
Multifunctional Analysis of Chia Seed (Salvia hispanica L.) Bioactive Peptides Using Peptidomics and Molecular Dynamics Simulations Approaches




Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Preparation of Chia Seed Peptides
3.3. Peptidomics Analysis of Low Molecular Weight Peptides by Liquid Chromatography-Mass Spectrometry
3.4. In Silico Analysis of Identified Peptides for their Potential Bioactivities
3.5. Construction of Human Molecular Targets and Identified Peptides
3.6. Molecular Dynamics and Ensemble Docking Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vidal-Limon, A.; Aguilar-Toalá, J.E.; Liceaga, A.M. Integration of molecular docking analysis and molecular dynamics simulations for studying food proteins and bioactive peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; Cheng, S.; Lu, W.; Du, M. Advancement and prospects of bioinformatics analysis for studying bioactive peptides from food-derived protein: Sequence, structure, and functions. TrAC Trends Anal. Chem. 2018, 105, 7–17. [Google Scholar] [CrossRef]
- Agyei, D.; Tsopmo, A.; Udenigwe, C.C. Bioinformatics and peptidomics approaches to the discovery and analysis of food-derived bioactive peptides. Anal. Bioanal. Chem. 2018, 410, 3463–3472. [Google Scholar] [CrossRef] [PubMed]
- Zettel, V.; Hitzmann, B. Applications of chia (Salvia hispanica L.) in food products. Trends Food Sci. Technol. 2018, 80, 43–50. [Google Scholar] [CrossRef]
- Kulczyński, B.; Kobus-Cisowska, J.; Taczanowski, M.; Kmiecik, D.; Gramza-Michałowska, A. The chemical composition and ntritional value of Chia seeds-Current state of knowledge. Nutrients 2019, 11, 1242. [Google Scholar] [CrossRef] [Green Version]
- Melo, D.; Machado, T.B.; Oliveira, M.B.P.P. Chia seeds: An ancient grain trending in modern human diets. Food Funct. 2019, 10, 3068–3089. [Google Scholar] [CrossRef]
- Urbizo-Reyes, U.; San Martin-González, M.F.; Garcia-Bravo, J.; López Malo Vigil, A.; Liceaga, A.M. Physicochemical characteristics of chia seed (Salvia hispanica) protein hydrolysates produced using ultrasonication followed by microwave-assisted hydrolysis. Food Hydrocoll. 2019, 97, 105187. [Google Scholar] [CrossRef]
- Aguilar-Toalá, J.E.; Deering, A.J.; Liceaga, A.M. New insights into the antimicrobial properties of hydrolysates and peptide fractions derived from Chia seed (Salvia hispanica L.). Probiotics Antimicrob. Proteins 2020, 12, 1571–1581. [Google Scholar] [CrossRef]
- Aguilar-Toalá, J.E.; Liceaga, A.M. Identification of chia seed (Salvia hispanica L.) peptides with enzyme inhibition activity towards skin-aging enzymes. Amino Acids 2020, 52, 1149–1159. [Google Scholar] [CrossRef]
- Cotabarren, J.; Rosso, A.M.; Tellechea, M.; García-Pardo, J.; Rivera, J.L.; Obregón, W.D.; Parisi, M.G. Adding value to the chia (Salvia hispanica L.) expeller: Production of bioactive peptides with antioxidant properties by enzymatic hydrolysis with Papain. Food Chem. 2019, 274, 848–856. [Google Scholar] [CrossRef]
- Coelho, M.S.; Soares-Freitas, R.A.M.; Arêas, J.A.G.; Gandra, E.A.; Salas-Mellado, M.d.l.M. Peptides from Chia present atibacterial ativity and inhibit colesterol synthesis. Plant Foods Hum. Nutr. 2018, 73, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Grancieri, M.; Martino, H.S.D.; Gonzalez de Mejia, E. Digested total protein and protein fractions from chia seed (Salvia hispanica L.) had high scavenging capacity and inhibited 5-LOX, COX-1-2, and iNOS enzymes. Food Chem. 2019, 289, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yang, C.; Xie, Y.; Wang, Y.; Li, X.; Wang, K.; Huang, J.; Yan, W. GM-Pep: A high efficiency strategy to de novo design functional peptide sequences. J. Chem. Inf. Model. 2022, 62, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Panyayai, T.; Ngamphiw, C.; Tongsima, S.; Mhuantong, W.; Limsripraphan, W.; Choowongkomon, K.; Sawatdichaikul, O. FeptideDB: A web application for new bioactive peptides from food protein. Heliyon 2019, 5, e02076. [Google Scholar] [CrossRef] [Green Version]
- Kartal, C.; Kaplan Türköz, B.; Otles, S. Prediction, identification and evaluation of bioactive peptides from tomato seed proteins using in silico approach. J. Food Meas. Charact. 2020, 14, 1865–1883. [Google Scholar] [CrossRef]
- Coscueta, E.R.; Batista, P.; Gomes, J.E.; da Silva, R.; Pintado, M.M. Screening of novel bioactive peptides from goat casein: In silico to in vitro validation. Int. J. Mol. Sci. 2022, 23, 2439. [Google Scholar] [CrossRef]
- Xiao, C.; Zhao, M.; Zhou, F.; Gallego, M.; Toldrá, F.; Mora, L. Data on bioactive peptides derived from chicken hydrolysate with potential alcohol dehydrogenase stabilizing activity and in silico analysis of their potential activity and applicability. Data Brief 2020, 29, 105163. [Google Scholar] [CrossRef]
- Kitts, D.D.; Weiler, K. Bioactive proteins and peptides from food sources. Applications of bioprocesses used in isolation and recovery. Curr. Pharm. Des. 2003, 9, 1309–1323. [Google Scholar] [CrossRef]
- Udenigwe, C.C.; Aluko, R.E. Food protein-derived bioactive peptides: Production, processing, and potential health benefits. J. Food Sci. 2012, 77, R11–R24. [Google Scholar] [CrossRef]
- Pripp, A.H.; Isaksson, T.; Stepaniak, L.; Sørhaug, T.; Ardö, Y. Quantitative structure activity relationship modelling of peptides and proteins as a tool in food science. Trends Food Sci. Technol. 2005, 16, 484–494. [Google Scholar] [CrossRef]
- Stockner, T.; Sterk, H.; Kaptein, R.; Bonvin, A.M.J.J. Molecular dynamics studies of a molecular switch in the glucocorticoid receptor. J. Mol. Biol. 2003, 328, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Khandelwal, R.; Mudgal, U.; Srinitha, S.; Khandekar, N.; Nayarisseri, A.; Vuree, S.; Singh, S.K. Identification of potent VEGF inhibitors for the clinical treatment of glioblastoma, a virtual screening approach. Asian Pac. J. Cancer Prev. 2019, 20, 2681–2692. [Google Scholar] [CrossRef] [PubMed]
- Kamal, H.; Mudgil, P.; Bhaskar, B.; Fisayo, A.F.; Gan, C.-Y.; Maqsood, S. Amaranth proteins as potential source of bioactive peptides with enhanced inhibition of enzymatic markers linked with hypertension and diabetes. J. Cereal Sci. 2021, 101, 103308. [Google Scholar] [CrossRef]
- Mudgil, P.; Baby, B.; Ngoh, Y.-Y.; Kamal, H.; Vijayan, R.; Gan, C.-Y.; Maqsood, S. Molecular binding mechanism and identification of novel anti-hypertensive and anti-inflammatory bioactive peptides from camel milk protein hydrolysates. LWT Food Sci. Technol. 2019, 112, 108193. [Google Scholar] [CrossRef]
- Amorim, F.G.; Coitinho, L.B.; Dias, A.T.; Friques, A.G.F.; Monteiro, B.L.; Rezende, L.C.D.d.; Pereira, T.d.M.C.; Campagnaro, B.P.; De Pauw, E.; Vasquez, E.C.; et al. Identification of new bioactive peptides from Kefir milk through proteopeptidomics: Bioprospection of antihypertensive molecules. Food Chem. 2019, 282, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Saldívar-González, F.I. Cheminformatics to characterize pharmacologically active natural products. Biomolecules 2020, 10, 1566. [Google Scholar] [CrossRef]
- Miao, Y.; McCammon, J.A. Chapter Six—Gaussian Accelerated Molecular Dynamics: Theory, Implementation, and Applications. In Annual Reports in Computational Chemistry; Dixon, D.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 13, pp. 231–278. [Google Scholar]
- Miao, Y.; Bhattarai, A.; Wang, J. Ligand Gaussian accelerated molecular dynamics (LiGaMD): Characterization of ligand binding thermodynamics and kinetics. J. Chem. Theory Comput. 2020, 16, 5526–5547. [Google Scholar] [CrossRef]
- Edman, K.; Hosseini, A.; Bjursell, M.K.; Aagaard, A.; Wissler, L.; Gunnarsson, A.; Kaminski, T.; Köhler, C.; Bäckström, S.; Jensen, T.J.; et al. Ligand binding mechanism in steroid receptors: From conserved plasticity to differential evolutionary constraints. Structure 2015, 23, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett Kizzmekia, S.; Goldsmith Jory, A.; Hsieh, C.-L.; Abiona, O.; Graham Barney, S.; McLellan Jason, S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger. QikProp v 2022-2; LLC: New York, NY, USA, 2021. [Google Scholar]
- Chang, Y.-C.; Chen, C.-P.; Chen, C.-C. Predicting skin permeability of chemical substances using a quantitative structure-activity relationship. Procedia Eng. 2012, 45, 875–879. [Google Scholar] [CrossRef]
- Zeng, R.; Deng, J.; Dang, L.; Yu, X. Correlation between the structure and skin permeability of compounds. Sci. Rep. 2021, 11, 10076. [Google Scholar] [CrossRef] [PubMed]
- Mudgil, P.; Kamal, H.; Priya Kilari, B.; Mohd Salim, M.A.S.; Gan, C.-Y.; Maqsood, S. Simulated gastrointestinal digestion of camel and bovine casein hydrolysates: Identification and characterization of novel anti-diabetic bioactive peptides. Food Chem. 2021, 353, 129374. [Google Scholar] [CrossRef] [PubMed]
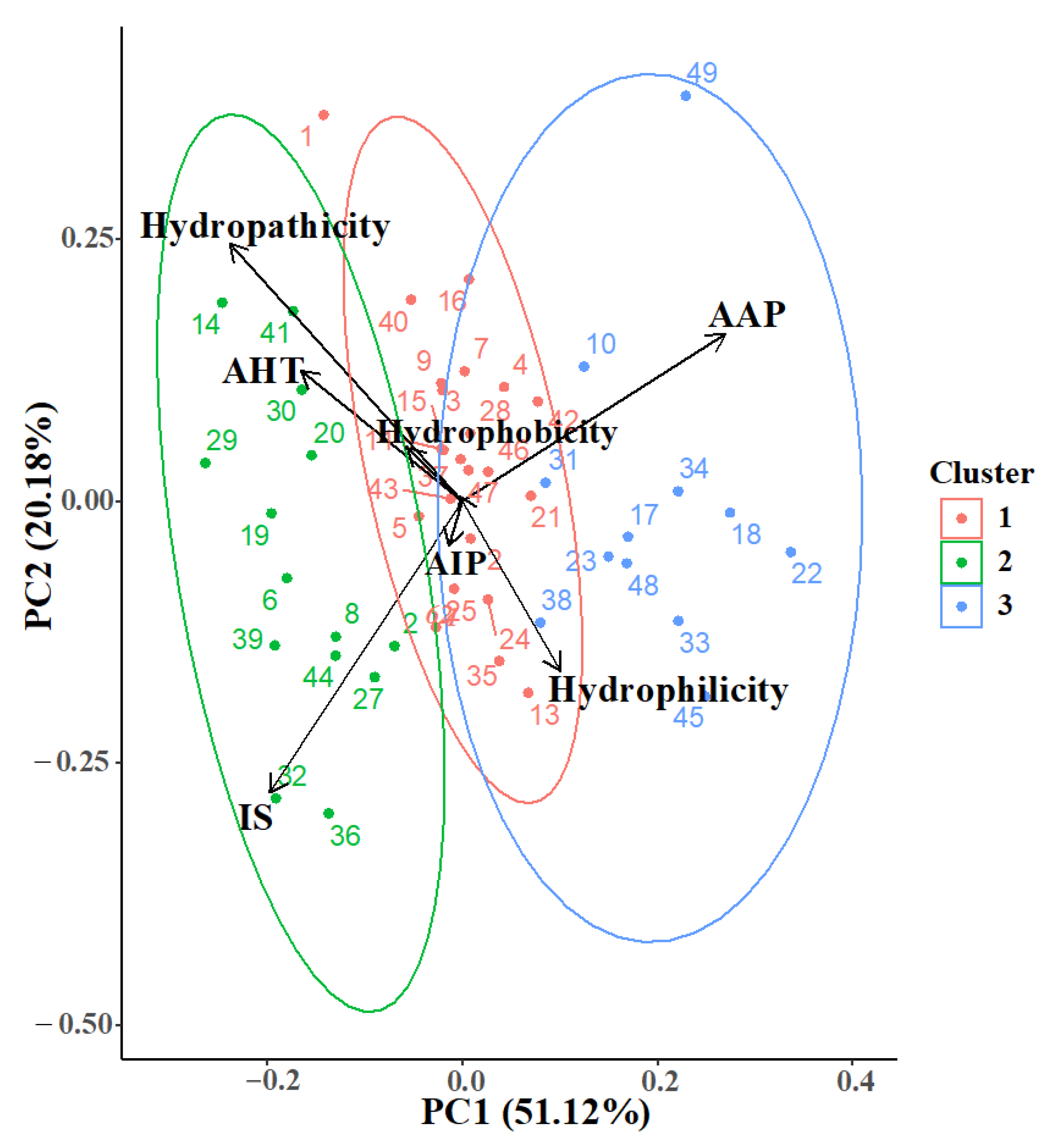
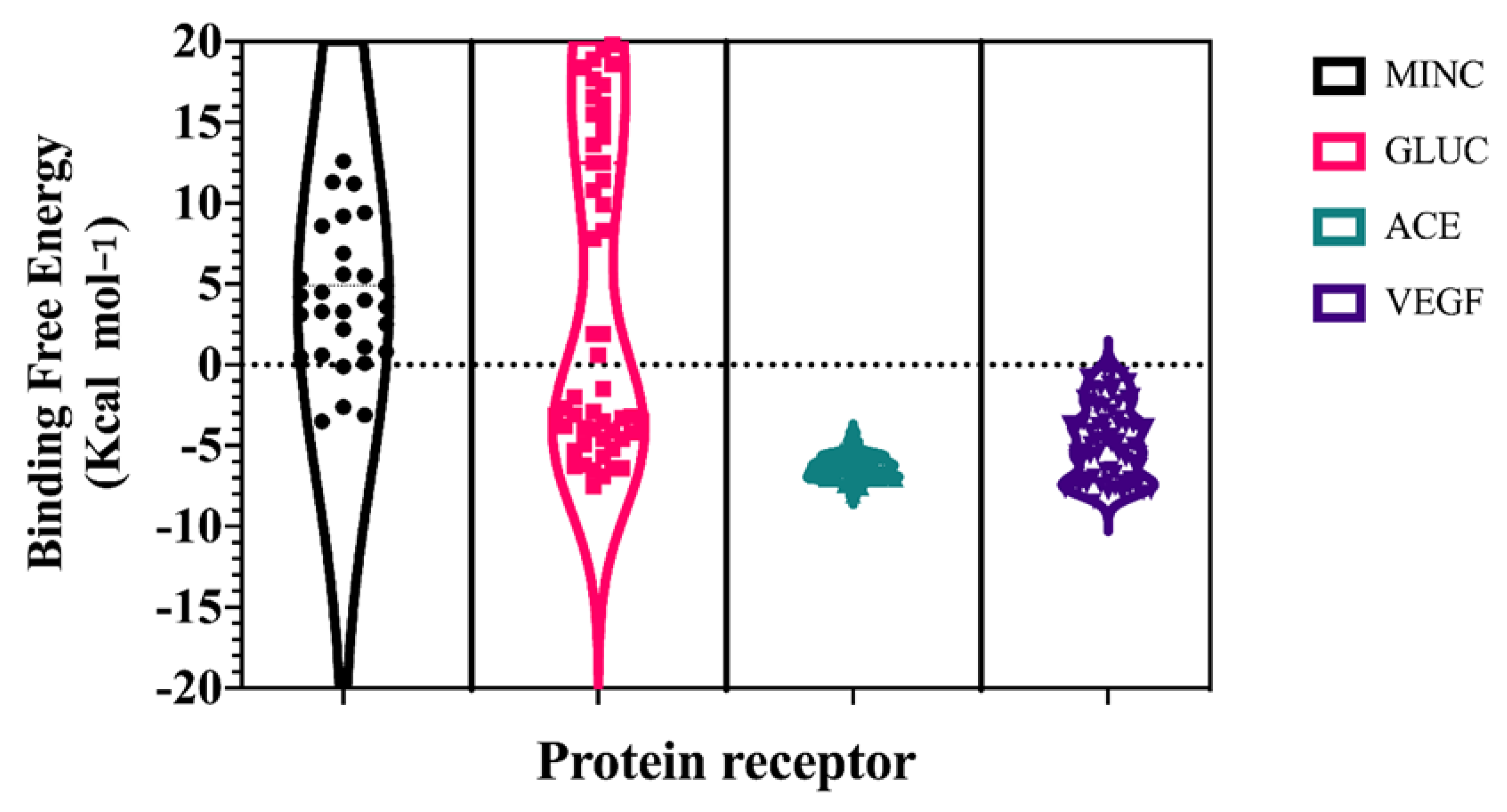
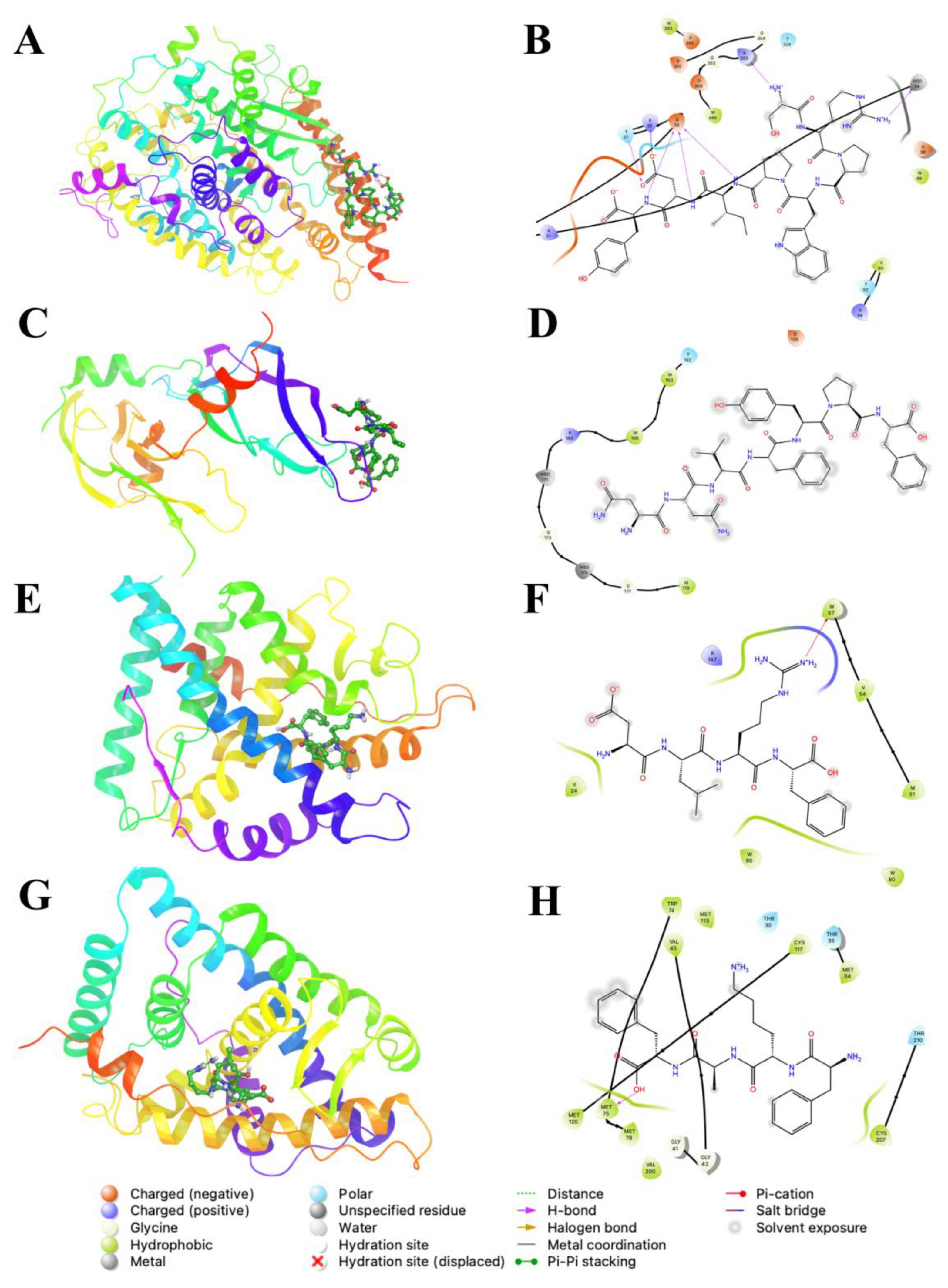


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Peptide Sequence | Peptide Ranker Score | PreAIP | AntiAngio-Pred | AHTpin | |||
|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | Score | Prediction | |||
| Database peptides | ||||||||
| 1 | FNLVFFLL | 0.951 | 0.416 | AIP | −0.21 | Non-AAP | −0.58 | Non-AHT |
| 2 | EGDVFWIPRF | 0.940 | 0.418 | AIP | −0.5 | Non-AAP | −0.12 | Non-AHT |
| 3 | DHFPFIY | 0.933 | 0.518 | AIP | 0.04 | AAP | 0.47 | AHT |
| 4 | EGGIWPF | 0.929 | 0.344 | AIP | 0.48 | AAP | −0.1 | Non-AHT |
| 5 | GFEWITF | 0.922 | 0.57 | AIP | 0.47 | AAP | −0.89 | Non-AHT |
| 6 | GLDFPELPLGM | 0.919 | 0.481 | AIP | −0.61 | Non-AAP | 1.31 | AHT |
| 7 | GQTPLFPRIF | 0.912 | 0.412 | AIP | 0.41 | AAP | 0.65 | AHT |
| 8 | GDAHYDPLFPF | 0.909 | 0.323 | Non-AIP | −1.23 | Non-AAP | 0.95 | AHT |
| 9 | NNVFYPF | 0.903 | 0.344 | AIP | −0.34 | Non-AAP | 0.22 | AHT |
| 10 | EYPPLGRF | 0.901 | 0.395 | AIP | 1.01 | AAP | 1.04 | AHT |
| 11 | KPLPFELF | 0.898 | 0.409 | AIP | 0.6 | AAP | 0.55 | AHT |
| 12 | DVWDPFQDFPL | 0.895 | 0.461 | AIP | 0.11 | AAP | 0.41 | AHT |
| 13 | SDKNGYFF | 0.883 | 0.418 | AIP | −0.82 | Non-AAP | −1.16 | Non-AHT |
| 14 | VPIPVPLPF | 0.883 | 0.318 | Non-AIP | −0.24 | Non-AAP | 2.29 | AHT |
| 15 | SNVFDPF | 0.876 | 0.338 | Non-AIP | −0.87 | Non-AAP | −0.06 | Non-AHT |
| 16 | TPLFPRIF | 0.876 | 0.393 | AIP | 1.03 | AAP | 0.58 | AHT |
| 17 | DQNPRSFFL | 0.873 | 0.444 | AIP | 1 | AAP | −0.67 | Non-AHT |
| 18 | QLQRWFR | 0.871 | 0.519 | AIP | 2.22 | AAP | −0.62 | Non-AHT |
| 19 | GFEWVAF | 0.868 | 0.59 | AIP | −0.94 | Non-AAP | 0.3 | AHT |
| 20 | SFNLPIL | 0.867 | 0.408 | AIP | −0.4 | Non-AAP | −0.15 | Non-AHT |
| 21 | QEGGIWPF | 0.863 | 0.37 | AIP | 0.39 | AAP | −0.52 | Non-AHT |
| 22 | GSRFDWTR | 0.858 | 0.488 | AIP | 2.17 | AAP | −1.36 | Non-AHT |
| 23 | ADFYNPR | 0.853 | 0.303 | Non-AIP | 0.92 | AAP | 0.44 | AHT |
| 24 | APSKDAPMF | 0.851 | 0.452 | AIP | −0.16 | Non-AAP | −0.24 | Non-AHT |
| 25 | GFEWITFK | 0.847 | 0.578 | AIP | 0.28 | AAP | −0.66 | Non-AHT |
| 26 | NGFEWITF | 0.842 | 0.549 | AIP | 0.4 | AAP | −1.03 | Non-AHT |
| 27 | VNEGDVFWIPRF | 0.841 | 0.414 | AIP | −1.1 | Non-AAP | −0.52 | Non-AHT |
| 28 | SSNVFDPF | 0.841 | 0.304 | Non-AIP | −0.93 | Non-AAP | −0.17 | Non-AHT |
| 29 | FNIVFPG | 0.839 | 0.385 | AIP | −1.68 | Non-AAP | 0.76 | AHT |
| 30 | VPVFPPPLN | 0.837 | 0.435 | Non-AIP | −0.26 | Non-AAP | 2 | AHT |
| 31 | GIDIPPPR | 0.835 | 0.316 | Non-AIP | 0.55 | AAP | 0.47 | AHT |
| 32 | APAEKGFAGF | 0.832 | 0.402 | AIP | −1.39 | Non-AAP | 0.23 | AHT |
| 33 | DQNPRSFF | 0.830 | 0.433 | AIP | 1.05 | AAP | −0.92 | Non-AHT |
| 34 | SRPWPIDY | 0.827 | 0.486 | AIP | 2.26 | AAP | −0.04 | Non-AHT |
| 35 | QNGFEWITF | 0.825 | 0.573 | AIP | 0.46 | AAP | −1.47 | Non-AHT |
| 36 | RPGDVFVFPR | 0.825 | 0.383 | AIP | −0.98 | Non-AAP | 0.22 | AHT |
| 37 | DNGIIYPW | 0.823 | 0.32 | Non-AIP | −0.36 | Non-AAP | 0.15 | AHT |
| 38 | NPQAGRF | 0.822 | 0.376 | AIP | −0.43 | Non-AAP | 0.03 | AHT |
| 39 | APVGSPVGSTGGNFGVF | 0.817 | 0.476 | AIP | −1.1 | Non-AAP | 0.39 | AHT |
| 40 | APPPVLAL | 0.816 | 0.396 | AIP | 0.61 | AAP | 0.07 | AHT |
| 41 | FPLLNYL | 0.813 | 0.554 | AIP | −0.18 | Non-AAP | 1.31 | AHT |
| 42 | RNNVFYPF | 0.811 | 0.378 | AIP | 0.61 | AAP | 0.49 | AHT |
| 43 | GNIFRGL | 0.811 | 0.452 | AIP | −0.34 | Non-AAP | −0.6 | Non-AHT |
| 44 | FPGLADRM | 0.810 | 0.333 | Non-AIP | −1.01 | Non-AAP | 0.67 | AHT |
| 45 | SNEWDPSFR | 0.806 | 0.393 | AIP | 0.81 | AAP | −1.34 | Non-AHT |
| 46 | SMLSPHW | 0.806 | 0.42 | AIP | 0.22 | AAP | −0.24 | Non-AHT |
| 47 | SLDVWDPFQDFPL | 0.804 | 0.464 | AIP | 0.44 | AAP | 0.59 | AHT |
| 48 | SPDLIRRM | 0.803 | 0.399 | AIP | 1.12 | AAP | −0.74 | Non-AHT |
| 49 | FGNVFKGM | 0.803 | 0.32 | Non-AIP | −1.68 | Non-AAP | −0.8 | Non-AHT |
| De novo peptides | ||||||||
| 1 | QFRF | 0.980 | 0.361 | AIP | ND | - | −0.05 | Non-AHT |
| 2 | FDRF | 0.978 | 0.301 | Non-AIP | ND | - | −0.79 | Non-AHT |
| 3 | GRPW | 0.971 | 0.266 | Non-AIP | ND | - | 0.84 | AHT |
| 4 | FWDR | 0.964 | 0.347 | AIP | ND | - | −0.74 | Non-AHT |
| 5 | FRSF | 0.962 | 0.339 | Non-AIP | ND | - | −0.76 | Non-AHT |
| 6 | GPHW | 0.958 | 0.354 | AIP | ND | - | 1 | AHT |
| 7 | KPPF | 0.955 | 0.294 | Non-AIP | ND | - | 2.09 | AHT |
| 8 | WLPR | 0.943 | 0.306 | Non-AIP | ND | - | 1.13 | AHT |
| 9 | FWDH | 0.938 | 0.318 | Non-AIP | ND | - | −0.61 | Non-AHT |
| 10 | FDKF | 0.937 | 0.32 | Non-AIP | ND | - | −0.87 | Non-AHT |
| 11 | FRGL | 0.937 | 0.318 | Non-AIP | ND | - | −0.38 | Non-AHT |
| 12 | DFKF | 0.932 | 0.336 | Non-AIP | ND | - | −0.87 | Non-AHT |
| 13 | KDFLFP | 0.929 | 0.352 | AIP | −0.03 | Non-AAP | −0.79 | Non-AHT |
| 14 | EFRF | 0.922 | 0.289 | Non-AIP | ND | - | 0.99 | AHT |
| 15 | APHW | 0.918 | 0.389 | AIP | ND | - | 0.03 | AHT |
| 16 | RPAF | 0.909 | 0.28 | Non-AIP | ND | - | 0.89 | AHT |
| 17 | ARGW | 0.908 | 0.317 | Non-AIP | ND | - | −0.76 | Non-AHT |
| 18 | FKAF | 0.907 | 0.31 | Non-AIP | ND | - | −0.67 | Non-AHT |
| 19 | WEFLTF | 0.907 | 0.329 | Non-AIP | 0.43 | AAP | −0.89 | Non-AHT |
| 20 | HVFF | 0.878 | 0.349 | AIP | ND | - | −0.21 | Non-AHT |
| 21 | WAPH | 0.873 | 0.313 | Non-AIP | ND | - | 0.99 | AHT |
| 22 | RPSF | 0.872 | 0.334 | Non-AIP | ND | - | 0.7 | AHT |
| 23 | HPAYW | 0.871 | 0.392 | AIP | 0.1 | AAP | 1.81 | AHT |
| 24 | DLRF | 0.863 | 0.319 | Non-AIP | ND | - | −0.56 | Non-AHT |
| 25 | QLRF | 0.863 | 0.344 | AIP | ND | - | 0.21 | AHT |
| 26 | GKFL | 0.850 | 0.316 | Non-AIP | ND | - | −0.48 | Non-AHT |
| 27 | QRYF | 0.848 | 0.327 | Non-AIP | ND | - | 0.89 | AHT |
| 28 | FWDNH | 0.834 | 0.345 | AIP | −0.63 | Non-AAP | 0.05 | AHT |
| 29 | FPLK | 0.834 | 0.336 | Non-AIP | ND | - | 1.07 | AHT |
| 30 | RAFL | 0.831 | 0.349 | AIP | ND | - | −0.42 | Non-AHT |
| 31 | FPLLN | 0.820 | 0.483 | AIP | −0.1 | Non-AAP | 0.22 | AHT |
| 32 | WDPSYR | 0.816 | 0.325 | Non-AIP | 2.67 | AAP | 0.11 | AHT |
| 33 | GLKF | 0.810 | 0.348 | AIP | ND | - | −0.48 | Non-AHT |
| 34 | HPNPRL | 0.808 | 0.358 | AIP | 0.51 | AAP | 0.98 | AHT |
| No. | Peptide Sequence | Intestinal Stability | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Molecular Weight |
|---|---|---|---|---|---|---|---|
| Database peptides | |||||||
| 1 | FNLVFFLL | 0.73 | 0.42 | 2.56 | −1.78 | 0 | 1012.35 |
| 2 | EGDVFWIPRF | 2.391 | −0.02 | −0.01 | −0.27 | −1 | 1265.57 |
| 3 | DHFPFIY | 1.094 | 0.11 | 0.07 | −0.94 | −1 | 938.13 |
| 4 | EGGIWPF | 0.945 | 0.19 | 0.07 | −0.67 | −1 | 805 |
| 5 | GFEWITF | 2.512 | 0.24 | 0.66 | −1.09 | −1 | 899.11 |
| 6 | GLDFPELPLGM | 2.79 | 0.12 | 0.46 | −0.29 | −2 | 1188.54 |
| 7 | GQTPLFPRIF | 1.079 | −0.01 | 0.16 | −0.58 | 1 | 1175.52 |
| 8 | GDAHYDPLFPF | 2.144 | 0.02 | −0.35 | −0.37 | −2 | 1278.52 |
| 9 | NNVFYPF | 0.798 | 0.06 | −0.01 | −1.2 | 0 | 900.09 |
| 10 | EYPPLGRF | 0.392 | −0.15 | −0.79 | −0.07 | 0 | 978.21 |
| 11 | KPLPFELF | 1.859 | 0.05 | 0.32 | −0.33 | 0 | 990.3 |
| 12 | DVWDPFQDFPL | 1.608 | −0.03 | −0.41 | −0.23 | −3 | 1378.65 |
| 13 | SDKNGYFF | 1.183 | −0.17 | −0.98 | −0.1 | 0 | 977.14 |
| 14 | VPIPVPLPF | 2.283 | 0.3 | 1.46 | −1.01 | 0 | 978.35 |
| 15 | SNVFDPF | 0.585 | 0.01 | 0.06 | −0.43 | −1 | 824.97 |
| 16 | TPLFPRIF | 1.143 | 0.05 | 0.69 | −0.75 | 1 | 990.3 |
| 17 | DQNPRSFFL | 1.023 | −0.27 | −0.89 | −0.01 | 0 | 1123.34 |
| 18 | QLQRWFR | 1.066 | −0.48 | −1.47 | −0.19 | 2 | 1033.3 |
| 19 | GFEWVAF | 2.466 | 0.27 | 0.97 | −1.06 | −1 | 855.06 |
| 20 | SFNLPIL | 2.342 | 0.2 | 1.29 | −1.06 | 0 | 803.04 |
| 21 | QEGGIWPF | 1.213 | 0.08 | −0.38 | −0.56 | −1 | 933.15 |
| 22 | GSRFDWTR | 0.667 | −0.44 | −1.56 | 0.38 | 1 | 1024.21 |
| 23 | ADFYNPR | 1.132 | −0.33 | −1.4 | 0.13 | 0 | 882.02 |
| 24 | APSKDAPMF | 1.538 | −0.09 | −0.34 | 0.17 | 0 | 963.22 |
| 25 | GFEWITFK | 2.682 | 0.07 | 0.09 | −0.58 | 0 | 1027.3 |
| 26 | NGFEWITF | 2.499 | 0.13 | 0.14 | −0.93 | −1 | 1013.23 |
| 27 | VNEGDVFWIPRF | 2.24 | −0.02 | 0.05 | −0.33 | −1 | 1478.84 |
| 28 | SSNVFDPF | 0.2 | −0.02 | −0.05 | −0.34 | −1 | 912.06 |
| 29 | FNIVFPG | 2.136 | 0.28 | 1.26 | −1.16 | 0 | 793.02 |
| 30 | VPVFPPPLN | 1.974 | 0.14 | 0.57 | −0.79 | 0 | 979.3 |
| 31 | GIDIPPPR | 0.964 | −0.13 | −0.53 | 0.3 | 0 | 864.1 |
| 32 | APAEKGFAGF | 3.43 | 0.05 | 0.12 | −0.05 | 0 | 994.24 |
| 33 | DQNPRSFF | 1.044 | −0.36 | −1.48 | 0.21 | 0 | 1010.17 |
| 34 | SRPWPIDY | 1.413 | −0.22 | −1.21 | −0.15 | 0 | 1033.25 |
| 35 | QNGFEWITF | 2.539 | 0.04 | −0.27 | −0.8 | −1 | 1141.38 |
| 36 | RPGDVFVFPR | 3.379 | −0.19 | −0.21 | 0.1 | 1 | 1189.51 |
| 37 | DNGIIYPW | 0.959 | 0.07 | −0.28 | −0.76 | −1 | 977.19 |
| 38 | NPQAGRF | 0.929 | −0.31 | −1.27 | 0.06 | 1 | 788.95 |
| 39 | APVGSPVGSTGGNFGVF | 2.935 | 0.14 | 0.53 | −0.56 | 0 | 1549.95 |
| 40 | APPPVLAL | 1.457 | 0.24 | 1.32 | −0.76 | 0 | 777.06 |
| 41 | FPLLNYL | 1.841 | 0.22 | 1.11 | −1.43 | 0 | 879.14 |
| 42 | RNNVFYPF | 0.769 | −0.17 | −0.58 | −0.67 | 1 | 1056.29 |
| 43 | GNIFRGL | 1.294 | −0.03 | 0.33 | −0.41 | 1 | 775.99 |
| 44 | FPGLADRM | 2.43 | −0.09 | 0.04 | −0.01 | 0 | 906.16 |
| 45 | SNEWDPSFR | 0.992 | −0.37 | −1.81 | 0.43 | −1 | 1137.29 |
| 46 | SMLSPHW | 1.339 | 0.02 | −0.23 | −0.91 | 0 | 857.09 |
| 47 | SLDVWDPFQDFPL | 1.558 | 0 | −0.12 | −0.31 | −3 | 1578.91 |
| 48 | SPDLIRRM | 1.23 | −0.38 | −0.59 | 0.55 | 1 | 987.27 |
| 49 | FGNVFKGM | 1.854 | 0.07 | 0.44 | −0.57 | 1 | 899.19 |
| De novo peptides | |||||||
| 1 | QFRF | ND | −0.31 | −0.6 | −0.45 | 1 | 596.73 |
| 2 | FDRF | ND | −0.32 | −0.6 | 0.25 | 0 | 583.68 |
| 3 | GRPW | ND | −0.33 | −1.85 | −0.1 | 1 | 514.64 |
| 4 | FWDR | ND | −0.38 | −1.52 | 0.02 | 0 | 622.73 |
| 5 | FRSF | ND | −0.2 | 0.07 | −0.42 | 1 | 555.67 |
| 6 | GPHW | ND | 0.01 | −1.53 | −0.97 | 0 | 495.6 |
| 7 | KPPF | ND | −0.16 | −1.07 | 0.12 | 1 | 487.64 |
| 8 | WLPR | ND | −0.23 | −0.8 | −0.55 | 1 | 570.74 |
| 9 | FWDH | ND | −0.04 | −1.2 | −0.85 | −1 | 603.69 |
| 10 | FDKF | ND | −0.15 | −0.45 | 0.25 | 0 | 555.67 |
| 11 | FRGL | ND | −0.11 | 0.42 | −0.33 | 1 | 491.63 |
| 12 | DFKF | ND | −0.15 | −0.45 | 0.25 | 0 | 555.67 |
| 13 | KDFLFP | 1.208 | −0.29 | −0.6 | 0.25 | 0 | 597.71 |
| 14 | EFRF | ND | 0.04 | −0.97 | −1.1 | 0 | 509.62 |
| 15 | APHW | ND | −0.02 | 0.07 | −0.13 | 0 | 765.97 |
| 16 | RPAF | ND | −0.24 | −0.38 | 0 | 1 | 489.61 |
| 17 | ARGW | ND | −0.25 | −1 | −0.22 | 1 | 488.6 |
| 18 | FKAF | ND | 0.09 | 0.88 | −0.62 | 1 | 511.66 |
| 19 | WEFLTF | 1.16 | 0.22 | 0.72 | −1.27 | −1 | 842.04 |
| 20 | HVFF | ND | 0.34 | 1.65 | −1.75 | 0 | 548.69 |
| 21 | WAPH | ND | 0.04 | −0.98 | −1.1 | 0 | 509.62 |
| 22 | RPSF | ND | −0.37 | −1.02 | 0.2 | 1 | 505.61 |
| 23 | HPAYW | 1.398 | 0.03 | −1.04 | −1.34 | 0 | 672.81 |
| 24 | DLRF | ND | −0.33 | −0.35 | 0.43 | 0 | 549.66 |
| 25 | QLRF | ND | −0.33 | −0.35 | −0.28 | 1 | 562.71 |
| 26 | GKFL | ND | 0.05 | 0.57 | −0.33 | 1 | 463.62 |
| 27 | QRYF | ND | −0.46 | −1.63 | −0.4 | 1 | 612.73 |
| 28 | FWDNH | 1.425 | −0.16 | −1.66 | −0.64 | −1 | 717.81 |
| 29 | FPLK | ND | −0.01 | 0.28 | −0.32 | 1 | 503.68 |
| 30 | RAFL | ND | −0.09 | 0.97 | −0.45 | 1 | 505.65 |
| 31 | FPLLN | 1.991 | 0.19 | 1.06 | −1.18 | 0 | 602.78 |
| 32 | WDPSYR | 1.486 | −0.4 | −2.1 | 0.1 | 0 | 822.95 |
| 33 | GLKF | ND | 0.05 | 0.57 | −0.33 | 1 | 463.62 |
| 34 | HPNPRL | 1.534 | −0.4 | −1.77 | 0.15 | 1 | 732.91 |
| Parameters | NNVFYPF | FNIVFPG | SRPWPIDY | QLQRWFR | GSRFDWTR | DFKF | DLRF | FKAF | FRSF | QFRF |
|---|---|---|---|---|---|---|---|---|---|---|
| MW (g/mol) | 899.9 | 792.93 | 899.07 | 10.33.19 | 1024.1 | 555.63 | 549.62 | 511.62 | 555.63 | 596.68 |
| H-bond donors | 8 | 6 | 8.5 | 17.5 | 14.5 | 5.75 | 7.75 | 5.75 | 7.75 | 9.75 |
| H-bond acceptors | 19.25 | 17 | 19 | 22.5 | 22.4 | 10.25 | 11.25 | 9.25 | 10.95 | 12.75 |
| logP o/w a | −3.56 | −1.83 | −2.79 | −5.68 | −4.64 | −1.06 | −1.83 | −1.43 | −1.28 | −3.14 |
| logS wat b | −1.87 | −3.77 | −0.49 | −1.18 | −0.24 | −0.68 | −0.64 | −0.79 | −2.72 | −0.96 |
| NlogK has Serum Protein Binding c | −2.39 | −1.88 | −2.53 | −3.35 | −3.18 | −1.23 | −1.54 | −1.02 | −1.26 | -1.75 |
| Apparent Caco-2 Permeability (nm/s) d | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Apparent MDCK Permeability (nm/s) e | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| logKp for skin permeability f | −9.04 | −6.92 | −8.53 | −14.29 | −13.53 | −8.69 | −9.95 | −8.26 | −8.62 | −10.60 |
| Qualitative Model for Human Oral Absorption | Low | Low | Low | Low | Low | Low | Low | Low | Low | Low |
| Most similar pharmaceutical drugs | Troxerutin, Voglibose, Monoxerutin | Lymecycline, Troxerutin, Proglumetacin | Razoxane, Hexoprenaline, Dihydralazine | Everolimus, Amiodarone, Fenethylline | Everolimus, Droxidopa, Polaprezinc | Hexoprenaline, Lisinopril, Lymecycline | Hexoprenaline, Voglibose, Lymecycline | Hexoprenaline, Lisinopril, Lymecycline | Aminopterin, Lymecycline, Hexobendine | Hexobendine, Hexoprenaline, Lymecycline |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Toalá, J.E.; Vidal-Limon, A.; Liceaga, A.M. Multifunctional Analysis of Chia Seed (Salvia hispanica L.) Bioactive Peptides Using Peptidomics and Molecular Dynamics Simulations Approaches. Int. J. Mol. Sci. 2022, 23, 7288. https://doi.org/10.3390/ijms23137288
Aguilar-Toalá JE, Vidal-Limon A, Liceaga AM. Multifunctional Analysis of Chia Seed (Salvia hispanica L.) Bioactive Peptides Using Peptidomics and Molecular Dynamics Simulations Approaches. International Journal of Molecular Sciences. 2022; 23(13):7288. https://doi.org/10.3390/ijms23137288
Chicago/Turabian StyleAguilar-Toalá, José E., Abraham Vidal-Limon, and Andrea M. Liceaga. 2022. "Multifunctional Analysis of Chia Seed (Salvia hispanica L.) Bioactive Peptides Using Peptidomics and Molecular Dynamics Simulations Approaches" International Journal of Molecular Sciences 23, no. 13: 7288. https://doi.org/10.3390/ijms23137288
APA StyleAguilar-Toalá, J. E., Vidal-Limon, A., & Liceaga, A. M. (2022). Multifunctional Analysis of Chia Seed (Salvia hispanica L.) Bioactive Peptides Using Peptidomics and Molecular Dynamics Simulations Approaches. International Journal of Molecular Sciences, 23(13), 7288. https://doi.org/10.3390/ijms23137288