
Tumor-Suppressor Role of the α1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma †

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
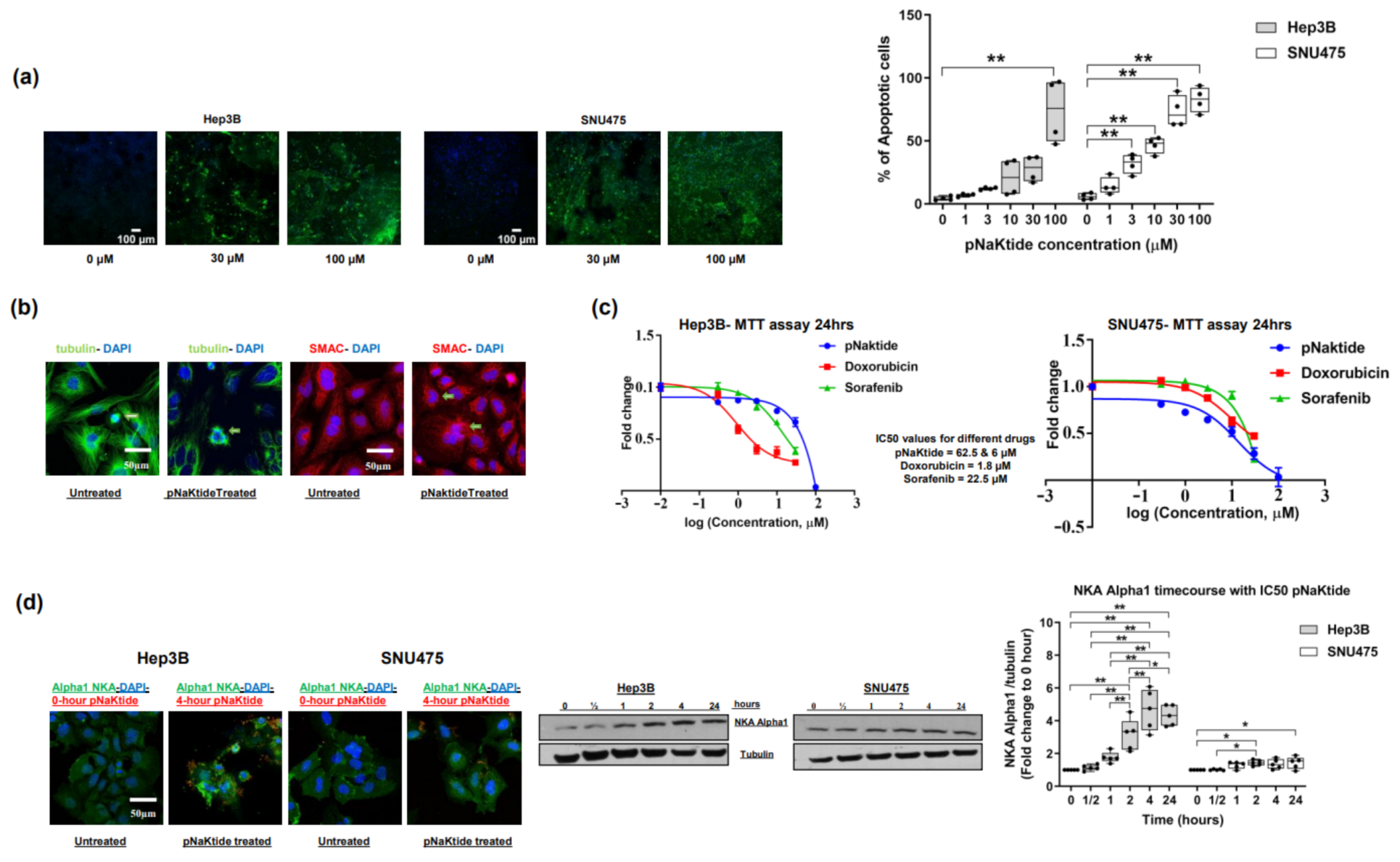
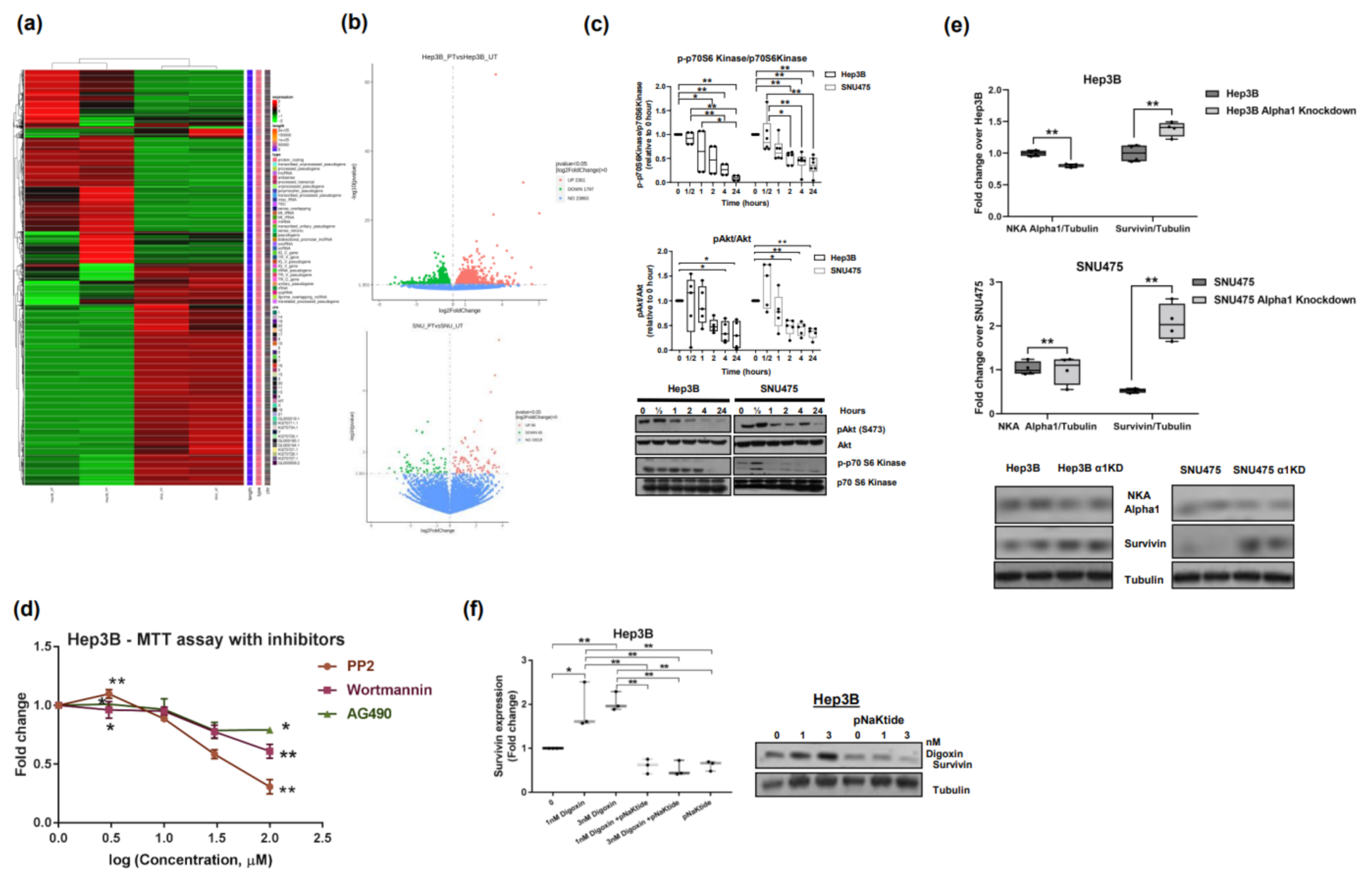
2.1. Effect of α1-Na/K-ATPase Receptor Complex Normalization in Two Human HCC Cell Lines (Hep3B and SNU475)
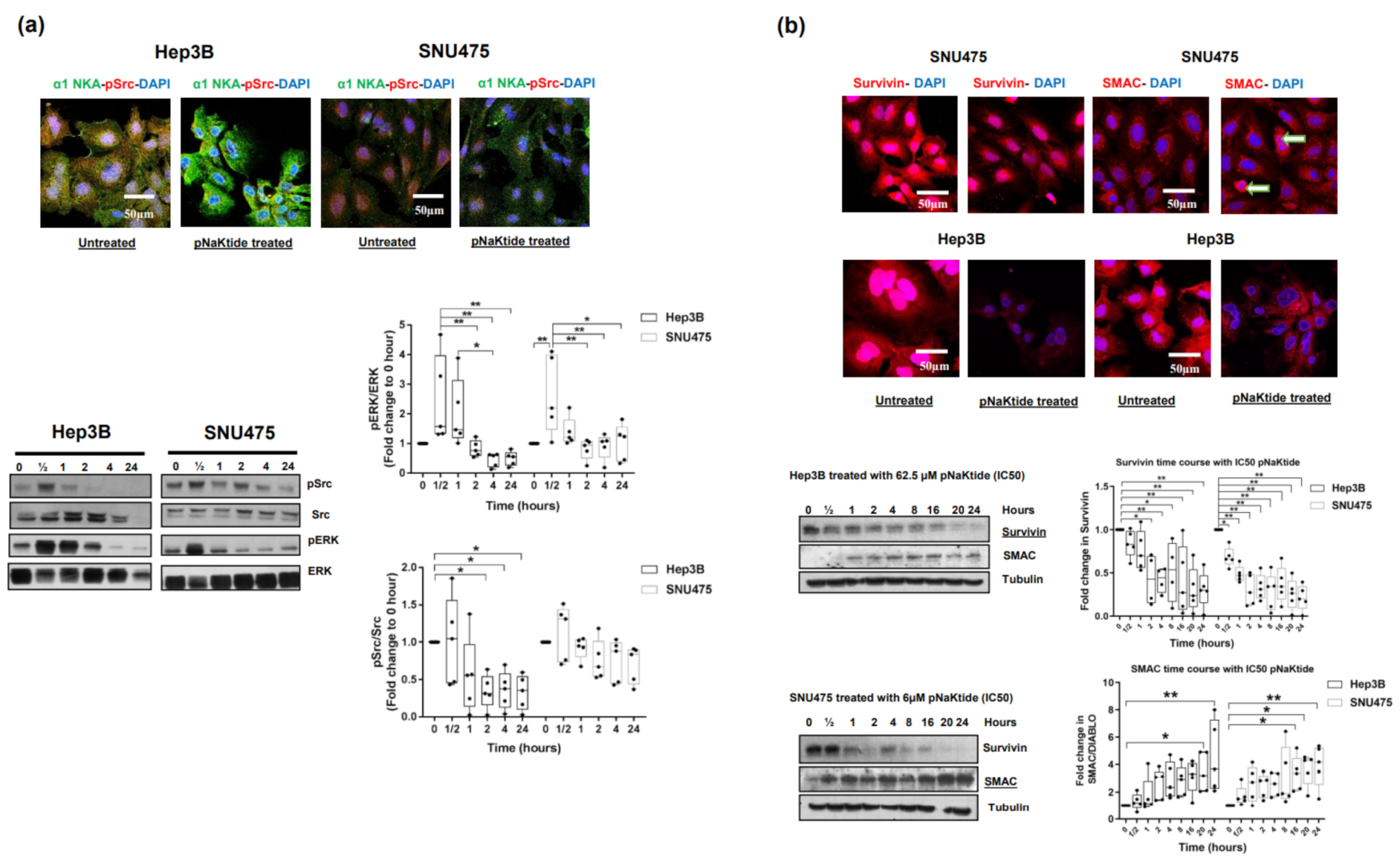
2.2. Effect of α1-Na/K-ATPase Receptor Complex Normalization on α1-Subunit, Src/ERK Kinases and Survivin/SMAC Protein Expressions in Two Human HCC Cell Lines
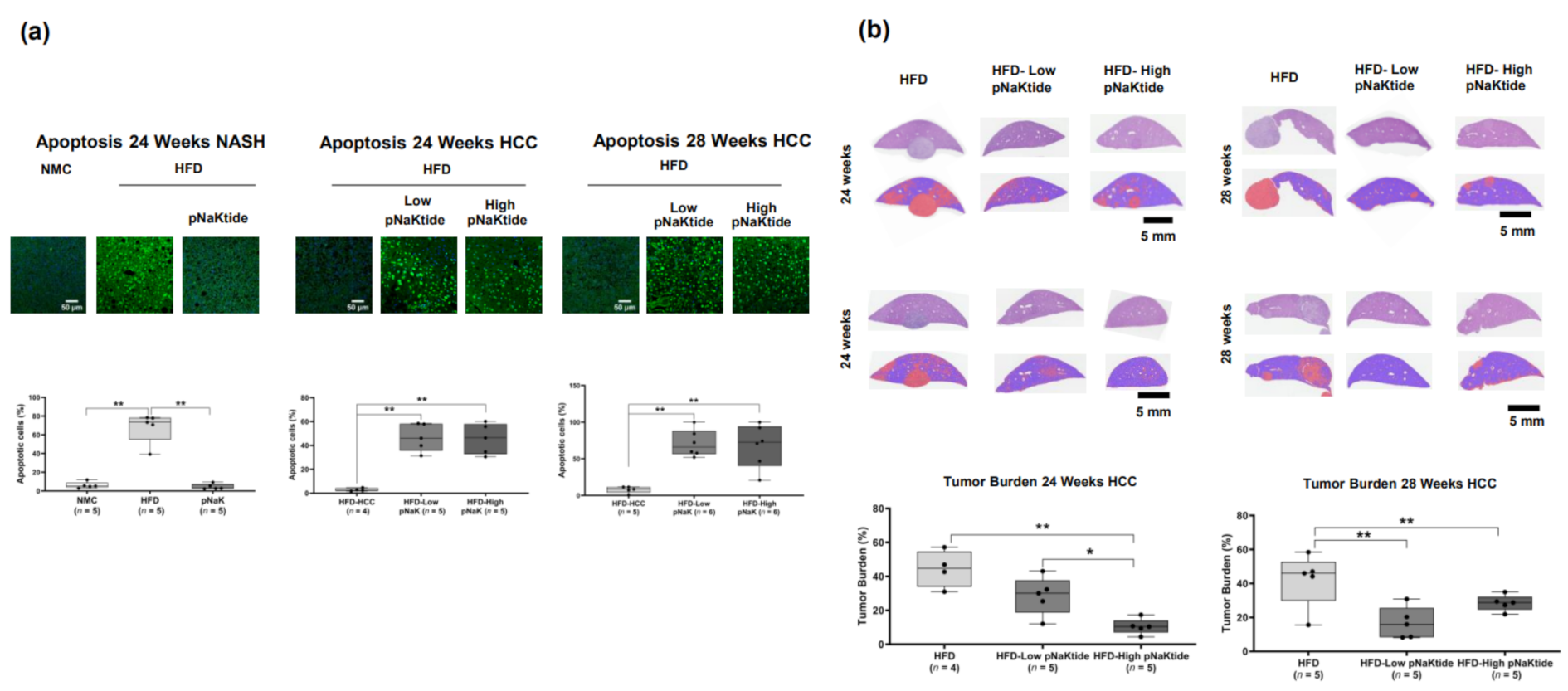
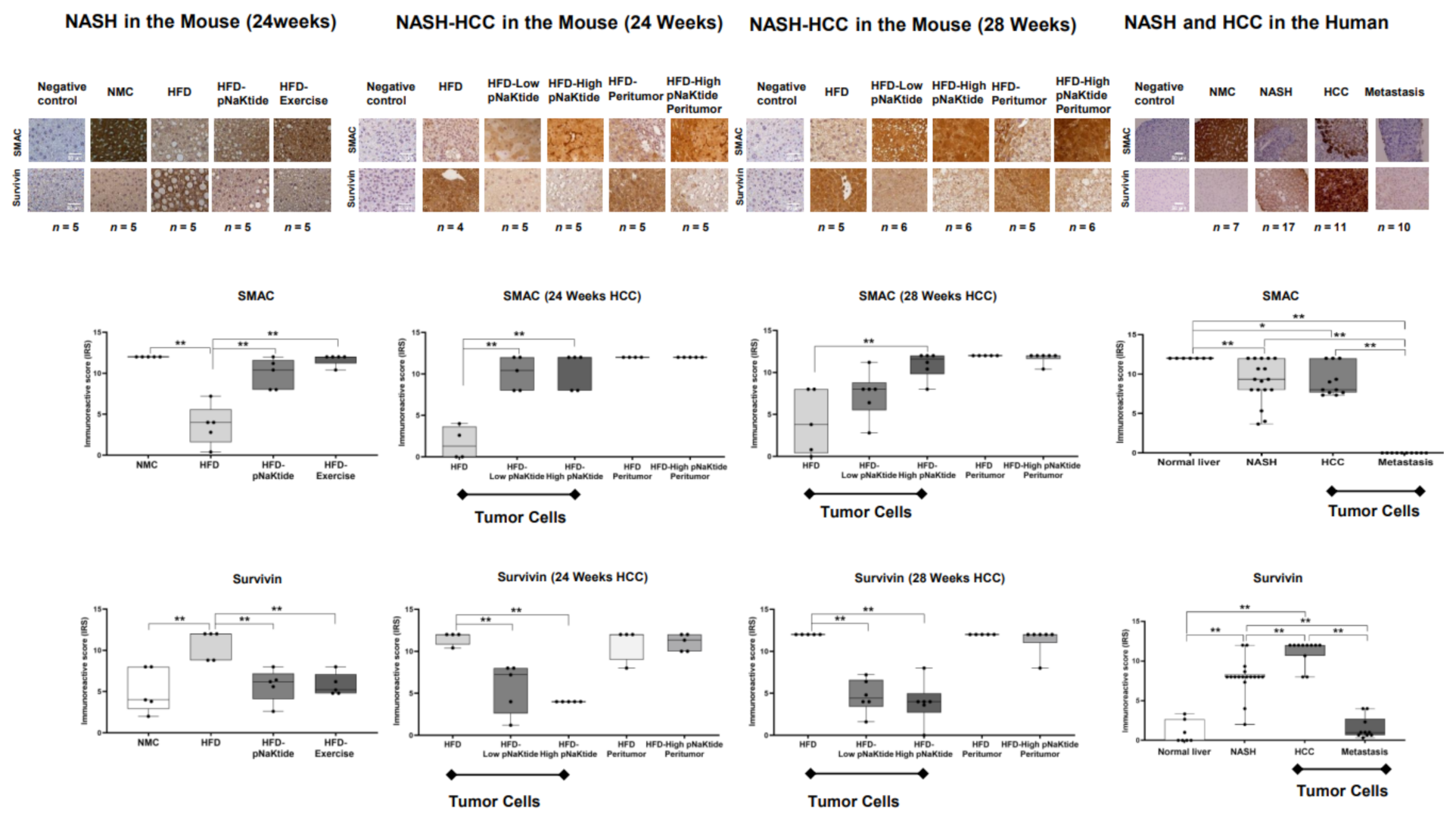
2.3. Effect of α1-Na/K-ATPase Receptor Complex Normalization on Apoptosis in Liver and Tumor Cells from Mouse Models of NASH & NASH-HCC
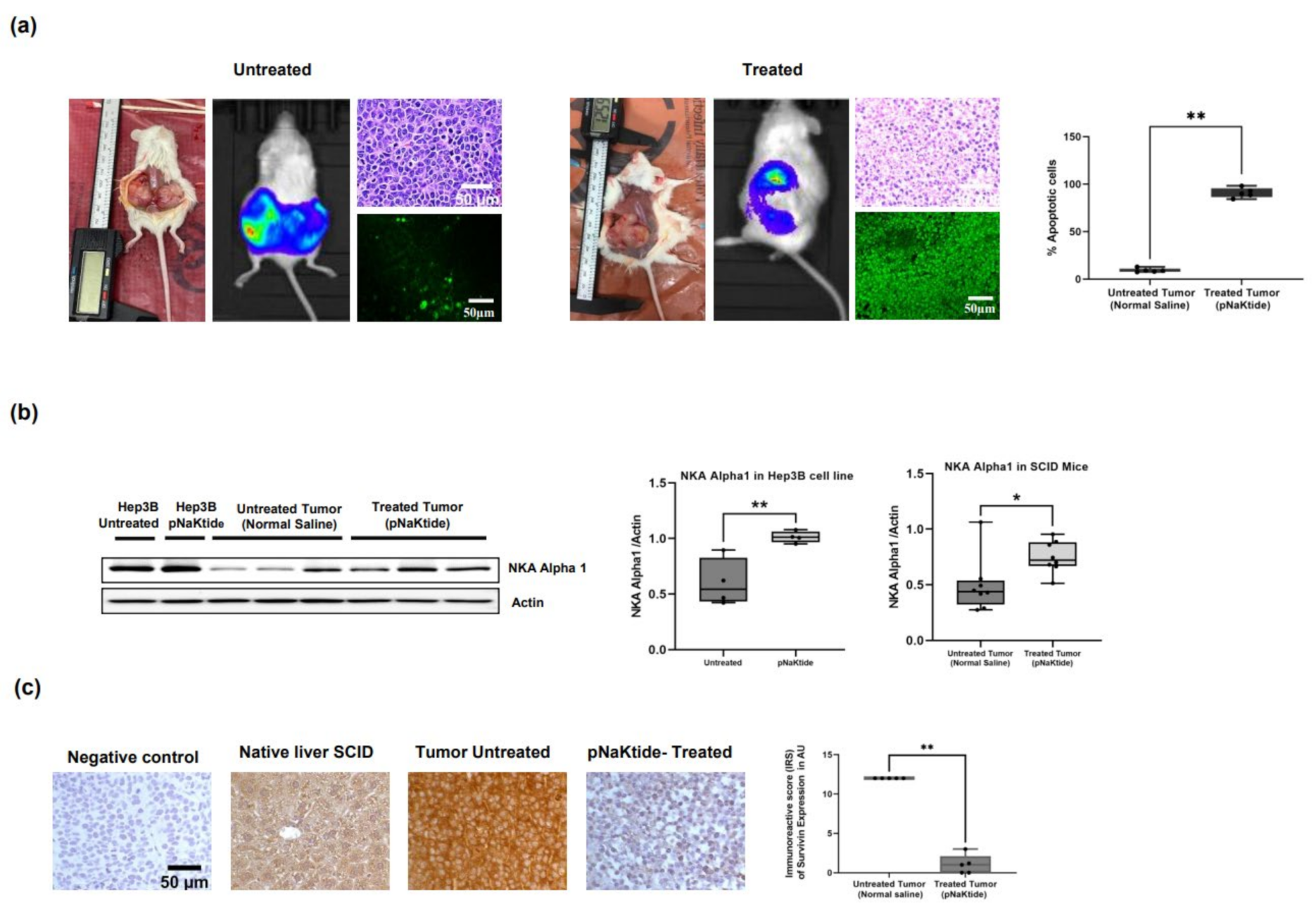
2.4. Effect of α1-Na/K-ATPase Receptor Complex Normalization in the SCID Mice Xenografts Tumors
2.5. Effects of α1-Na/K-ATPase Receptor Complex Normalization in Mouse Models of NASH & NASH-HCC
2.6. Survivin and SMAC Proteins Expression in Human Subjects
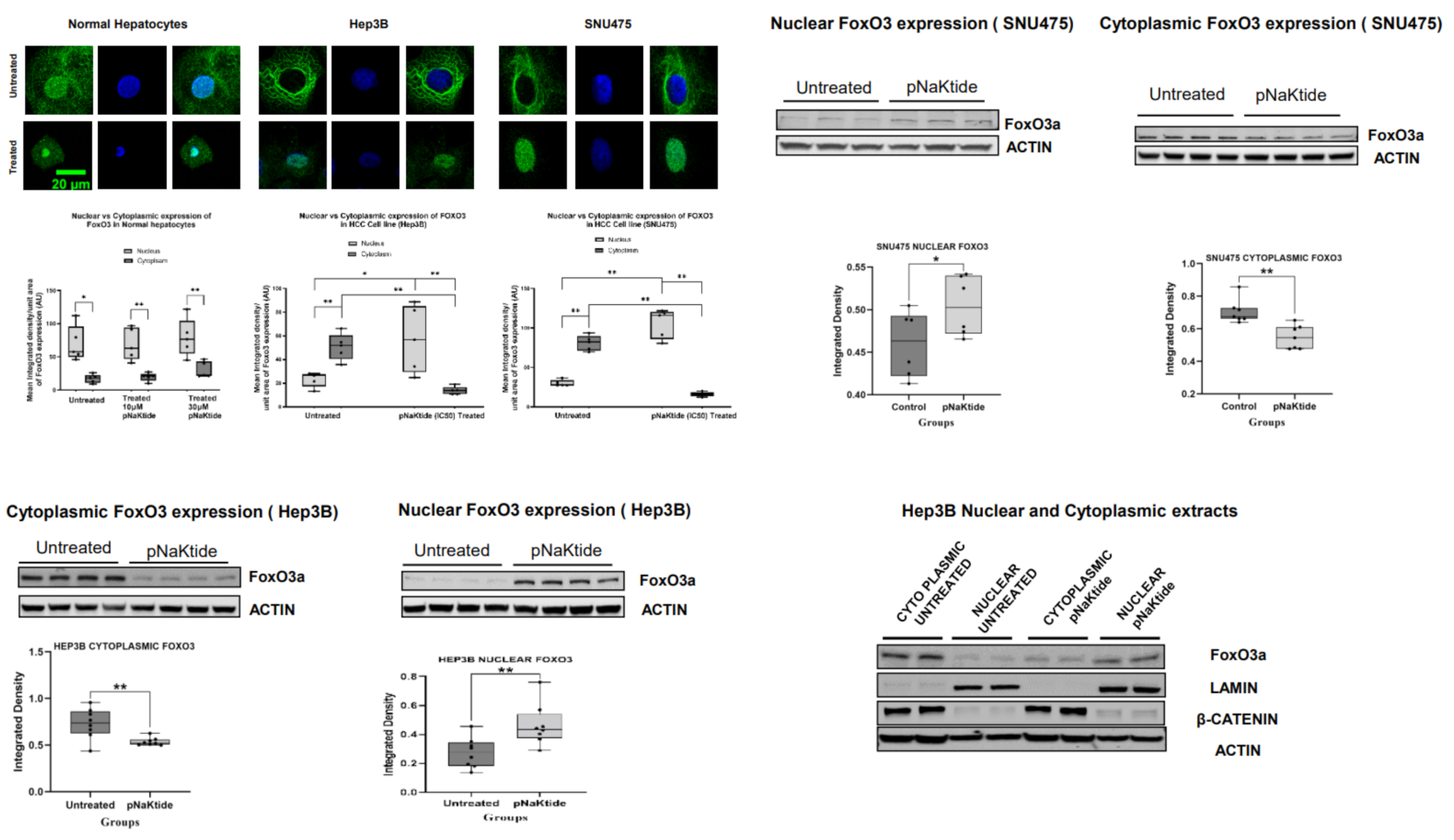
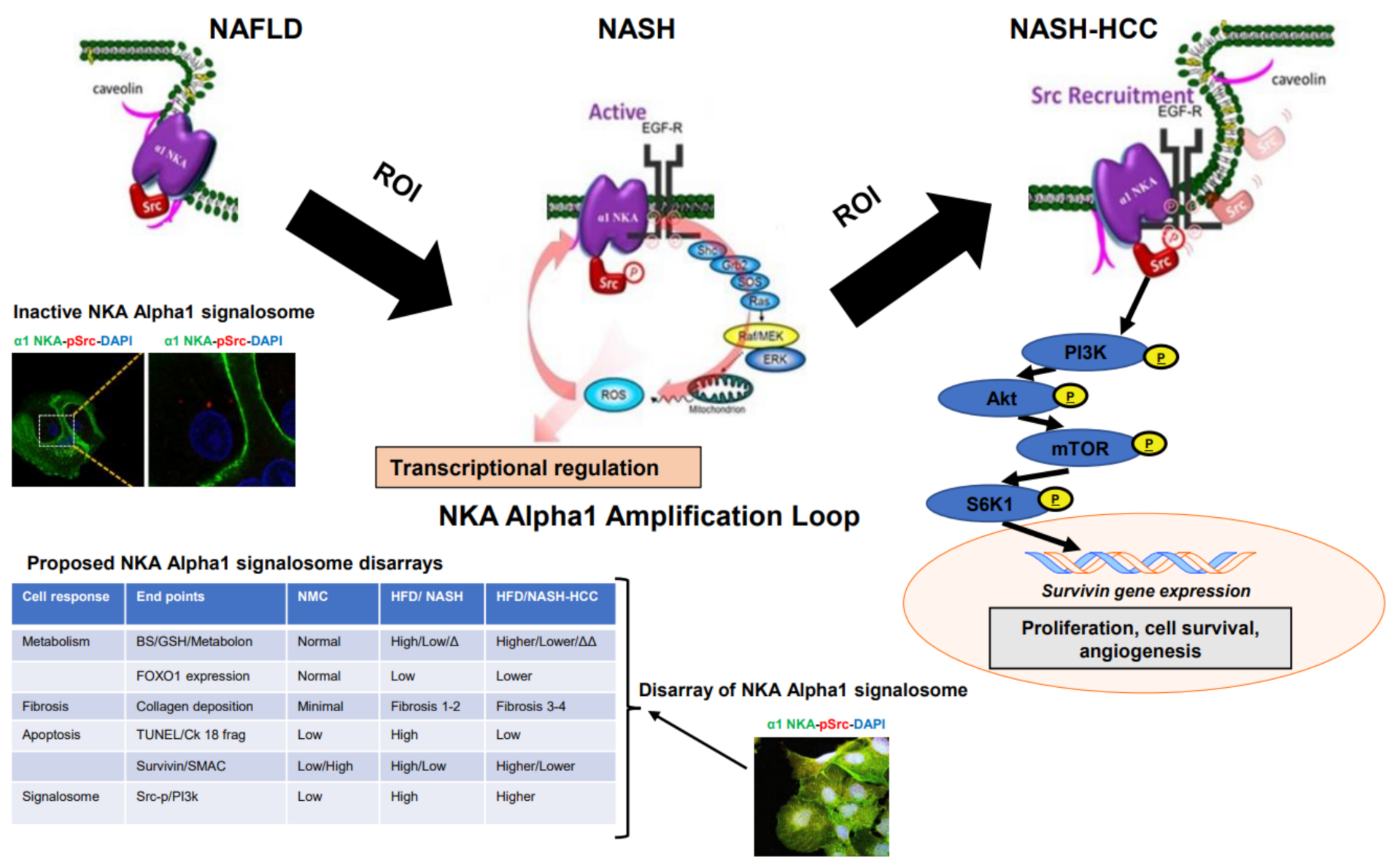
2.7. Signalosome of the α1-NKA
3. Discussion
4. Materials and Methods
4.1. In-Vitro Studies
4.2. NASH and NASH Related HCC Mouse Models
4.3. Tumor Xenograft Model
4.4. Human Liver Tissues
4.5. Treatment Agents
4.6. Liver Apoptotic Activity Assessment
4.7. Tumor Burden in Mouse Liver
4.8. SCID Mouse Xenograft Model
4.9. Confocal Microscopy Assessment on Immuno-Stained Cells/Liver Tissue
4.10. Quantitation of Protein Expression
4.11. Western Blotting
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | protein kinase B |
| CRM1 | chromosomal region maintenance 1 |
| Smac/DIABLO | second mitochondria-derived activator of caspases/direct inhibitor of apoptosis-binding protein with low pI |
| EGFR, | growth factor receptor |
| ESLD | end stage liver disease |
| FoxO | forkhead box O family of transcription factors |
| FoxO3 | Forkhead box O3 |
| CC | hepatocellular carcinoma |
| H&E | hematoxylin and eosin |
| HFD | high fat diet |
| IVIS | In vivo Imaging System |
| mTOR | mammalian target of rapamycin |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NKA | Na/K-ATPase |
| NMC | standard mouse chow |
| NS | normal saline |
| pAkt | phosphorylated protein kinase B |
| pERK | phosphorylated extracellular signal-regulated kinase |
| PI3K | phosphatidylinositol-3-kinase |
| pSrc | phosphorylation of Src kinase |
| PTEN | phosphatase and tensin homolog deleted on Chromosome 10 |
| RIPA buffer | Radioimmunoprecipitation assay buffer |
| ROI | reactive oxygen intermediates |
| siRNA | Small interfering RNA |
| Smac | second mitochondria-derived activator of caspases |
| Src | Src kinase |
| S6K1 | ribosomal protein S6 kinase beta-1 |
| α1-Na/K-ATPase | Na/K-ATPase Alpha1 subunit |
| α1-NKA-KND | α1-NKA knock down HCC cell lines |
References
- Takeda, M.; Sakaguchi, T.; Hiraide, T.; Shibasaki, Y.; Morita, Y.; Kikuchi, H.; Ikegami, K.; Setou, M.; Konno, H.; Takeuchi, H. Role of caveolin-1 in hepatocellular carcinoma arising from non-alcoholic fatty liver disease. Cancer Sci. 2018, 109, 2401–2411. [Google Scholar] [CrossRef] [PubMed]
- Beyoglu, D.; Idle, J.R. The metabolomic window into hepatobiliary disease. J. Hepatol. 2013, 59, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Hepatocellular Carcinoma in Obesity: Finding a Needle in the Haystack? Adv. Exp. Med. Biol. 2018, 1061, 63–77. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22, 990. [Google Scholar] [CrossRef]
- Cai, T.; Wang, H.; Chen, Y.; Liu, L.; Gunning, W.T.; Quintas, L.E.; Xie, Z.J. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J. Cell Biol. 2008, 182, 1153–1169. [Google Scholar] [CrossRef]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.J. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef]
- Xie, Z.; Cai, T. Na+-K+--ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef]
- Kinoshita, P.F.; Yshii, L.M.; Vasconcelos, A.R.; Orellana, A.M.; Lima Lde, S.; Davel, A.P.; Rossoni, L.V.; Kawamoto, E.M.; Scavone, C. Signaling function of Na,K-ATPase induced by ouabain against LPS as an inflammation model in hippocampus. J. Neuroinflammation 2014, 11, 218. [Google Scholar] [CrossRef]
- Bejcek, J.; Spiwok, V.; Kmonickova, E.; Rimpelova, S. Na+/K+-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation. Molecules 2021, 26, 1905. [Google Scholar] [CrossRef]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar] [CrossRef]
- Wang, X.; Cai, L.; Xie, J.X.; Cui, X.; Zhang, J.; Wang, J.; Chen, Y.; Larre, I.; Shapiro, J.I.; Pierre, S.V.; et al. A caveolin binding motif in Na/K-ATPase is required for stem cell differentiation and organogenesis in mammals and C. elegans. Sci. Adv. 2020, 6, eaaw5851. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef]
- Holmes, D. PI3K pathway inhibitors approach junction. Nat. Rev. Drug. Discov. 2011, 10, 563–564. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50. [Google Scholar] [CrossRef]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and Tumorigenesis: Molecular Mechanisms and Therapeutic Strategies. J Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef]
- Liu, W.; Yue, F.; Zheng, M.; Merlot, A.; Bae, D.H.; Huang, M.; Lane, D.; Jansson, P.; Lui, G.Y.; Richardson, V.; et al. The proto-oncogene c-Src and its downstream signaling pathways are inhibited by the metastasis suppressor, NDRG1. Oncotarget 2015, 6, 8851–8874. [Google Scholar] [CrossRef]
- Iskandar, Z.A.; Al-Joudi, F.S. Expression of survivin in fetal and adult normal tissues of rat. Malays. J. Pathol. 2006, 28, 101–105. [Google Scholar]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef]
- Barrett, R.M.; Osborne, T.P.; Wheatley, S.P. Phosphorylation of survivin at threonine 34 inhibits its mitotic function and enhances its cytoprotective activity. Cell Cycle 2009, 8, 278–283. [Google Scholar] [CrossRef]
- Townley, A.R.; Wheatley, S.P. Mitochondrial survivin reduces oxidative phosphorylation in cancer cells by inhibiting mitophagy. J. Cell Sci. 2020, 133, jcs247379. [Google Scholar] [CrossRef]
- Colnaghi, R.; Connell, C.M.; Barrett, R.M.; Wheatley, S.P. Separating the anti-apoptotic and mitotic roles of survivin. J. Biol. Chem. 2006, 281, 33450–33456. [Google Scholar] [CrossRef]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J. Clin. Investig. 2004, 114, 1117–1127. [Google Scholar] [CrossRef]
- Dohi, T.; Xia, F.; Altieri, D.C. Compartmentalized phosphorylation of IAP by protein kinase A regulates cytoprotection. Mol. Cell 2007, 27, 17–28. [Google Scholar] [CrossRef]
- McKenzie, J.A.; Grossman, D. Role of the apoptotic and mitotic regulator survivin in melanoma. Anticancer Res. 2012, 32, 397–404. [Google Scholar]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Cui, X.; Li, Z.; Yu, H.; Cai, L.; Jia, X.; He, D.; Wang, C.; Gao, T.; Xie, Z. Na/K-ATPase Y260 Phosphorylation-mediated Src Regulation in Control of Aerobic Glycolysis and Tumor Growth. Sci. Rep. 2018, 8, 12322. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Cancino, G.; Espinosa, M.; Maldonado, V.; Melendez-Zajgla, J. Regulation of mitochondrial Smac/DIABLO-selective release by survivin. Oncogene 2007, 26, 7569–7575. [Google Scholar] [CrossRef] [PubMed]
- McNeish, I.A.; Lopes, R.; Bell, S.J.; McKay, T.R.; Fernandez, M.; Lockley, M.; Wheatley, S.P.; Lemoine, N.R. Survivin interacts with Smac/DIABLO in ovarian carcinoma cells but is redundant in Smac-mediated apoptosis. Exp. Cell Res. 2005, 302, 69–82. [Google Scholar] [CrossRef]
- Song, Z.; Yao, X.; Wu, M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J. Biol. Chem. 2003, 278, 23130–23140. [Google Scholar] [CrossRef]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Hirsova, P.; Bohm, F.; Dohnalkova, E.; Nozickova, B.; Heikenwalder, M.; Gores, G.J.; Weber, A. Hepatocyte apoptosis is tumor promoting in murine nonalcoholic steatohepatitis. Cell Death Dis. 2020, 11, 80. [Google Scholar] [CrossRef]
- Mallick, A.; Sanabria, J.A.; Schade, M.; Aguilar, R.; Andryka, M.; Schlatzer, D.; Li, X.; Hazlett, F.E.; Kachman, M.; Raskind, A.; et al. PNaKtide abrogates accelerated liver cells aging/senescenceinduced by western diet in the rodent. HPB 2019, 21, S139–S140. [Google Scholar] [CrossRef]
- Boidot, R.; Vegran, F.; Lizard-Nacol, S. Transcriptional regulation of the survivin gene. Mol. Biol. Rep. 2014, 41, 233–240. [Google Scholar] [CrossRef]
- Liu, L.; Wu, J.; Kennedy, D.J. Regulation of Cardiac Remodeling by Cardiac Na+/K+-ATPase Isoforms. Front. Physiol. 2016, 7, 382. [Google Scholar] [CrossRef]
- Xie, J.X.; Li, X.; Xie, Z. Regulation of renal function and structure by the signaling Na/K-ATPase. IUBMB Life 2013, 65, 991–998. [Google Scholar] [CrossRef]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Onda, M.; Uchida, E.; Yokomuro, S.; Hayashi, T.; Kobayashi, T.; Sasajima, K.; Shirota, T.; Tajiri, T.; Egami, K.; et al. Immunohistochemical localization of Na+, K+-ATPase in human normal and malignant pancreatic tissues. Nihon Ika Daigaku Zasshi 1989, 56, 579–583. [Google Scholar] [CrossRef][Green Version]
- Seligson, D.B.; Rajasekaran, S.A.; Yu, H.; Liu, X.; Eeva, M.; Tze, S.; Ball, W., Jr.; Horvath, S.; deKernion, J.B.; Rajasekaran, A.K. Na,K-adenosine triphosphatase alpha1-subunit predicts survival of renal clear cell carcinoma. J. Urol. 2008, 179, 338–345. [Google Scholar] [CrossRef]
- Suzuki, A.; Ito, T.; Kawano, H.; Hayashida, M.; Hayasaki, Y.; Tsutomi, Y.; Akahane, K.; Nakano, T.; Miura, M.; Shiraki, K. Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene 2000, 19, 1346–1353. [Google Scholar] [CrossRef]
- Su, C. Survivin in survival of hepatocellular carcinoma. Cancer Lett. 2016, 379, 184–190. [Google Scholar] [CrossRef]
- Giodini, A.; Kallio, M.J.; Wall, N.R.; Gorbsky, G.J.; Tognin, S.; Marchisio, P.C.; Symons, M.; Altieri, D.C. Regulation of microtubule stability and mitotic progression by survivin. Cancer Res. 2002, 62, 2462–2467. [Google Scholar]
- Zaffaroni, N.; Pennati, M.; Daidone, M.G. Survivin as a target for new anticancer interventions. J. Cell. Mol. Med. 2005, 9, 360–372. [Google Scholar] [CrossRef]
- O’Connor, D.S.; Schechner, J.S.; Adida, C.; Mesri, M.; Rothermel, A.L.; Li, F.; Nath, A.K.; Pober, J.S.; Altieri, D.C. Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am. J. Pathol. 2000, 156, 393–398. [Google Scholar] [CrossRef]
- Dan, H.C.; Jiang, K.; Coppola, D.; Hamilton, A.; Nicosia, S.V.; Sebti, S.M.; Cheng, J.Q. Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene 2004, 23, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Meng, Q.; Liu, L.Z.; You, Y.P.; Liu, N.; Jiang, B.H. Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem. Biophys Res. Commun. 2010, 395, 219–224. [Google Scholar] [CrossRef]
- Beretta, G.L.; Corno, C.; Zaffaroni, N.; Perego, P. Role of FoxO Proteins in Cellular Response to Antitumor Agents. Cancers 2019, 11, 90. [Google Scholar] [CrossRef]
- Yang, S.; Pang, L.; Dai, W.; Wu, S.; Ren, T.; Duan, Y.; Zheng, Y.; Bi, S.; Zhang, X.; Kong, J. Role of Forkhead Box O Proteins in Hepatocellular Carcinoma Biology and Progression (Review). Front. Oncol. 2021, 11, 667730. [Google Scholar] [CrossRef]
- Guha, M.; Plescia, J.; Leav, I.; Li, J.; Languino, L.R.; Altieri, D.C. Endogenous tumor suppression mediated by PTEN involves survivin gene silencing. Cancer Res. 2009, 69, 4954–4958. [Google Scholar] [CrossRef]
- Obexer, P.; Hagenbuchner, J.; Unterkircher, T.; Sachsenmaier, N.; Seifarth, C.; Bock, G.; Porto, V.; Geiger, K.; Ausserlechner, M. Repression of BIRC5/survivin by FOXO3/FKHRL1 sensitizes human neuroblastoma cells to DNA damage-induced apoptosis. Mol. Biol. Cell 2009, 20, 2041–2048. [Google Scholar] [CrossRef]
- Hay, N. Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta 2011, 1813, 1965–1970. [Google Scholar] [CrossRef]
- Kroh, A.; Ivanova, V.; Drescher, H.; Andruszkow, J.; Longerich, T.; Nolting, J.; Eickhoff, R.; Heise, D.; Rheinwalt, K.P.; Neumann, U.P.; et al. Mouse Models of Nonalcoholic Steatohepatitis: Head-to-Head Comparison of Dietary Models and Impact on Inflammation and Animal Welfare. Gastroenterol. Res. Pract. 2020, 2020, 7347068. [Google Scholar] [CrossRef]
- Takakura, K.; Oikawa, T.; Tomita, Y.; Mizuno, Y.; Nakano, M.; Saeki, C.; Torisu, Y.; Saruta, M. Mouse models for investigating the underlying mechanisms of nonalcoholic steatohepatitis-derived hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 1989–1994. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Hudson, W.A.; Li, Q.; Le, C.; Kersey, J.H. Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia 1998, 12, 2029–2033. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brennan, T.V.; Lin, L.; Huang, X.; Yang, Y. Generation of Luciferase-expressing Tumor Cell Lines. Bio. Protoc. 2018, 8, e2817. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef]
- Marisi, G.; Cucchetti, A.; Ulivi, P.; Canale, M.; Cabibbo, G.; Solaini, L.; Foschi, F.G.; De Matteis, S.; Ercolani, G.; Valgiusti, M.; et al. Ten years of sorafenib in hepatocellular carcinoma: Are there any predictive and/or prognostic markers? World J. Gastroenterol. 2018, 24, 4152–4163. [Google Scholar] [CrossRef]
- Cervello, M.; Bachvarov, D.; Lampiasi, N.; Cusimano, A.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 2012, 11, 2843–2855. [Google Scholar] [CrossRef]
- Buschauer, S.; Koch, A.; Wiggermann, P.; Muller, M.; Hellerbrand, C. Hepatocellular carcinoma cells surviving doxorubicin treatment exhibit increased migratory potential and resistance to doxorubicin re-treatment in vitro. Oncol. Lett. 2018, 15, 4635–4640. [Google Scholar] [CrossRef]
- Hanke, J.H.; Gardner, J.P.; Dow, R.L.; Changelian, P.S.; Brissette, W.H.; Weringer, E.J.; Pollok, B.A.; Connelly, P.A. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 1996, 271, 695–701. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, Y.; Hou, W.; Ma, L.; Tao, Y.; Li, D.; Xu, C.; Bao, J.; Fan, W. The JAK2/STAT3 pathway inhibitor, AG490, suppresses the abnormal behavior of keloid fibroblasts in vitro. Int. J. Mol. Med. 2020, 46, 191–200. [Google Scholar] [CrossRef]
- Guan, H.P.; Chen, G. Factors affecting insulin-regulated hepatic gene expression. Prog. Mol. Biol. Transl. Sci. 2014, 121, 165–215. [Google Scholar] [CrossRef]
- Taubert, H.; Heidenreich, C.; Holzhausen, H.J.; Schulz, A.; Bache, M.; Kappler, M.; Eckert, A.W.; Wurl, P.; Melcher, I.; Hauptmann, K.; et al. Expression of survivin detected by immunohistochemistry in the cytoplasm and in the nucleus is associated with prognosis of leiomyosarcoma and synovial sarcoma patients. BMC Cancer 2010, 10, 65. [Google Scholar] [CrossRef]
- Kawasaki, H.; Altieri, D.C.; Lu, C.D.; Toyoda, M.; Tenjo, T.; Tanigawa, N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998, 58, 5071–5074. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udoh, U.-A.S.; Banerjee, M.; Rajan, P.K.; Sanabria, J.D.; Smith, G.; Schade, M.; Sanabria, J.A.; Nakafuku, Y.; Sodhi, K.; Pierre, S.V.; et al. Tumor-Suppressor Role of the α1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 7359. https://doi.org/10.3390/ijms23137359
Udoh U-AS, Banerjee M, Rajan PK, Sanabria JD, Smith G, Schade M, Sanabria JA, Nakafuku Y, Sodhi K, Pierre SV, et al. Tumor-Suppressor Role of the α1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(13):7359. https://doi.org/10.3390/ijms23137359
Chicago/Turabian StyleUdoh, Utibe-Abasi S., Moumita Banerjee, Pradeep K. Rajan, Juan D. Sanabria, Gary Smith, Mathew Schade, Jacqueline A. Sanabria, Yuto Nakafuku, Komal Sodhi, Sandrine V. Pierre, and et al. 2022. "Tumor-Suppressor Role of the α1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 13: 7359. https://doi.org/10.3390/ijms23137359
APA StyleUdoh, U.-A. S., Banerjee, M., Rajan, P. K., Sanabria, J. D., Smith, G., Schade, M., Sanabria, J. A., Nakafuku, Y., Sodhi, K., Pierre, S. V., Shapiro, J. I., & Sanabria, J. R. (2022). Tumor-Suppressor Role of the α1-Na/K-ATPase Signalosome in NASH Related Hepatocellular Carcinoma. International Journal of Molecular Sciences, 23(13), 7359. https://doi.org/10.3390/ijms23137359