
Intra- and Intermolecular Hydrogen Bonding in Miscible Blends of CO2/Epoxy Cyclohexene Copolymer with Poly(Vinyl Phenol)



Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of LH and LZn2OAc2
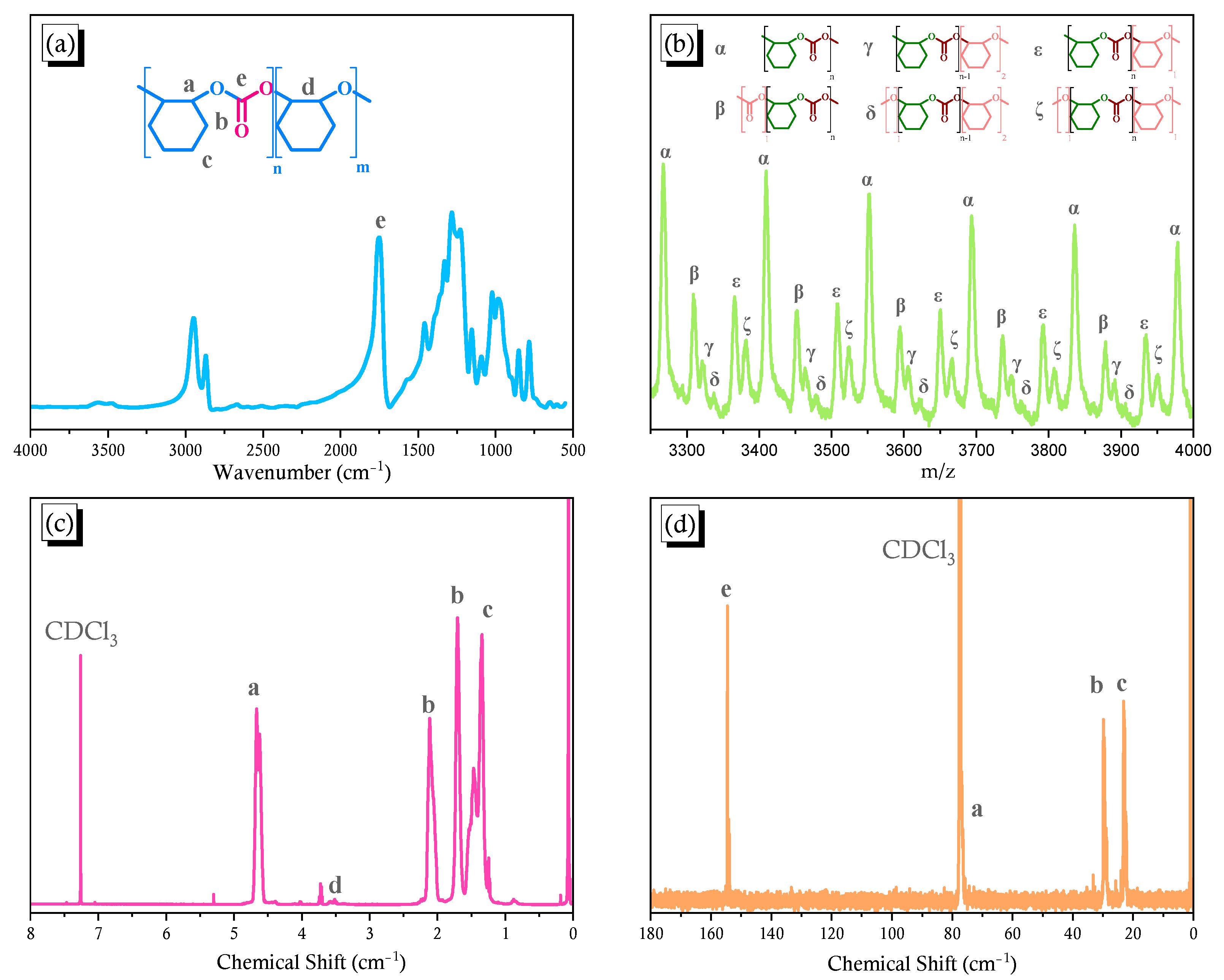
2.2. Synthesis of PCHC
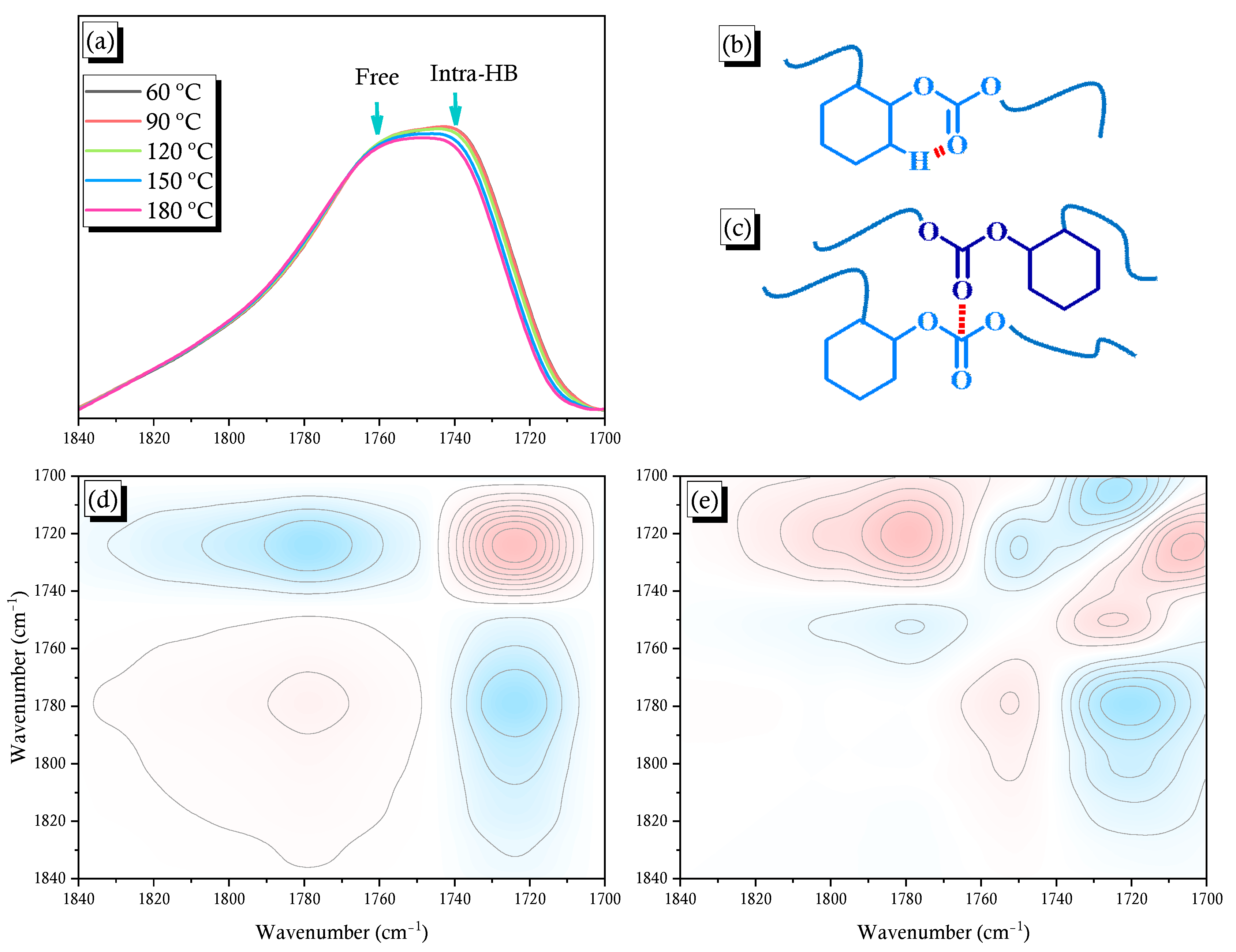
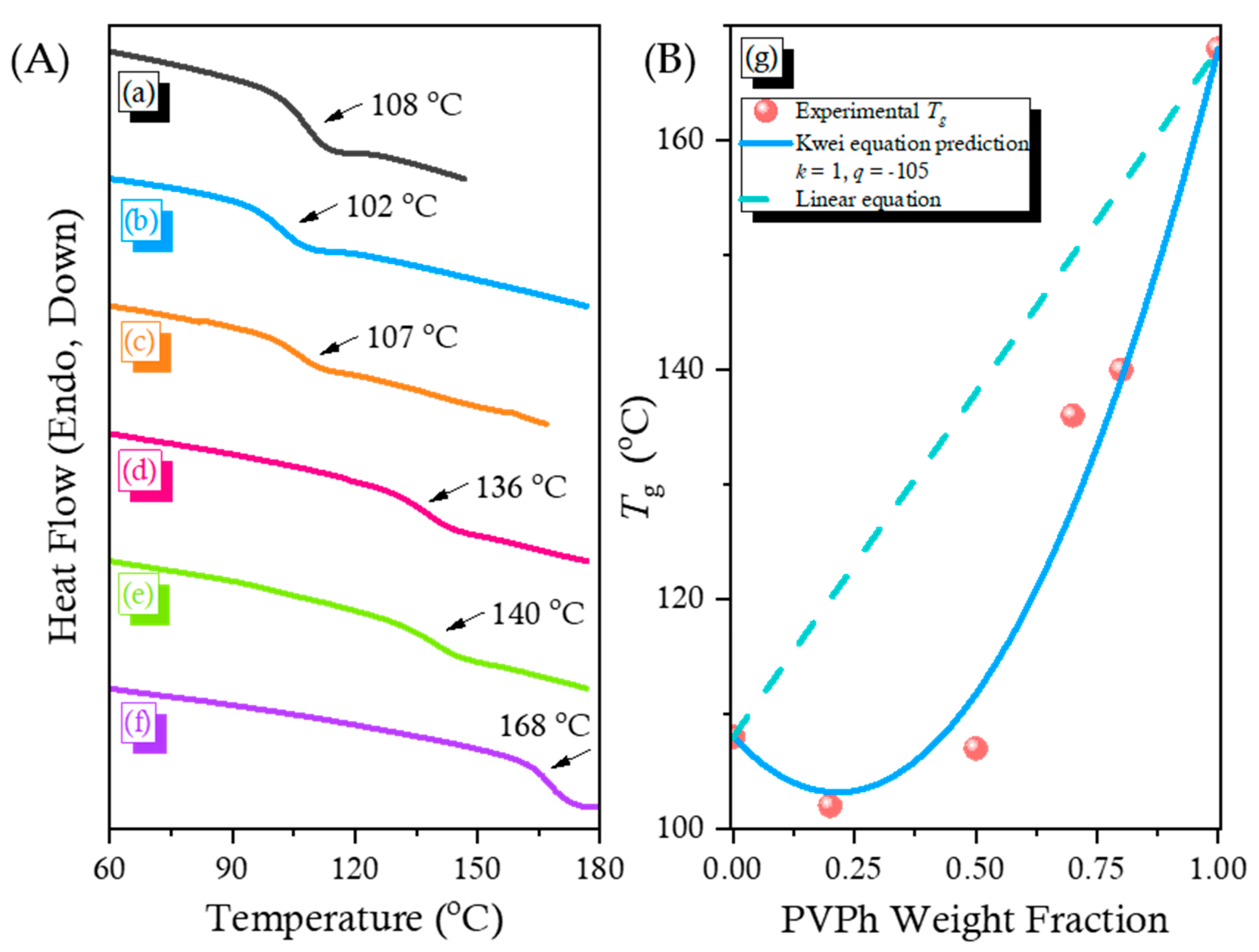
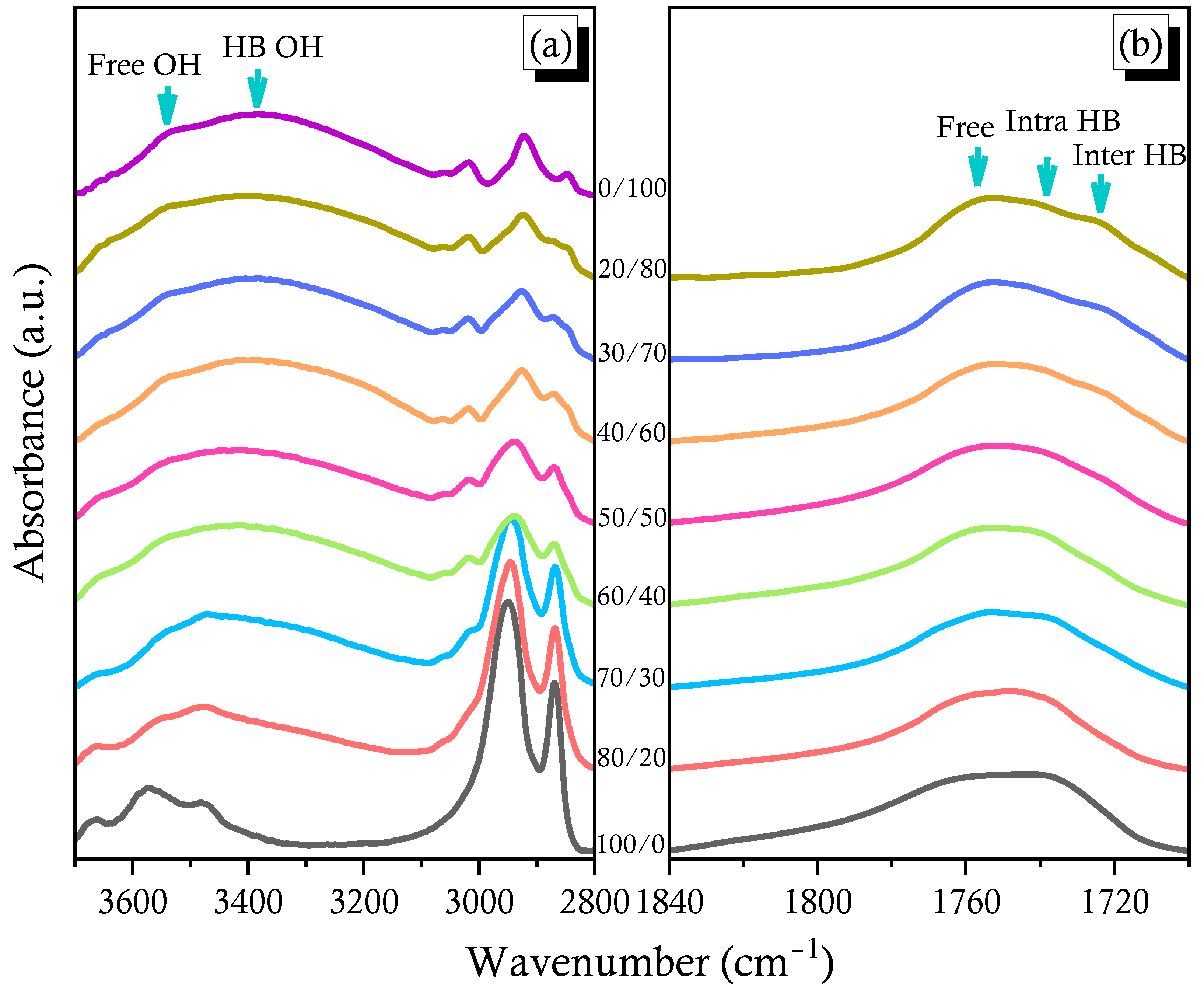
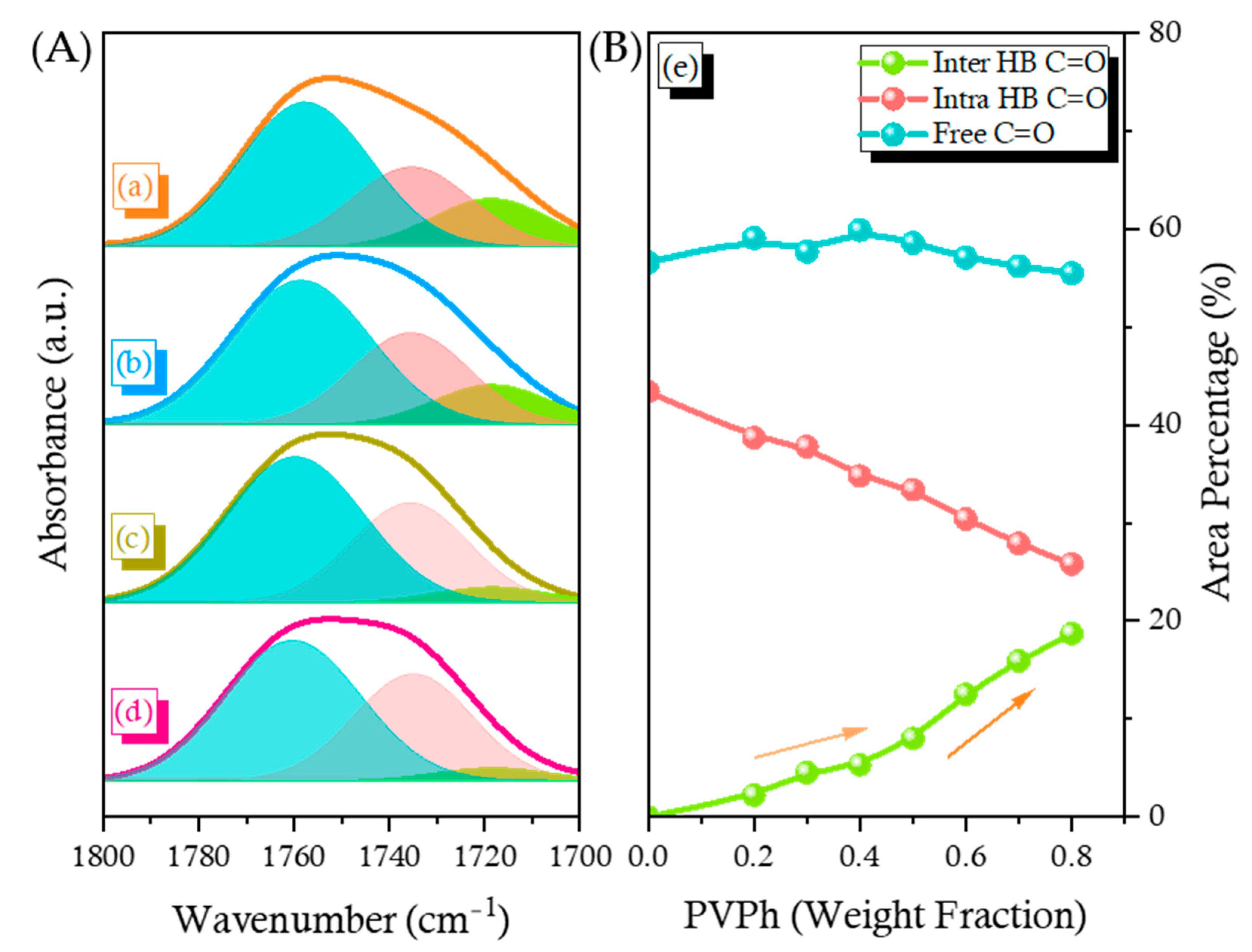
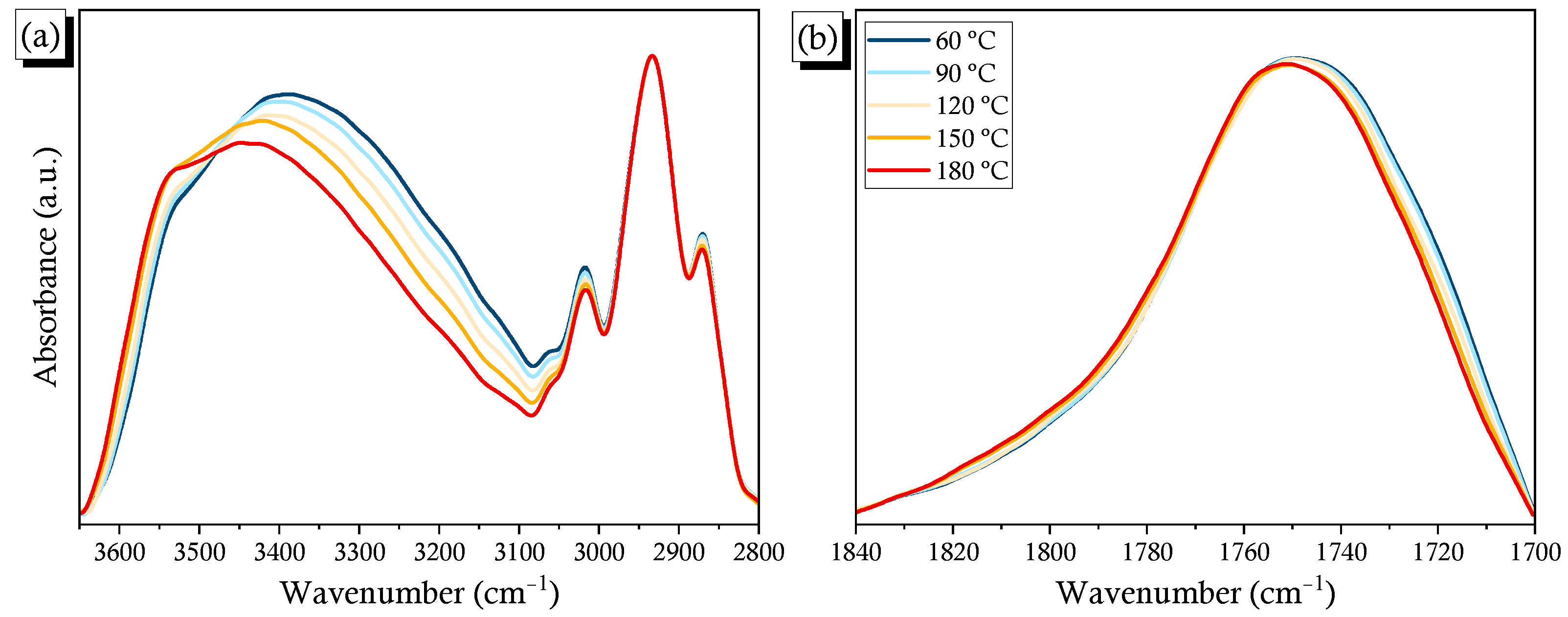
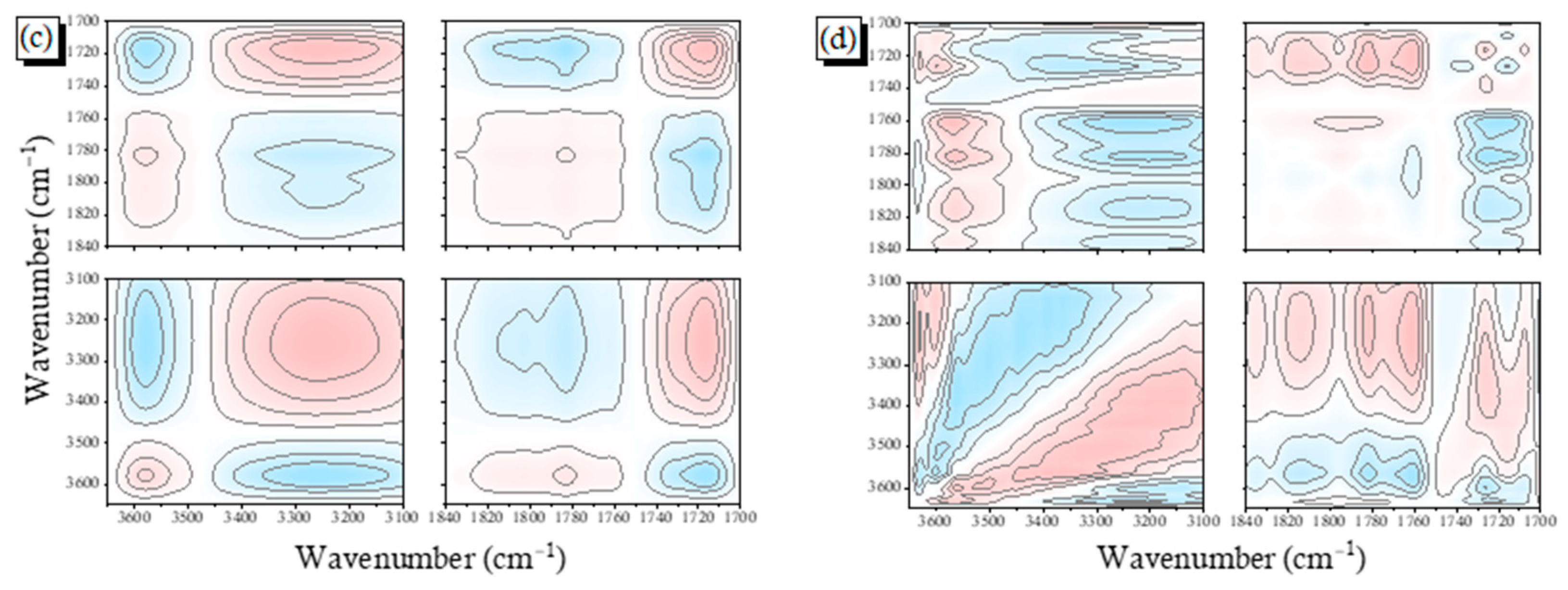
2.3. PCHC/PVPh Binary Blends
2.4. Solid State NMR Spectra
3. Materials and Methods
3.1. Materials
3.2. Copolymerization of CO2 and CHO
3.3. PVPh/PCHC Binary Blends
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peter, S.C. Reduction of CO2 to Chemicals and Fuels: A Solution to Global Warming and Energy Crisis. ACS Energy Lett. 2018, 3, 1557–1561. [Google Scholar] [CrossRef]
- Tackett, B.M.; Gomez, E.; Chen, J.G. Net reduction of CO2 via its thermocatalytic and electrocatalytic transformation reactions in standard and hybrid processes. Nat. Catal. 2019, 2, 381–386. [Google Scholar] [CrossRef]
- Mohamed, M.G.; Chen, T.Z.; Kuo, S.W. Solid-state chemical transformations to enhance gas capture in benzoxazine-linked conjugated microporous polymers. Macromolecules 2021, 54, 5866–5877. [Google Scholar] [CrossRef]
- Bera, R.; Ansari, M.; Alam, A.; Das, D. Triptycene, Phenolic-OH, and Azo-Functionalized Porous Organic Polymers: Efficient and Selective CO2 Capture. ACS Appl. Polym. Mater. 2019, 1, 959–968. [Google Scholar] [CrossRef]
- Mohamed, M.G.; Samy, M.M.; Mansoure, T.H.; Li, C.-J.; Li, W.-C.; Chen, J.-H.; Zhang, K.; Kuo, S.-W. Microporous Carbon and Carbon/Metal Composite Materials Derived from Bio-Benzoxazine-Linked Precursor for CO2 Capture and Energy Storage Applications. Int. J. Mol. Sci. 2022, 23, 347. [Google Scholar] [CrossRef]
- Gamal Mohamed, M.; Tsai, M.-Y.; Wang, C.-F.; Huang, C.-F.; Danko, M.; Dai, L.; Chen, T.; Kuo, S.-W. Multifunctional Polyhedral Oligomeric Silsesquioxane (POSS) Based Hybrid Porous Materials for CO2 Uptake and Iodine Adsorption. Polymers 2021, 13, 221. [Google Scholar] [CrossRef]
- Hill, M.R.; Tang, S.; Masada, K.; Hirooka, Y.; Nozaki, K. Incorporation of CO2-Derived Bicyclic Lactone into Conventional Vinyl Polymers. Macromolecules 2022, 55, 3311–3316. [Google Scholar] [CrossRef]
- Allen, S.D.; Moore, D.R.; Lobkovsky, E.B.; Coates, G.W. High-Activity, Single-Site Catalysts for the Alternating Copolymerization of CO2 and Propylene Oxide. J. Am. Chem. Soc. 2002, 124, 14284–14285. [Google Scholar] [CrossRef]
- Deacy, A.C.; Durr, C.B.; Garden, J.A.; White, J.P.; Williams, C.K. Groups 1, 2 and Zn(II) Heterodinuclear Catalysts for Epoxide/CO2 Ring-Opening Copolymerization. Inorg. Chem. 2018, 57, 15575–15583. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Duan, R.; Yang, S.; Pang, X.; Chen, X. CO2 Controlled Catalysis: Switchable Homopolymerization and Copolymerization. Macromolecules 2018, 51, 4699–4704. [Google Scholar] [CrossRef]
- Han, B.; Liu, B.; Ding, H.; Duan, Z.; Wang, X.; Theato, P. CO2-Tuned Sequential Synthesis of Stereoblock Copolymers Comprising a Stereoregularity-Adjustable Polyester Block and an Atactic CO2-Based Polycarbonate Block. Macromolecules 2017, 50, 9207–9215. [Google Scholar] [CrossRef]
- Wang, E.; Liu, S.; Lam, J.W.Y.; Tang, B.Z.; Wang, X.; Wang, F. Deciphering Structure–Functionality Relationship of Polycarbonate-Based Polyelectrolytes by AIE Technology. Macromolecules 2020, 53, 5839–5846. [Google Scholar] [CrossRef]
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerization of carbon dioxide and epoxide. J. Polym. Sci. Polym. Lett. 1969, 7, 287–292. [Google Scholar] [CrossRef]
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerization of carbon dioxide and epoxide with organometallic compounds. Makromol. Chem. 1969, 130, 210–220. [Google Scholar] [CrossRef]
- Muthuraj, R.; Mekonnen, T. Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: Co-polymers and polymer blends. Polymer 2018, 145, 348–373. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, L.; Xiao, M.; Wang, S.; Smith, A.T.; Sun, L.; Meng, Y. Synthesis and properties of CO2-based plastics: Environmentally-friendly, energy-saving and biomedical polymeric materials. Prog. Polym. Sci. 2018, 80, 163–182. [Google Scholar] [CrossRef]
- Li, Y.; Shimizu, H. Compatibilization by Homopolymer: Significant Improvements in the Modulus and Tensile Strength of PPC/PMMA Blends by the Addition of a Small Amount of PVAc. ACS Appl. Mater. Interface 2009, 1, 1650–1655. [Google Scholar] [CrossRef]
- Meereboer, K.W.; Pal, A.K.; Misra, M.; Mohanty, A.K. Green Composites from a Bioplastic Blend of Poly(3-hyroxybutyrate-co-3-hydroxyvalerate) and Carbon Dioxide-Derived Poly(propylene carbonate) and Filled with a Corn Ethanol-Industry Co-product. ACS Omega 2021, 6, 20103–20111. [Google Scholar] [CrossRef]
- Song, L.; Li, Y.; Meng, X.; Wang, T.; Shi, Y.; Wang, Y.; Shi, S.; Liu, L.-Z. Crystallization, Structure and Significantly Improved Mechanical Properties of PLA/PPC Blends Compatibilized with PLA-PPC Copolymers Produced by Reactions Initiated with TBT or TDI. Polymers 2021, 13, 3245. [Google Scholar] [CrossRef]
- Zuo, H.; Liu, J.; Huang, D.; Bai, Y.; Cui, L.; Pan, L.; Zhang, K.; Wang, H. Sustainable and high-performance ternary blends from polylactide, CO2-based polyester and microbial polyesters with different chemical structure. J. Polym. Sci. 2021, 59, 1578–1595. [Google Scholar] [CrossRef]
- Dong, X.; Liu, L.; Wang, Y.; Li, T.; Wu, Z.; Yuan, H.; Ma, P.; Shi, D.; Chen, M.; Dong, W. The compatibilization of poly (propylene carbonate)/poly (lactic acid) blends in presence of core-shell starch nanoparticles. Carbohydr. Polym. 2021, 254, 117321. [Google Scholar] [CrossRef]
- Yu, T.; Zhou, Y.; Zhao, Y.; Liu, K.P.; Chen, E.Q.; Wang, D.J.; Wang, F.S. Hydrogen-bonded thermostable liquid crystalline complex formed by biodegradable polymer and amphiphilic molecules. Macromolecules 2008, 41, 3175–3180. [Google Scholar] [CrossRef]
- Cui, S.; Li, L.; Wang, Q. Enhancing glass transition temperature and mechanical properties of poly (propylene carbonate) by intermacromolecular complexation with poly (vinyl alcohol). Compos. Sci. Technol. 2016, 127, 177–184. [Google Scholar] [CrossRef]
- Fei, B.; Chen, C.; Peng, S.; Zhao, X.; Wang, X.; Dong, L. FTIR study of poly(propylene carbonate)/bisphenol A blends. Polym. Int. 2004, 53, 2092–2908. [Google Scholar] [CrossRef]
- Zhang, Z.; Mo, Z.; Zhang, H.; Zhang, Y.; Na, T.; An, Y.; Wang, X.; Zhao, X. Miscibility and Hydrogen-Bonding Interactions in Blends of Carbon Dioxide/Epoxy Propane Copolymer with Poly(p-vinylphenol). J. Polym. Sci. Part B Polym. Sci. 2002, 40, 1957–1964. [Google Scholar] [CrossRef]
- Qiu, F.; Chen, S.; Tan, L.; Ping, Z. The study of the miscibility and morphology of poly(styrene-co-4-vinylphenol)/poly(propylene carbonate) blends. Polym. Adv. Technol. 2004, 15, 453–458. [Google Scholar] [CrossRef]
- Kuo, S.W.; Chang, F.C. Studies of miscibility behavior and hydrogen bonding in blends of poly (vinylphenol) and poly (vinylpyrrolidone). Macromolecules 2001, 34, 5224–5228. [Google Scholar] [CrossRef]
- Tsou, C.T.; Kuo, S.W. Competing Hydrogen Bonding Interaction Creates Hierarchically Ordered Self-Assembled Structures of PMMA-b-P4VP/PVPh-b-PS Mixtures. Macromolecules 2019, 52, 8374–8383. [Google Scholar] [CrossRef]
- Tseng, T.C.; Kuo, S.W. Hydrogen-Bonding Strength Influences Hierarchical Self-Assembled Structures in Unusual Miscible/Immiscible Diblock Copolymer Blends. Macromolecules 2018, 51, 6451–6459. [Google Scholar] [CrossRef]
- Kuo, S.W. Hydrogen Bonding Mediated Self-Assembled Structures from Block Copolymer Mixtures to Mesoporous Materials. Polym. Int. 2022, 71, 393–410. [Google Scholar] [CrossRef]
- Lin, C.L.; Chen, W.C.; Liao, C.S.; Su, Y.C.; Haung, C.F.; Kuo, S.W.; Chang, F.C. Sequence distribution and polydispersity index affect the hydrogen-bonding strength of poly (vinylphenol-co-methyl methacrylate) copolymers. Macromolecules 2005, 38, 6435–6444. [Google Scholar] [CrossRef]
- Kuo, S.W. Hydrogen bonding interactions in polymer/polyhedral oligomeric silsesquioxane nanomaterials. J. Polym. Res. 2022, 29, 69. [Google Scholar] [CrossRef]
- Kuo, S.W. Hydrogen Bonding in Polymeric Materials; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Pan, P.; Bao, J.; Han, L.; Xie, Q.; Shan, G.; Bao, Y. Stereocomplexation of high-molecular-weight enantiomeric poly (lactic acid) s enhanced by miscible polymer blending with hydrogen bond interactions. Polymer 2016, 98, 80–87. [Google Scholar] [CrossRef]
- Zhang, L.; Goh, S.H.; Lee, S.Y. Miscibility and crystallization behaviour of poly(l-lactide)/poly(p-vinylphenol) blends. Polymer 1998, 39, 4841–4847. [Google Scholar] [CrossRef]
- Lodge, T.P.; McLeish, T.C.B. Self-concentrations and effective glass transition temperatures in polymer blends. Macromolecules 2002, 33, 5278–5284. [Google Scholar] [CrossRef]
- Yang, Z.; Han, C.D. Rheology of miscible polymer blends with hydrogen bonding. Macromolecules 2008, 41, 2104–2118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.H.; Jin, X.; Painter, P.C.; Runt, J. Dynamical heterogeneity in the thermodynamically miscible polymer blend of poly(vinyl ethyl ether) and styrene-co-p-hydroxystyrene copolymer. Macromolecules 2003, 36, 5710–5718. [Google Scholar] [CrossRef]
- Talibuddin, S.; Wu, L.; Runt, J.; Lin, J.S. Microstructure of melt-miscible, semicrystalline polymer blends. Macromolecules 1996, 29, 7527–7535. [Google Scholar] [CrossRef]
- Thevenon, A.; Garden, J.A.; White, A.J.P.; Williams, C.K. Dinuclear Zinc Salen Catalysts for the Ring Opening Copolymerization of Epoxides and Carbon Dioxide or Anhydrides. Inorg. Chem. 2015, 54, 11906–11915. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Y.; Yang, G.-W.; Wang, Y.; Lu, X.-Y.; Wu, G.-P.; Zhang, Z.-S.; Wang, K.; Zhang, R.-Y.; Nealey, P.F.; Darensbourg, D.J.; et al. Synthesis of CO2-Based Block Copolymers via Chain Transfer Polymerization Using Macroinitiators: Activity, Blocking Efficiency, and Nanostructure. Macromolecules 2018, 51, 791–800. [Google Scholar] [CrossRef]
- Mohamed, M.G.; Hsu, K.C.; Hong, J.L.; Kuo, S.W. Unexpected fluorescence from maleimide-containing polyhedral oligomeric silsesquioxanes: Nanoparticle and sequence distribution analyses of polystyrene-based alternating copolymers. Polym. Chem. 2016, 7, 135–145. [Google Scholar] [CrossRef]
- Du, W.-T.; Orabi, E.A.; Mohamed, M.G.; Kuo, S.-W. Inter/intramolecular hydrogen bonding mediate miscible blend formation between near-perfect alternating Poly(styrene-alt- hydroxyphenylmaleimide) copolymers and Poly(vinyl pyrrolidone). Polymer 2021, 219, 123542. [Google Scholar] [CrossRef]
- Du, W.-T.; Kuo, S.-W. Varying the sequence distribution and hydrogen bonding strength provides highly Heat-Resistant PMMA copolymers. Eur. Polym. J. 2022, 170, 111165. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Yang, G.-W.; Wu, G.-P. A Bifunctional β-Diiminate Zinc Catalyst with CO2/Epoxides Copolymerization and RAFT Polymerization Capacities for Versatile Block Copolymers Construction. Macromolecules 2018, 51, 3640–3646. [Google Scholar] [CrossRef]
- Zhang, J.; Sato, H.; Tsuji, H.; Noda, I.; Ozaki, Y. Infrared Spectroscopic Study of CH3-O=C Interaction during Poly(L-lactide)/Poly(D-lactide) Stereocomplex Formation. Macromolecules 2005, 38, 1822–1828. [Google Scholar] [CrossRef]
- Noda, I. Two-dimensional infrared spectroscopy. J. Am. Chem. Soc. 1989, 111, 8116–8118. [Google Scholar] [CrossRef]
- Chen, S.C.; Kuo, S.W.; Liao, C.S.; Chang, F.C. Syntheses, specific interactions, and pH-sensitive micellization behavior of poly [vinylphenol-b-2-(dimethylamino) ethyl methacrylate] diblock copolymers. Macromolecules 2008, 41, 8865–8876. [Google Scholar] [CrossRef]
- Wang, L.; Di, S.; Wang, W.; Chen, H.; Yang, X.; Gong, T.; Zhou, S. Tunable Temperature Memory Effect of Photo-Cross-Linked Star PCL–PEG Networks. Macromolecules 2014, 47, 1828–1836. [Google Scholar] [CrossRef]
- Kuo, S.W.; Huang, W.J.; Huang, C.F.; Chan, S.C.; Chang, F.C. Miscibility, specific interactions, and spherulite growth rates of binary poly (acetoxystyrene)/poly (ethylene oxide) blends. Macromolecules 2004, 37, 4164–4173. [Google Scholar] [CrossRef]
- Noda, I.; Ozaki, Y. Two-Dimensional Correlation Spectroscopy; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Kwei, T.K. The effect of hydrogen bonding on the glass transition temperatures of polymer mixtures. J. Polym. Sci. Polym. Lett. Ed. 1984, 22, 307–313. [Google Scholar] [CrossRef]
- Coleman, M.M.; Painter, P.C. Hydrogen Bonded Polymer Blends. Prog. Polym. Sci. 1995, 20, 1–59. [Google Scholar] [CrossRef]
- Asano, A.; Eguchi, M.; Shimizu, M.; Kurotsu, T. Miscibility and Molecular Motion of PMAA/PVAc Blends Investigated by High-Resolution Solid-State CPMAS 13C NMR. Macromolecules 2002, 35, 8819. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tung, P.H.; Chang, F.C. Syntheses and the study of strongly hydrogen-bonded poly (vinylphenol-b-vinylpyridine) diblock copolymer through anionic polymerization. Macromolecules 2006, 39, 9388–9395. [Google Scholar] [CrossRef]
- Lau, C.; Mi, Y. A study of blending and complexation of poly (acrylic acid)/poly (vinyl pyrrolidone). Polymer 2002, 43, 823. [Google Scholar] [CrossRef]
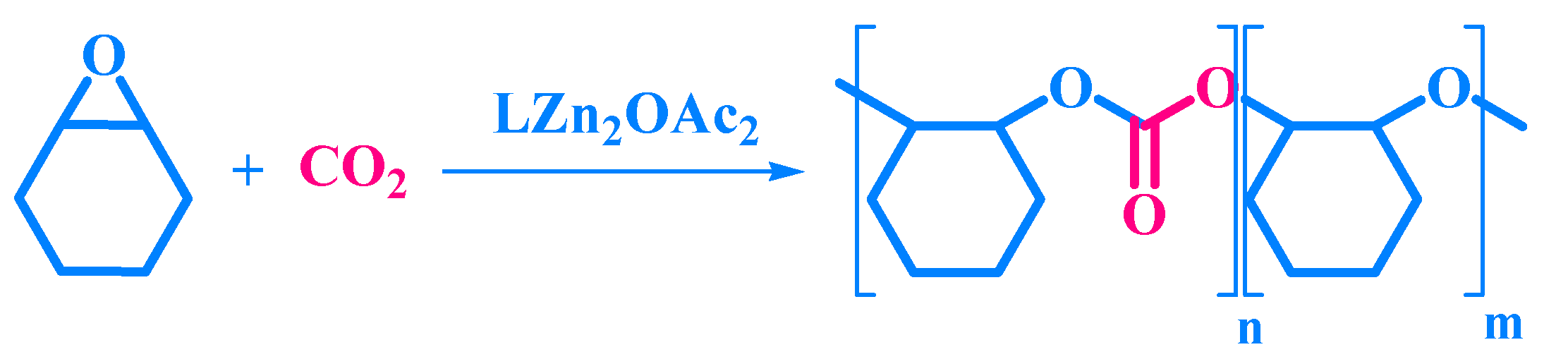



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Molar Volume (mL mol–1) | Molecular Weight (g mol–1) | Solubility Parameter (cal mL–1) | DP | Equilibrium Constants | ||
|---|---|---|---|---|---|---|---|
| K2 | KB | KA | |||||
| PVPh | 100.0 | 120.0 | 10.6 | 201 | 21.0 | 66.8 | – |
| PCHC | 94.5 | 142.0 | 10.2 | 154 | – | – | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, W.-T.; Kuan, Y.-L.; Kuo, S.-W. Intra- and Intermolecular Hydrogen Bonding in Miscible Blends of CO2/Epoxy Cyclohexene Copolymer with Poly(Vinyl Phenol). Int. J. Mol. Sci. 2022, 23, 7018. https://doi.org/10.3390/ijms23137018
Du W-T, Kuan Y-L, Kuo S-W. Intra- and Intermolecular Hydrogen Bonding in Miscible Blends of CO2/Epoxy Cyclohexene Copolymer with Poly(Vinyl Phenol). International Journal of Molecular Sciences. 2022; 23(13):7018. https://doi.org/10.3390/ijms23137018
Chicago/Turabian StyleDu, Wei-Ting, Yen-Ling Kuan, and Shiao-Wei Kuo. 2022. "Intra- and Intermolecular Hydrogen Bonding in Miscible Blends of CO2/Epoxy Cyclohexene Copolymer with Poly(Vinyl Phenol)" International Journal of Molecular Sciences 23, no. 13: 7018. https://doi.org/10.3390/ijms23137018
APA StyleDu, W.-T., Kuan, Y.-L., & Kuo, S.-W. (2022). Intra- and Intermolecular Hydrogen Bonding in Miscible Blends of CO2/Epoxy Cyclohexene Copolymer with Poly(Vinyl Phenol). International Journal of Molecular Sciences, 23(13), 7018. https://doi.org/10.3390/ijms23137018