I.; Ann Jacobs, M.; Milligan, S.; Parker, M. The Nuclear Receptor Co-Repressor Nrip1 (RIP140) Is Essential for Female Fertility. Nat. Med. 2000, 6, 1368–1374. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative Studies of the Growth of Mouse Embryo Cells in Culture and Their Development into Established Lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Rodier, G.; Kirsh, O.; Baraibar, M.; Houlès, T.; Lacroix, M.; Delpech, H.; Hatchi, E.; Arnould, S.; Severac, D.; Dubois, E.; et al. The Transcription Factor E4F1 Coordinates CHK1-Dependent Checkpoint and Mitochondrial Functions. Cell Rep. 2015, 11, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Palassin, P.; Lapierre, M.; Pyrdziak, S.; Wagner, A.; Stehle, R.; Corsini, C.; Duffour, J.; Bonnet, S.; Boulahtouf, A.; Rodriguez, C.; et al. A Truncated NRIP1 Mutant Amplifies Microsatellite Instability of Colorectal Cancer by Regulating MSH2/MSH6 Expression, and Is a Prognostic Marker of Stage III Tumors. Cancers 2021, 13, 4449. [Google Scholar] [CrossRef] [PubMed]
- Quantitative Measurement of Cancer Cell Proliferation Using CellPlayerTM Kinetic Proliferation Assay. Available online: https://www.news-medical.net/whitepaper/20161003/Quantitative-Measurement-of-Cancer-Cell-Proliferation-Using-CellPlayer-Kinetic-Proliferation-Assay.aspx (accessed on 24 June 2022).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquier, V.; Gitenay, D.; Cavaillès, V.; Teyssier, C. The Transcription Coregulator RIP140 Inhibits Cancer Cell Proliferation by Targeting the Pentose Phosphate Pathway. Int. J. Mol. Sci. 2022, 23, 7419. https://doi.org/10.3390/ijms23137419
Jacquier V, Gitenay D, Cavaillès V, Teyssier C. The Transcription Coregulator RIP140 Inhibits Cancer Cell Proliferation by Targeting the Pentose Phosphate Pathway. International Journal of Molecular Sciences. 2022; 23(13):7419. https://doi.org/10.3390/ijms23137419
Chicago/Turabian StyleJacquier, Valentin, Delphine Gitenay, Vincent Cavaillès, and Catherine Teyssier. 2022. "The Transcription Coregulator RIP140 Inhibits Cancer Cell Proliferation by Targeting the Pentose Phosphate Pathway" International Journal of Molecular Sciences 23, no. 13: 7419. https://doi.org/10.3390/ijms23137419
APA StyleJacquier, V., Gitenay, D., Cavaillès, V., & Teyssier, C. (2022). The Transcription Coregulator RIP140 Inhibits Cancer Cell Proliferation by Targeting the Pentose Phosphate Pathway. International Journal of Molecular Sciences, 23(13), 7419. https://doi.org/10.3390/ijms23137419