
Synthesis and Biological Evaluation of Small Molecules as Potential Anticancer Multitarget Agents

 , and
, and
Abstract
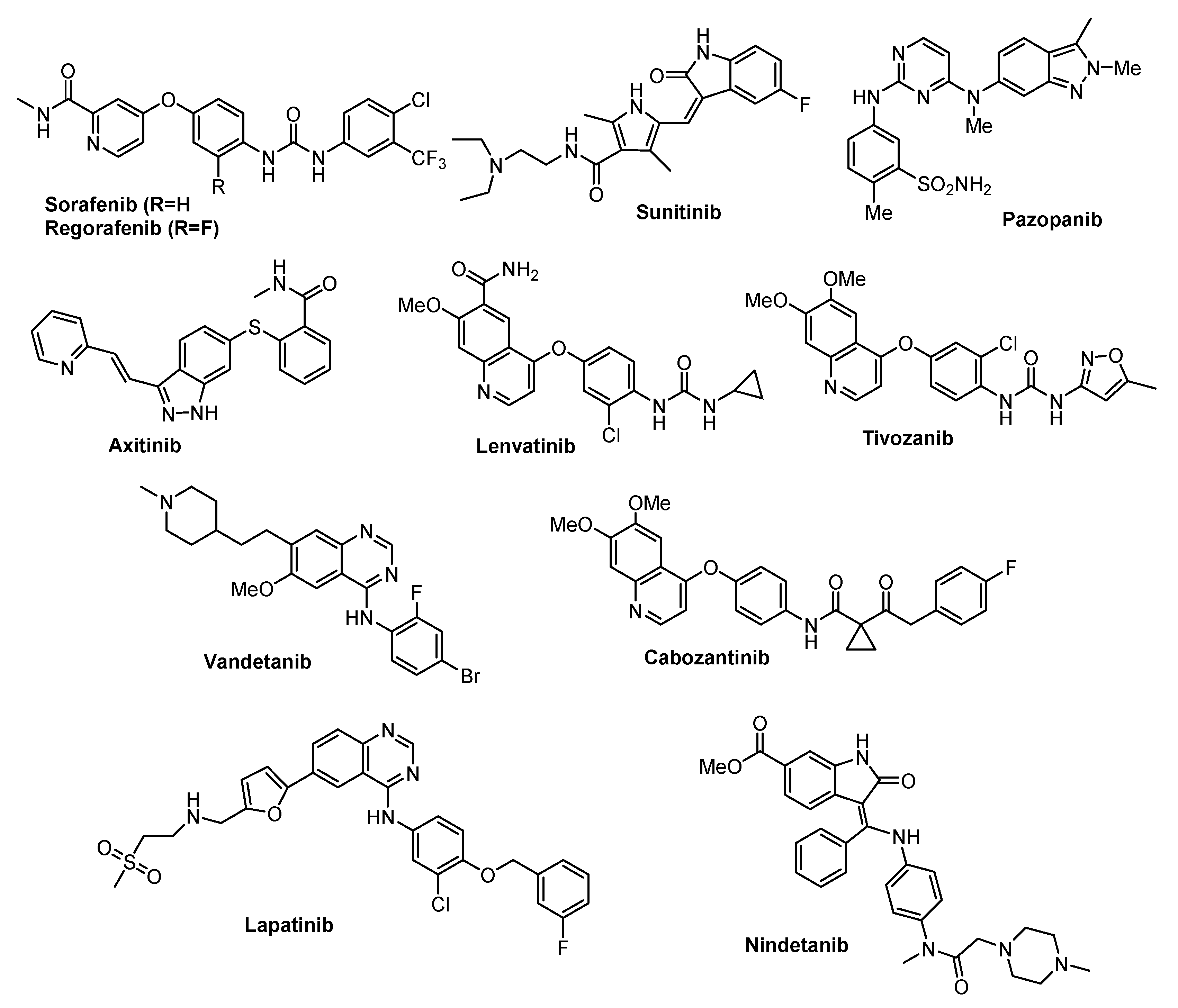
:1. Introduction
2. Results
2.1. Docking Studies
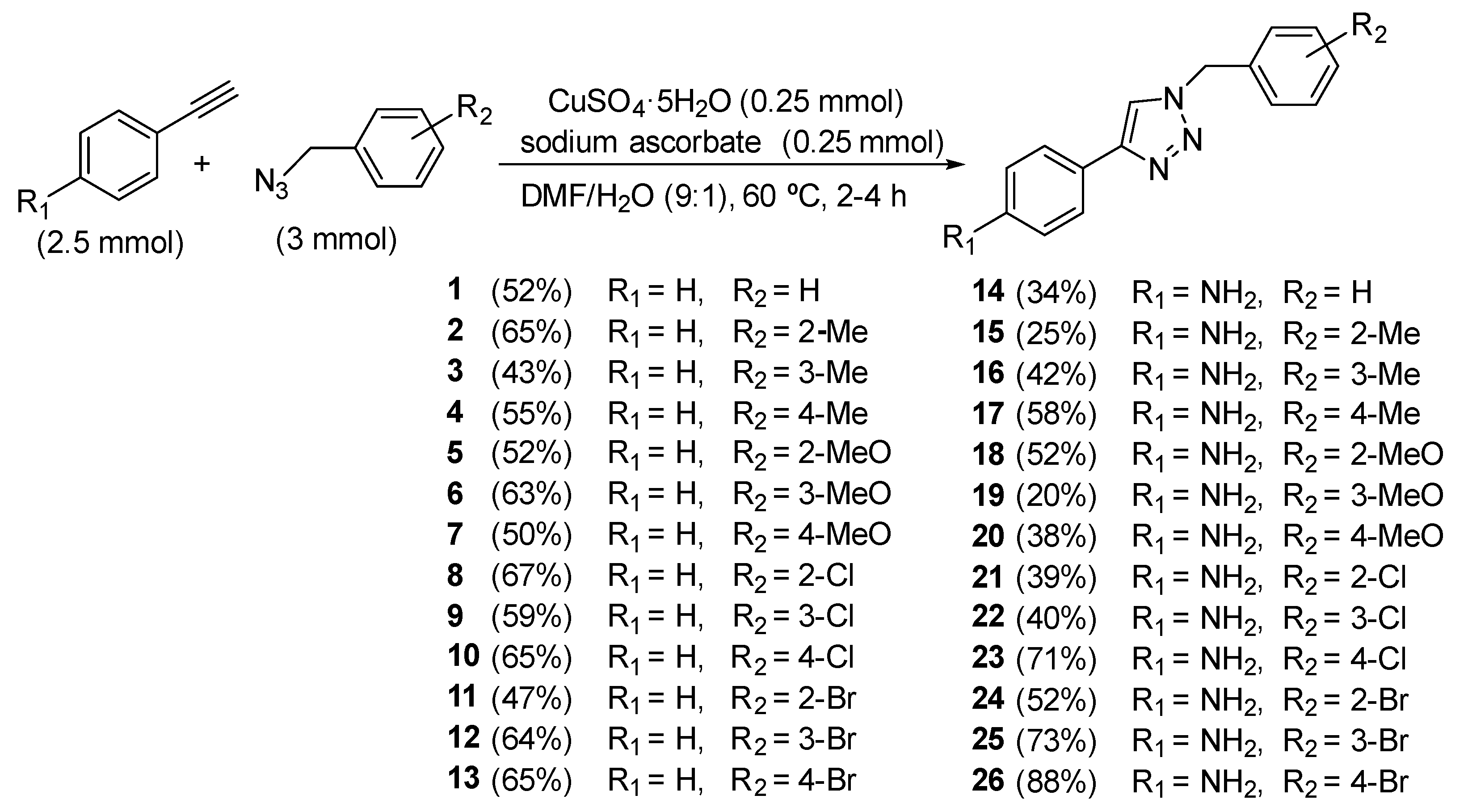
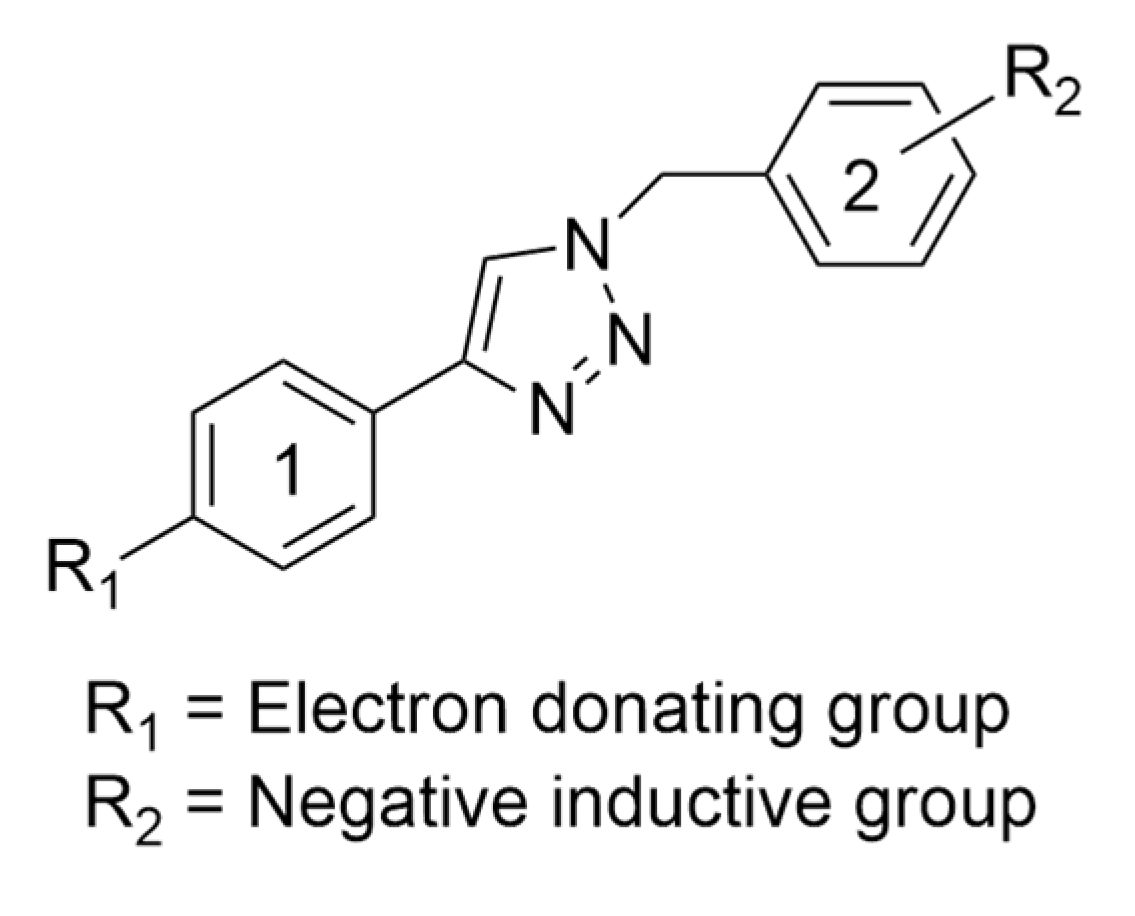
2.2. Synthesis
2.3. Biological Evaluation
2.3.1. Study of Cell Viability
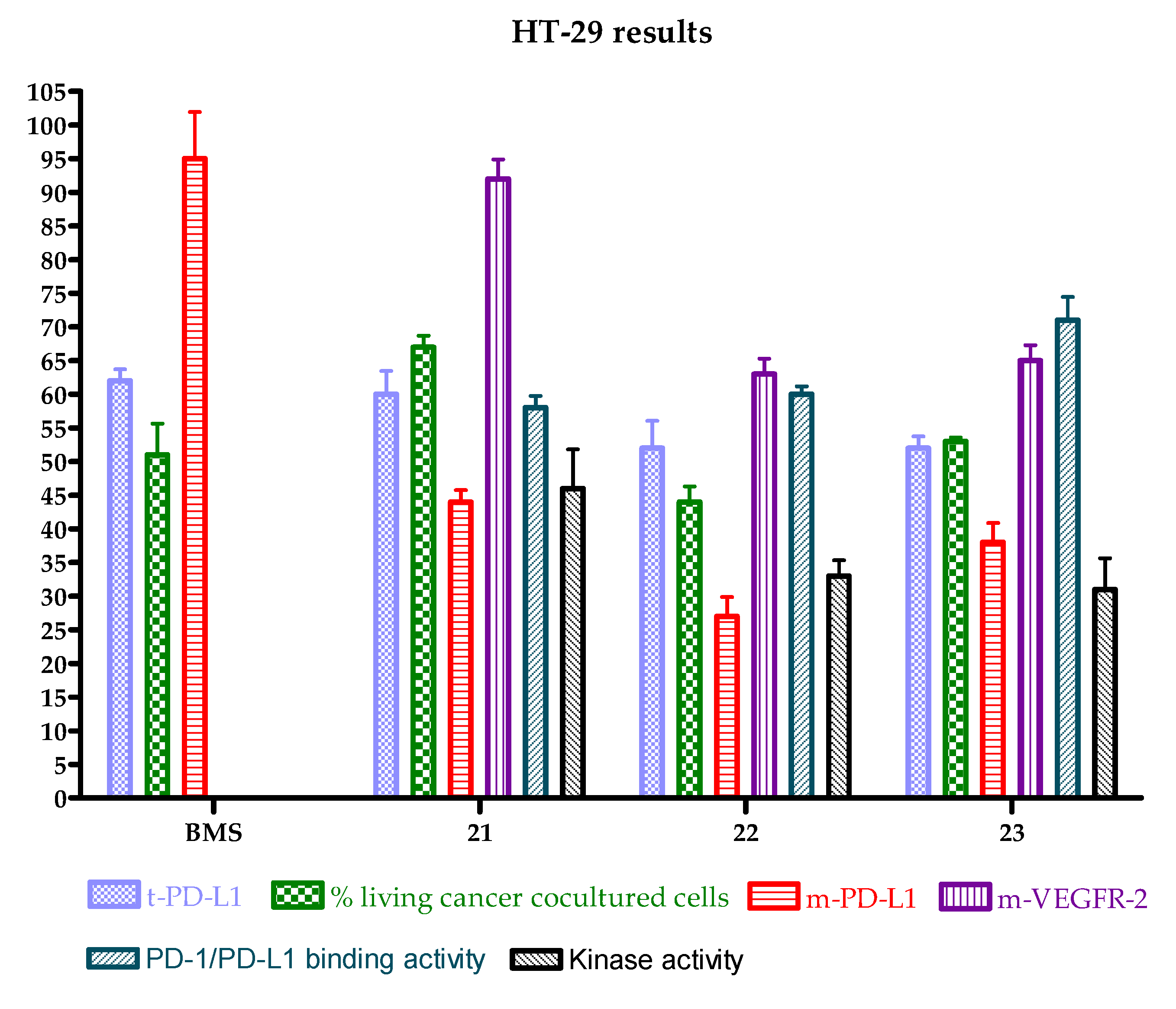
2.3.2. Effect of Derivatives 14–26 on Membrane PD-L1 and VEGFR-2 in Cancer Cell Lines
2.3.3. Effect of Derivatives 14–26 on Total PD-L1 and c-Myc in Cancer Cell Lines
2.3.4. Study of Cellular PD-1/PD-L1 Blocking Activity in Co-Cultures: Effect on Cancer Cell Viability
2.3.5. Study of the Direct Interaction with PD-L1 Protein
2.3.6. Study of PD-1/PD-L1 Blocking Activity by Competitive ELISA Assay
2.3.7. Study of Potential Kinase Inhibitory Activity by ADP Assay
2.3.8. Study of Antivascular and Antiangiogenic Effect on HMEC-1 by Microtubes Formation on Matrigel® Assay
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedures
4.1.2. Experimental Procedure for the Synthesis of Triazole Compounds 1–26
4.2. Biological Studies
4.2.1. Cell Culture
4.2.2. Cell Proliferation Assay
4.2.3. PD-L1, VEGFR-2, and c-Myc Relative Quantification by Flow Cytometry
4.2.4. Protein Thermal Shift for Studying the Interaction between the Compounds and PD-L1
4.2.5. Competitive ELISA Assay
4.2.6. ADP Assay
4.2.7. Tube Disruption and Formation on Matrigel Assay
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201–249. [Google Scholar] [CrossRef]
- Van der Zanden, S.Y.; Luimstra, J.J.; Neefjes, J.; Borst, J.; Ovaa, H. Opportunities for Small Molecules in Cancer Immunotherapy. Trends Immun 2020, 41, 154–196. [Google Scholar] [CrossRef]
- Huck, B.; Kötzner, L.; Urbahns, K. Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem. Int. Ed. Engl. 2018, 57, 4412–4428. [Google Scholar] [CrossRef] [Green Version]
- Shulun, C.; Zilan, S.; Zhang, Z. Small-Molecule Immuno-Oncology Therapy: Advances, Challenges and New Directions Curr. Top. Med. Chem. 2019, 19, 180–185. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Casacuberta-Serra, L.; Soucek, K. Myc and Ras, the Bonnie and Clyde of immune evasion. Transl. Cancer Res. 2018, 7, S457–S459. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC Regulates the Anti-Tumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Chatterjee, S.; Heukamp, L.C.; Siobal, M.; Schöttle, J.; Wieczorek, C.; Peifer, M.; Frasca, D.; Koker, M.; König, K.; Meder, L.; et al. Tumor VEGF:VEGFR-2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Investig. 2013, 123, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharma, B.; Mehra, V.; Kumar, V. Recent accomplishments on the synthetic/biological facets of pharmacologically active 1H-1,2,3-triazoles. Eur. J. Med. Chem. 2021, 212, 113069. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Alminderej, F.M.; Mounier, M.M.; Nossier, E.S.; Saleh, S.M.; Kassem, A.F. Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2. Molecules 2022, 27, 2047. [Google Scholar] [CrossRef]
- Wang, D.P.; Liu, K.L.; Li, X.Y.; Lu, G.Q.; Xue, W.H.; Qian, X.H.; Mohamed, O.K.; Meng, F.H. Design, synthesis, and in vitro and in vivo anti-angiogenesis study of a novel vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor based on 1,2,3-triazole scaffold. Eur. J. Med. Chem. 2021, 211, 113083. [Google Scholar] [CrossRef]
- Pan, X.; Liang, L.; Si, R.; Wang, J.; Zhang, Q.; Zhou, H.; Zhang, L.; Zhang, J. Discovery of novel anti-angiogenesis agents. Part 10: Multi-target inhibitors of VEGFR-2, Tie-2 and EphB4 incorporated with 1,2,3-triazol. Eur. J. Med. Chem. 2019, 163, 1–9. [Google Scholar] [CrossRef]
- Gilandoust, M.; Harsha, K.B.; Mohan, C.D.; Raquib, A.R.; Rangappa, S.; Pandey, V.; Lobie, P.E.; Basappa; Rangappa, K.S. Synthesis, characterization and cytotoxicity studies of 1,2,3-triazoles and 1,2,4-triazolo [1,5-a] pyrimidines in human breast cancer cells. Bioorg. Med. Chem. Lett. 2018, 28, 2314–2319. [Google Scholar] [CrossRef]
- Abd-Rabou, A.A.; Abdel-Wahab, B.F.; Bekheit, M.S. Synthesis, molecular docking, and evaluation of novel bivalent pyrazolinyl-1,2,3-triazoles as potential VEGFR TK inhibitors and anti-cancer agents. Chem. Pap. 2018, 72, 2225–2237. [Google Scholar] [CrossRef]
- Vojticková, M.; Dobiaš, J.; Hanquet, G.; Addová, G.; Cetin-Atalay, R.; Yildirim, D.C.; Bohác, A. Ynamide Click chemistry in development of triazole VEGFR2 TK modulators. Eur. J. Med. Chem. 2015, 103, 105–122. [Google Scholar] [CrossRef]
- Kiselyov, A.S.; Semenova, M.; Semenov, V.V. (1,2,3-Triazol-4-yl)benzenamines: Synthesis and activity against VEGF receptors 1 and 2. Bioorg. Med. Chem. Lett. 2009, 19, 1344–1348. [Google Scholar] [CrossRef]
- Conesa-Milián, L.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Novel multitarget inhibitors with antiangiogenic and immunomodulator properties. Eur. J. Med. Chem. 2019, 148, 87–98. [Google Scholar] [CrossRef]
- Martín-Beltrán, C.; Gil-Edo, R.; Hernández-Ribelles, G.; Agut, R.; Marí-Mezquita, P.; Carda, M.; Falomir, E. Aryl Urea Based Scaffolds for Multitarget Drug Discovery in Anticancer Immunotherapies. Pharmaceuticals 2021, 14, 337. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [Green Version]
- Schaftenaar, G. Molden. 2018. Available online: https://www3.cmbi.umcn.nl/molden/ (accessed on 24 September 2021).
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Liu, C.; Seeran, N.P.; Ma, H. Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: A review. Cancer Cell Int. 2021, 21, 239. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Mabrouk, R.R.; Elnagar, M.R.; Farrag, A.M.; Kalaba, M.H.; Sharaf, M.H.; El-Fakharany, E.M.; Bakhotmah, D.A.; Elkaeed, E.B.; Al Ward, M.M.S. New Series of VEGFR-2 Inhibitors and Apoptosis Enhancers: Design, Synthesis and Biological Evaluation. Drug Des. Dev. Ther. 2022, 16, 587–607. [Google Scholar] [CrossRef]
- Martin, J.L.; Baxter, R.C. Expression of Insulin-Like Growth Factor Binding Protein-2 by MCF-7 Breast Cancer Cells Is Regulated through the Phosphatidylinositol 3-Kinase/AKT/Mammalian Target of Rapamycin Pathway. Endocrinology 2007, 148, 2532–2541. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, L.; Chen, H.; Kong, R.; Pan, S.; Hu, J.; Wang, Y.; Li, Y.; Sun, B. Silencing IGFBP-2 decreases pancreatic cancer metastasis and enhances chemotherapeutic sensitivity. Oncotarget 2017, 37, 61674–61686. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | HT-29 | A-549 | MCF-7 | HEK-293 |
|---|---|---|---|---|---|
| 1 | 14 (NH2-H) | 2 ± 1 | >100 | 1,0 ± 0,5 | >100 |
| 2 | 15 (NH2-o-CH3) | 7 ± 1 | 4 ± 3 | 4 ± 1 | >100 |
| 3 | 16 (NH2-m-CH3) | 9 ± 6 | 0,7 ± 0,4 | 2,1 ± 0,6 | 63 ± 34 |
| 4 | 17 (NH2-p-CH3) | 8 ± 2 | 1,7 ± 0,7 | 4 ± 1 | 4 ± 2 |
| 5 | 18 (NH2-o-OCH3) | >100 | >100 | >100 | >100 |
| 6 | 19 (NH2-m-OCH3) | >100 | >100 | 12 ± 5 | >100 |
| 7 | 20 (NH2-p-OCH3) | >100 | >100 | 6 ± 4 | >100 |
| 8 | 21 (NH2-o-Cl) | >100 | >100 | 11 ± 2 | >100 |
| 9 | 22 (NH2-m-Cl) | >100 | >100 | 8 ± 2 | >100 |
| 10 | 23 (NH2-p-Cl) | >100 | >100 | 9 ± 6 | >100 |
| 11 | 24 (NH2-o-Br) | 3,7 ± 1,7 | 0,45 ± 0,16 | 5 ± 1 | >100 |
| 12 | 25 (NH2-m-Br) | 8,5 ± 1,3 | 2,7 ± 0.5 | 8 ± 4 | 4 ± 3 |
| 13 | 26 (NH2-p-Br) | 14 ± 6 | 8 ± 2 | 8 ± 1 | >100 |
| 14 | Sorafenib | 17 ± 4 | 27 ± 2 | 14 ± 4 | 5,0 ± 0,7 |
| 15 | BMS-8 | 19 ± 2 | 6 ± 1 | 20 ± 3 | 60 ± 10 |
| HT-29 | A-549 | MCF-7 | |||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | mPD-L1 | mVEGFR-2 | mPD-L1 | mVEGFR-2 | mPD-L1 | mVEGFR-2 |
| 1 | 14 (NH2-H) | >100 | 95 ± 6 | >100 | >100 | 94 ± 5 | >100 |
| 2 | 15 (NH2-o-CH3) | 74 ± 13 | 87 ± 1 | 90 ± 6 | >100 | 84 ± 11 | >100 |
| 3 | 16 (NH2-m-CH3) | 66 ± 21 | 93 ± 1 | 86 ± 17 | >100 | >100 | >100 |
| 4 | 17 (NH2-p-CH3) | 87 ± 14 | 81 ± 1 | 88 ± 21 | >100 | 89 ± 5 | >100 |
| 5 | 18 (NH2-o-OCH3) | 61 ± 22 | 93 ± 1 | 89 ± 9 | >100 | 81 ± 2 | >100 |
| 6 | 19 (NH2-m-OCH3) | >100 | 94 ± 1 | >100 | >100 | 96 ± 5 | >100 |
| 7 | 20 (NH2-p-OCH3) | >100 | 92 ± 14 | 95 ± 8 | >100 | 91 ± 9 | >100 |
| 8 | 21 (NH2-o-Cl) | 44 ± 3 | 92 ± 5 | 95 ± 5 | >100 | 83 ± 7 | >100 |
| 9 | 22 (NH2-m-Cl) | 27 ± 5 | 63 ± 4 | 82 ± 18 | >100 | 90 ± 2 | >100 |
| 10 | 23 (NH2-p-Cl) | 38 ± 5 | 65 ± 4 | >100 | >100 | 86 ± 3 | >100 |
| 11 | 24 (NH2-o-Br) | 86 ± 11 | 84 ± 8 | 88 ± 12 | >100 | 91 ± 10 | >100 |
| 12 | 25 (NH2-m-Br) | 87 ± 17 | >100 | >100 | >100 | 87 ± 17 | >100 |
| 13 | 26 (NH2-p-Br) | 94 ± 15 | 85 ± 7 | 76 ± 4 | >100 | 84 ± 23 | >100 |
| 14 | Sorafenib | --- | 85 ± 5 | --- | 80 ± 8 | --- | 85 ± 5 |
| 15 | BMS-8 | 95 ± 12 | --- | 99 ± 10 | --- | 90 ± 8 | --- |
| HT-29 | A-549 | MCF-7 | |||||
|---|---|---|---|---|---|---|---|
| Entry | Compound | tPD-L1 | c-Myc | tPD-L1 | c-Myc | tPD-L1 | c-Myc |
| 1 | 14 (NH2-H) | >100 | >100 | 98 ± 3 | >100 | >100 | 60 ± 3 |
| 2 | 15 (NH2-o-CH3) | >100 | >100 | >100 | >100 | >100 | >100 |
| 3 | 16 (NH2-m-CH3) | >100 | >100 | >100 | >100 | >100 | 78 ± 7 |
| 4 | 17 (NH2-p-CH3) | >100 | >100 | 56 ± 19 | >100 | >100 | 63 ± 2 |
| 5 | 18 (NH2-o-OCH3) | 70 ± 11 | >100 | 91 ± 4 | >100 | >100 | 53 ± 3 |
| 6 | 19 (NH2-m-OCH3) | 85 ± 17 | >100 | >100 | >100 | >100 | 55 ± 5 |
| 7 | 20 (NH2-p-OCH3) | 67 ± 3 | 74 ± 23 | 54 ± 6 | 69 ± 9 | >100 | 62 ± 7 |
| 8 | 21 (NH2-o-Cl) | 60 ± 6 | >100 | 78 ± 9 | >100 | >100 | 59 ± 6 |
| 9 | 22 (NH2-m-Cl) | 52 ± 7 | >100 | 75 ± 10 | >100 | 69 ± 7 | 59 ± 5 |
| 10 | 23 (NH2-p-Cl) | 52 ± 3 | 84 ± 5 | 81 ± 15 | >100 | 61 ± 14 | 51 ± 8 |
| 11 | 24 (NH2-o-Br) | 45 ± 3 | >100 | 56 ± 7 | >100 | >100 | 57 ± 8 |
| 12 | 25 (NH2-m-Br) | 48 ± 7 | >100 | 76 ± 1 | >100 | >100 | 56 ± 10 |
| 13 | 26 (NH2-p-Br) | 51 ± 2 | >100 | 80 ± 12 | 82 ± 4 | >100 | 64 ± 6 |
| 14 | BMS-8 | 62 ± 3 | 99 ± 7 | 66 ± 8 | 135 ± 15 | 68 ± 5 | 60 ± 7 |
| HT-29 + Jurkat T | |||
|---|---|---|---|
| Entry | Compound | HT-29 | Jurkat |
| 1 | 21 (NH2-o-Cl) | 67 ± 3% | 80 ± 3% |
| 2 | 22 (NH2-m-Cl) | 44 ± 4% | 79 ± 9% |
| 3 | 23 (NH2-p-Cl) | 53 ± 1% | 99 ± 11% |
| 14 | BMS-8 | 51 ± 8% | 110 ± 4% |
| PTS | HT-29 | ||||
|---|---|---|---|---|---|
| Entry | Compound | Tm (°C) | ΔTm (°C) | tPD-L1 (%) | % Living Cells in Co- Cultures with Jurkat |
| 1 | DMSO | 34 | 0 | ||
| 2 | 21 (NH2-o-Cl) | 30 | −4 | 60 ± 6 | 67 ± 3% |
| 3 | 22 (NH2-m-Cl) | 26 | −8 | 52 ± 7 | 44 ± 4% |
| 4 | 23 (NH2-p-Cl) | 29 | −5 | 52 ± 3 | 53 ± 1% |
| 5 | BMS-8 | 38 | +4 | 62 ± 3 | 51 ± 8 |
| Entry | Compound | HT-29 |
|---|---|---|
| 1 | 21 (NH2-o-Cl) | 46 ± 10% |
| 2 | 22 (NH2-m-Cl) | 33 ± 4% |
| 3 | 23 (NH2-p-Cl) | 31 ± 8% |
| Entry | Compound | HT-29 | A-549 | MCF-7 |
|---|---|---|---|---|
| 1 | 14 (NH2-H) | 100 | 100 | 100 |
| 2 | 15 (NH2-o-CH3) | 10 | 10 | 10 |
| 3 | 16 (NH2-m-CH3) | 10 | 10 | 10 |
| 4 | 17 (NH2-p-CH3) | 10 | 10 | 10 |
| 5 | 18 (NH2-o-OCH3) | 100 | 100 | 10 |
| 6 | 19 (NH2-m-OCH3) | 100 | 100 | 10 |
| 7 | 20 (NH2-p-OCH3) | 100 | 100 | 10 |
| 8 | 21 (NH2-o-Cl) | 100 | 100 | 10 |
| 9 | 22 (NH2-m-Cl) | 100 | 100 | 10 |
| 10 | 23 (NH2-p-Cl) | 100 | 100 | 10 |
| 11 | 24 (NH2-o-Br) | 10 | 10 | 10 |
| 12 | 25 (NH2-m-Br) | 10 | 10 | 10 |
| 13 | 26 (NH2-p-Br) | 10 | 10 | 10 |
| 14 | BMS-8 | 100 | 100 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pla-López, A.; Castillo, R.; Cejudo-Marín, R.; García-Pedrero, O.; Bakir-Laso, M.; Falomir, E.; Carda, M. Synthesis and Biological Evaluation of Small Molecules as Potential Anticancer Multitarget Agents. Int. J. Mol. Sci. 2022, 23, 7049. https://doi.org/10.3390/ijms23137049
Pla-López A, Castillo R, Cejudo-Marín R, García-Pedrero O, Bakir-Laso M, Falomir E, Carda M. Synthesis and Biological Evaluation of Small Molecules as Potential Anticancer Multitarget Agents. International Journal of Molecular Sciences. 2022; 23(13):7049. https://doi.org/10.3390/ijms23137049
Chicago/Turabian StylePla-López, Alberto, Raquel Castillo, Rocío Cejudo-Marín, Olaya García-Pedrero, Mariam Bakir-Laso, Eva Falomir, and Miguel Carda. 2022. "Synthesis and Biological Evaluation of Small Molecules as Potential Anticancer Multitarget Agents" International Journal of Molecular Sciences 23, no. 13: 7049. https://doi.org/10.3390/ijms23137049
APA StylePla-López, A., Castillo, R., Cejudo-Marín, R., García-Pedrero, O., Bakir-Laso, M., Falomir, E., & Carda, M. (2022). Synthesis and Biological Evaluation of Small Molecules as Potential Anticancer Multitarget Agents. International Journal of Molecular Sciences, 23(13), 7049. https://doi.org/10.3390/ijms23137049