
Pathophysiology and Emerging Molecular Therapeutic Targets in Heterotopic Ossification

 , , and
, , and
Abstract
1. Introduction
2. Overview of Normal Bone Formation
3. Cellular Origins of HO
3.1. Hematopoietic Cells
3.2. Endothelial Cells
3.3. Fibro-Adipogenic Cells
3.4. Myosatellite Cells
3.5. Other Cell Types

{kind=link}
{kind=link}
| Cell Type | Location | Description | Key Papers |
|---|---|---|---|
| Hematopoietic cells | Bone marrow | Contribute to inflammation and marrow-repopulating stages. Contribution to HO is unclear. | [19,23,25,60] |
| Endothelial cells | Blood and lymphatic vessels | Contribute to HO through EndMT route, but may be overestimated due to lack of surface marker endothelial cell-specificity. | [28,35] |
| FAPs | Muscle and related soft tissues; widely spread in other tissues | Support muscle regeneration. Contribute to a high percentage of HO. | [32,43,61] |
| Myosatellite cells | Muscle | BMP2-induced HO. Contribution low based on most lineage studies. | [32,48] |
| Pericytes | Vascular basement membrane | BMP-induced HO but assessment of contribution unclear due to high degree of heterogeneity. | [50,62,63,64] |
| Hoxa11+ Mesenchymal stromal cells | Tendon, muscle and skeletal tissue | Contribute to skeletal repair, express chondrogenic and osteogenic transcription profile following injury. | [56,57,58,59] |
| Tendon and ligament progenitor cells | Tendon Ligament | Account for 25 and 40% of heterotopic bone and cartilage, respectively, after bone/tendonectomy based on Scx-Cre labelling. Molecularly heterogeneous. | [39,43,65] |
| Sensory neurons | Dermis, epidermis, and muscle spindle | Mediate HO formation via substance P and calcitonin gene-related peptide. BMP2 may induce neurogenic inflammation to remodel nerve and release HO precursor cells. May explain how HO occurs following traumatic brain injury. Mice lacking sensory neurons cells do not develop HO. Tie2+ endoneurial progenitors the major HO cell contributors in a mice model; however, Tie2 marker is also expressed in endothelial and mesenchymal cells. | [66,67,68,69] |
| Transient brown adipocyte-like cells | Adipose | Specialized pool of brown adipocytes that contribute to HO. Associated to deposition of cartilage. Detected in human traumatic injury-induced HO. | [70,71] |
4. Signalling Pathways in HO
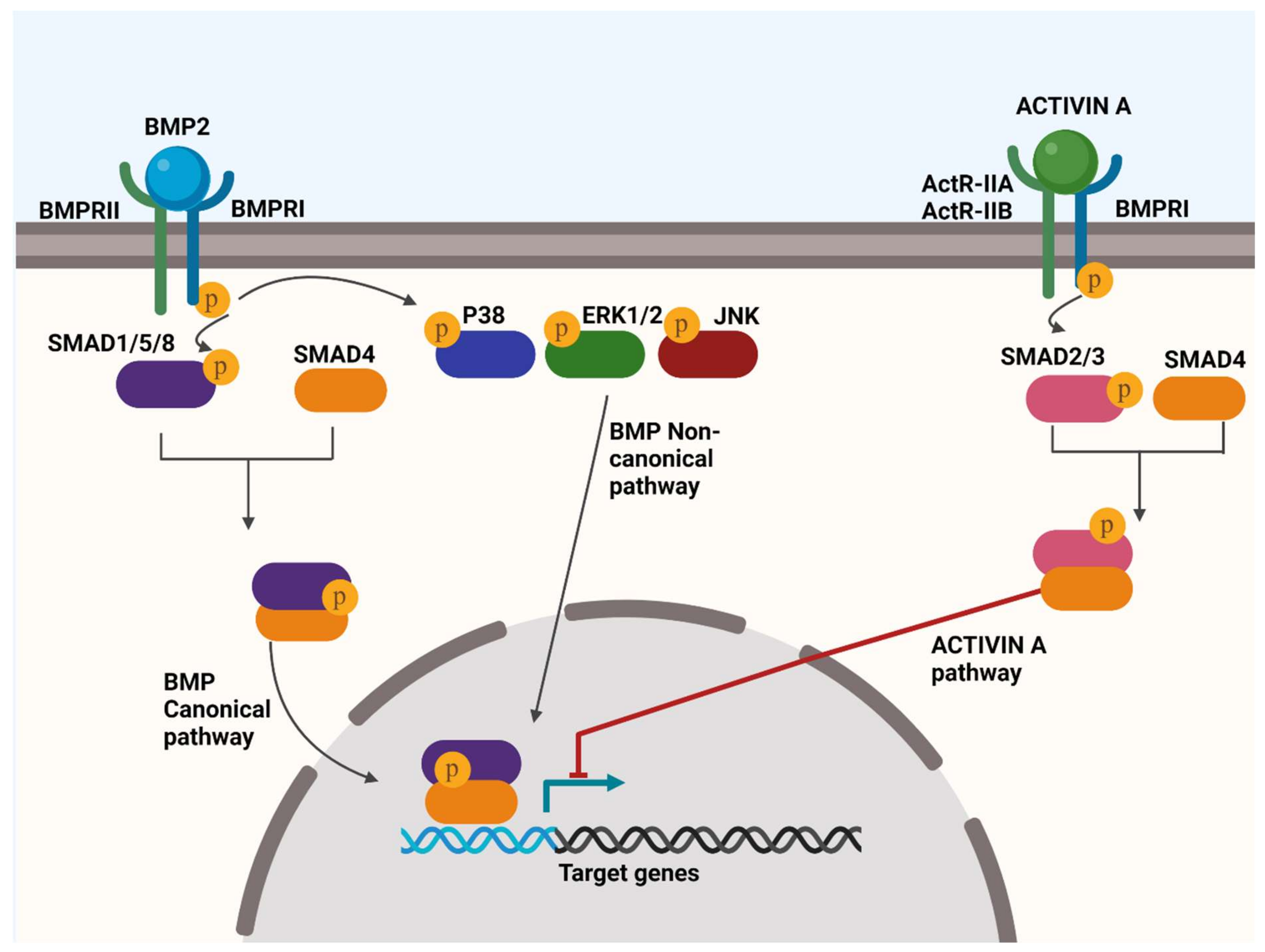
4.1. BMP Signalling
| Signalling Protein | Function | Key Papers |
|---|---|---|
| BMP1 | Bone formation and homeostasis. | [74] |
| BMP2 | Induces bone and cartilage development. Induces EndMT transition. Also involved in hedgehog pathway, cardiac cell differentiation, embryonic development. | [75,76,77,78] |
| BMP3 | Bone and cartilage development; antagonizes other BMPs in osteo-differentiation. | [79] |
| BMP4 | Potently induces chondro- and osteogenic differentiation; induces EndMT transition. Also involved in embryonic development, adipogenesis, neurogenesis. | [80,81,82,83] |
| BMP5 | Bone and cartilage development; may play a role in some cancer types; expressed in the visual apparatus. | [84,85,86] |
| BMP6 | Osteogenic differentiation; closely related to BMP5 and BMP7; regulates iron metabolism | [87,88,89] |
| BMP7 | Bone homeostasis; induces osteoblast differentiation through SMAD canonical pathway; involved in embryonic development, adipogenesis. | [90,91,92] |
| BMP8 | Expressed in developing skeleton; osteogenesis and germ cell generation. | [93,94,95,96] |
| BMP9/GDF2 | Induces chondro- and osteogenesis; cannot be blocked by BMP3 unlike most BMPs; involved in lymphatic development. | [97,98,99] |
| BMP10 | Involved in the trabeculation oof the heart and regulates monocyte recruitment to the vascular endothelium. | [100,101,102] |
| BMP11/GDF11 | Augments bone formation; induces embryonic development. | [103,104] |
| BMP12/GDF7 | Inhibits endochondral bone growth; induces tenogenic differentiation; regulates bone structure | [105] |
| BMP13/GDF6/CDMP2 | Establishes the boundaries between skeletal elements during development; induces tenogenic differentiation | [105,106] |
| BMP14/GDF5/CDMP1 | Regulates skeletal development and joint formation; promotes fracture healing. | [106,107,108] |
| BMP15 | Involved in fertilization and ovulation | [109,110] |
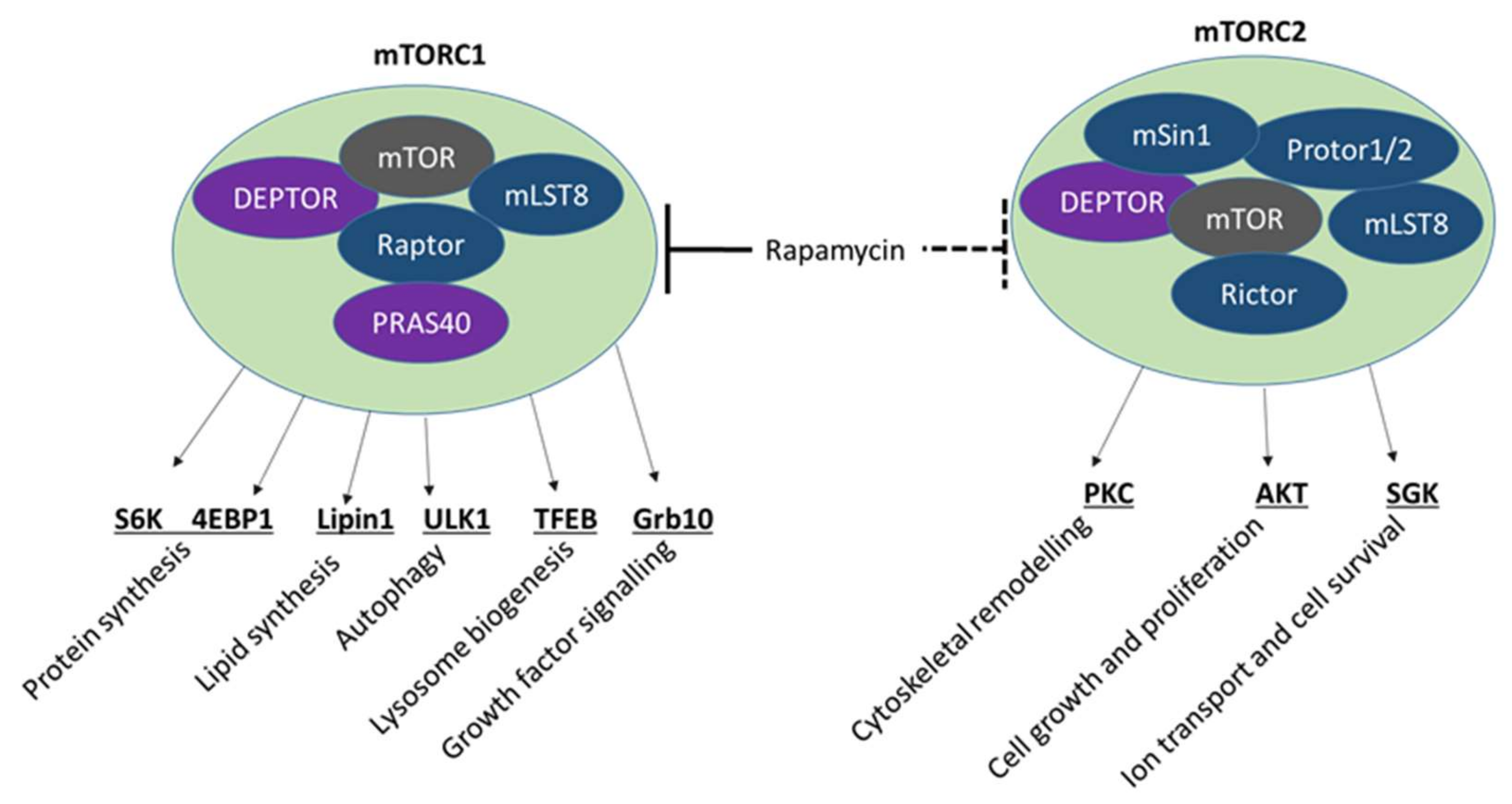
4.2. mTOR Signalling
4.3. Other Signalling Pathways
5. Therapeutic Strategies for HO
5.1. Palovarotene and Other RAR Agonists
5.2. Targeting ACVR1/ALK2 and Other Related Signalling Pathways
| Type of HO Pathways | Type of Molecule | Molecule | Description and Function | Key Papers |
|---|---|---|---|---|
| Antibody | REGN2477 (Garetosmab) | Anti-activin-A human monoclonal antibody in phase 2 clinical trial for FOP (LUMINA-1 study, NCT03188666). Blocks signalling of activin A, AB, and AC. Inhibits HO in animal model of FOP. | [179,185,186,187] | |
| FOP | Antibody | Perhexiline maleate (Pex) | Identified in screening of 1040 FDA-approved drugs for suppression of the Id1 promoter activated by mutant ACVR1/ALK2 in mouse C2C12 myoblasts. Pex reduced HO volume in BMP-induced mouse model, but failed to inhibit HO in an open-label clinical trial in FOP. | [188,189] |
| tHO | Antibody | Metformin | Regulates osteogenic differentiation via AMPK, and RUNX2/CBFA1 in vitro and in vivo. Prevents traumatic HO in mouse by decreasing ALK2 and AMPK regulation of Smad2. | [190,191,192] |
| FOP | Alpha-2 blocker | Fendiline hydrochloride | Identified in screen of 1040 FDA-approved drugs for suppression of the Id1 promoter activated by mutant ACVR1/ALK2. Mice administered with fendiline showed a slight reduction in HO. | [188] |
| FOP | Small molecule inhibitor | Dorsomorphin | Identified by chemical library screen for small molecules that dorsalise zebrafish embryos. Selectively inhibited ALK2 to block BMP-mediated SMAD1/5/8 phosphorylation. Preclinical use precluded by the inhibition of other ALKs (ALK3 and ALK6) and other kinases. | [176,193] |
| FOP, tHO | Small molecule inhibitor | LDN-193189 | An optimised version of dorsomorphin with greater potency and selectivity. Inhibits transcriptional activity of ALK2, ALK3, and constitutively active ALK2 mutant proteins. | [124] |
| FOP, tHO | Small molecule inhibitor | LDN-212854 | Derivative of dorsomorphin with increased selectivity for ALK2. LDN-212854 and LDN-193189 reduce osteogenic differentiation of tissue-resident MPCs from injured tissue following burn or tenotomy insult in animal model. In a blast-induced rat tHO model, LDN193189 and LDN212854 effective at limiting tHO. | [194,195] |
| FOP, tHO | Small molecule inhibitor | Other dorsomorphin derivatives | Currently undergoing investigation, including K02288, DMH-1, ML347, LDN 214117 and VU465350. | [196,197,198] |
| FOP | Small-molecule inhibitor | Saracatinib (AZD-0530) | Identified by screening compounds in an ALK2-mutated chondrogenic ATDC5 cell line. Inhibited both BMP and TGF-β signalling in vivo. Currently undergoing phase 2 clinical trial for FOP (NCT04307953). Well tolerated and potently inhibits the development of HO in inducible ALKQ207D transgenic and ACVR1R206H knock-in mouse. | [199,200,201,202] |
| FOP | Small-molecule inhibitor | PD 161570 | Identified by screening compounds in an ALK2-mutated chondrogenic ATDC5 cell line. Inhibits both BMP and TGF-β signalling in vivo. | [199] |
| FOP | Small-molecule inhibitor | TAK 165 | Identified by screening compounds in an ALK2-mutated chondrogenic ATDC5 cell line. Indirectly modulates mTOR signalling in vivo. | [199] |
| FOP | Ligand traps | sActR-IIA-Fc and sActR-IIB-Fc | ACVR1-Fc fusion proteins comprising the extracellular domain of human WT ACVR1 and the Fc portion of human immunoglobulin γ1. Inhibits dysregulated BMP signalling caused by FOP mutant ACVR1 and abrogates chondro-osseous differentiation in vitro. | [203,204,205] |
| FOP | Platelet inhibitor | Dipyridamole | Identified in screening of 1280 FDA-approved compounds for suppression of ACVR1 gene expression. Showed the highest inhibitory effect on SMAD signalling, chondrogenic and osteogenic differentiation in vitro. Reduced HO in BMP-induced model in mice. | [206,207] |
| FOP, tHO | Nucleotides | microRNAs | Altered expression of miRNA detected in HO. mir148b and mir365 down-regulate ACVR1/Alk-2 expression, whereas mir26a showed a positive effect on its mRNA. Inhibition of miRNAs, miR-146b-5p and -424 suppresses osteocyte maturation. Manipulating miR-574-3p levels both in vitro and in vivo inhibits chondrogenesis. miR-630 downregulated in early HO and used to distinguish HO from other processes in tHO. miR-17-5p upregulated in ankylosing spondylitis (AS) patients versus non-AS individuals. Knockdown and overexpression of miR-17-5p in fibroblasts derived from AS patients modulates osteogenesis. | [208,209,210,211,212,213,214] |
| FOP, tHO | Nucleotides | Antisense oligonucleotide (AON) | AON binds to specific exons in the primary mRNA transcript to prevent splicing and enable the skipping of specific exons. AONs designed to knockdown ALK2 expression in mice impair ALK2 signalling in both C2C12 end endothelial cells. However, AON affects both wild-type and mutated allele. | [215,216,217] |
| FOP, tHO | Nucleotides | RNA interference (RNAi) | Allele-specific siRNA (ASP-RNAi) duplexes tested for specific inhibition of mutant c.617A allele in mesenchymal progenitor cells from FOP patients. ASP-RNAi decreased BMP signalling to control cell levels. | [218,219] |
| tHO | Nucleotides | LncRNAs | Several lncRNAs regulate bone formation. Downregulation of MANCR inhibits osteoinduction in vitro. In a mouse in vivo tHO model, Brd4-Mancr signalling attenuated HO. | [220,221,222] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dejerine, A.; Ceillier, A. Paraosteoarthropathies of paraplegic patients by spinal cord lesion. Clinical and roentgenographic study. Clin. Orthop. Relat. Res. 1991, 263, 3–12. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Hsiao, E.C.; Baujat, G.; Lapidus, D.; Sherman, A.; Kaplan, F.S. Prevalence of fibrodysplasia ossificans progressiva (FOP) in the United States: Estimate from three treatment centers and a patient organization. Orphanet J. Rare Dis. 2021, 16, 350. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Kaplan, F.S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 2010, 6, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Craver, R.; MacEwen, G.D.; Gannon, F.H.; Finkel, G.; Hahn, G.; Tabas, J.; Gardner, R.J.; Zasloff, M.A. Progressive osseous heteroplasia: A distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. J. Bone Jt. Surg. Am. 1994, 76, 425–436. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Shore, E.M. Progressive osseous heteroplasia. J. Bone Miner. Res. 2000, 15, 2084–2094. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Zasloff, M.A.; Kitterman, J.A.; Shore, E.M.; Hong, C.C.; Rocke, D.M. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J. Bone Jt. Surg. Am. 2010, 92, 686–691. [Google Scholar] [CrossRef]
- Vanden Bossche, L.; Vanderstraeten, G. Heterotopic ossification: A review. J. Rehabil. Med. 2005, 37, 129–136. [Google Scholar] [CrossRef]
- Cipriano, C.A.; Pill, S.G.; Keenan, M.A. Heterotopic ossification following traumatic brain injury and spinal cord injury. J. Am. Acad. Orthop. Surg. 2009, 17, 689–697. [Google Scholar] [CrossRef]
- Brooker, A.F.; Bowerman, J.W.; Robinson, R.A.; Riley, L.H., Jr. Ectopic ossification following total hip arthroplasty. Incidence and method of classification. J. Bone Jt. Surg. Am. 1973, 55, 1629–1632. [Google Scholar] [CrossRef]
- Potter, B.K.; Burns, T.C.; Lacap, A.P.; Granville, R.R.; Gajewski, D.A. Heterotopic ossification following traumatic and combat-related amputations. Prevalence, risk factors, and preliminary results of excision. J. Bone Jt. Surg. Am. 2007, 89, 476–486. [Google Scholar] [CrossRef]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Buck, D.W., 2nd; Dumanian, G.A. Bone biology and physiology: Part I. The fundamentals. Plast. Reconstr. Surg. 2012, 129, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Shehab, D.; Elgazzar, A.H.; Collier, B.D. Heterotopic ossification. J. Nucl. Med. 2002, 43, 346–353. [Google Scholar] [PubMed]
- Chalmers, J.; Gray, D.H.; Rush, J. Observations on the induction of bone in soft tissues. J. Bone Jt. Surg. Br 1975, 57, 36–45. [Google Scholar] [CrossRef]
- Kan, L.; Kessler, J.A. Evaluation of the cellular origins of heterotopic ossification. Orthopedics 2014, 37, 329–340. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Goldhamer, D.J. Stem cells and heterotopic ossification: Lessons from animal models. Bone 2018, 109, 178–186. [Google Scholar] [CrossRef]
- Friedenstein, A.Y.; Lalykina, K.S. Lymphoid cell populations are competent systems for induced osteogenesis. Calcif. Tissue Res. 1970, 4, 105–106. [Google Scholar] [CrossRef]
- Shafritz, A.B.; Shore, E.M.; Gannon, F.H.; Zasloff, M.A.; Taub, R.; Muenke, M.; Kaplan, F.S. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N. Engl. J. Med. 1996, 335, 555–561. [Google Scholar] [CrossRef]
- Olmsted-Davis, E.A.; Gugala, Z.; Camargo, F.; Gannon, F.H.; Jackson, K.; Kienstra, K.A.; Shine, H.D.; Lindsey, R.W.; Hirschi, K.K.; Goodell, M.A.; et al. Primitive adult hematopoietic stem cells can function as osteoblast precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 15877–15882. [Google Scholar] [CrossRef]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [CrossRef]
- Jackson, K.A.; Majka, S.M.; Wang, H.; Pocius, J.; Hartley, C.J.; Majesky, M.W.; Entman, M.L.; Michael, L.H.; Hirschi, K.K.; Goodell, M.A. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J. Clin. Investig. 2001, 107, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Pritchard, C.; Garlits, J.E.; Hofmann, T.J.; Persons, D.A.; Horwitz, E.M. Hematopoietic cells and osteoblasts are derived from a common marrow progenitor after bone marrow transplantation. Proc. Natl. Acad. Sci. USA 2004, 101, 11761–11766. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Glaser, D.L.; Shore, E.M.; Pignolo, R.J.; Xu, M.; Zhang, Y.; Senitzer, D.; Forman, S.J.; Emerson, S.G. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J. Bone Jt. Surg. Am. 2007, 89, 347–357. [Google Scholar] [CrossRef]
- Otsuru, S.; Tamai, K.; Yamazaki, T.; Yoshikawa, H.; Kaneda, Y. Bone marrow-derived osteoblast progenitor cells in circulating blood contribute to ectopic bone formation in mice. Biochem. Biophys. Res. Commun. 2007, 354, 453–458. [Google Scholar] [CrossRef]
- Otsuru, S.; Tamai, K.; Yamazaki, T.; Yoshikawa, H.; Kaneda, Y. Circulating bone marrow-derived osteoblast progenitor cells are recruited to the bone-forming site by the CXCR4/stromal cell-derived factor-1 pathway. Stem Cells 2008, 26, 223–234. [Google Scholar] [CrossRef]
- Egan, K.P.; Duque, G.; Keenan, M.A.; Pignolo, R.J. Circulating osteogentic precursor cells in non-hereditary heterotopic ossification. Bone 2018, 109, 61–64. [Google Scholar] [CrossRef]
- Lounev, V.Y.; Ramachandran, R.; Wosczyna, M.N.; Yamamoto, M.; Maidment, A.D.; Shore, E.M.; Glaser, D.L.; Goldhamer, D.J.; Kaplan, F.S. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Jt. Surg. Am. 2009, 91, 652–663. [Google Scholar] [CrossRef]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- De Angelis, L.; Berghella, L.; Coletta, M.; Lattanzi, L.; Zanchi, M.; Cusella-De Angelis, M.G.; Ponzetto, C.; Cossu, G. Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J. Cell Biol. 1999, 147, 869–878. [Google Scholar] [CrossRef]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Loder, S.; Cholok, D.; Peterson, J.; Li, J.; Fireman, D.; Breuler, C.; Hsieh, H.S.; Ranganathan, K.; Hwang, C.; et al. Local and Circulating Endothelial Cells Undergo Endothelial to Mesenchymal Transition (EndMT) in Response to Musculoskeletal Injury. Sci. Rep. 2016, 6, 32514. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Li, T.; Shen, B.; Cao, X.; Zhong, H.; Zhang, L.; Zhou, F.; Ma, W.; Jiang, H.; Xie, P.; et al. Angiopoietin receptor Tie2 is required for vein specification and maintenance via regulating COUP-TFII. eLife 2016, 5, e21032. [Google Scholar] [CrossRef]
- Sato, A.; Iwama, A.; Takakura, N.; Nishio, H.; Yancopoulos, G.D.; Suda, T. Characterization of TEK receptor tyrosine kinase and its ligands, Angiopoietins, in human hematopoietic progenitor cells. Int. Immunol. 1998, 10, 1217–1227. [Google Scholar] [CrossRef]
- Yano, M.; Iwama, A.; Nishio, H.; Suda, J.; Takada, G.; Suda, T. Expression and function of murine receptor tyrosine kinases, TIE and TEK, in hematopoietic stem cells. Blood 1997, 89, 4317–4326. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef]
- Agarwal, S.; Loder, S.; Brownley, C.; Cholok, D.; Mangiavini, L.; Li, J.; Breuler, C.; Sung, H.H.; Li, S.; Ranganathan, K.; et al. Inhibition of Hif1alpha prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl. Acad. Sci. USA 2016, 113, E338–E347. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Loder, S.; Cholok, D.; Li, J.; Breuler, C.; Drake, J.; Brownley, C.; Peterson, J.; Li, S.; Levi, B. Surgical Excision of Heterotopic Ossification Leads to Re-Emergence of Mesenchymal Stem Cell Populations Responsible for Recurrence. Stem Cells Transl. Med. 2017, 6, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Loder, S.J.; Cholok, D.; Peterson, J.; Li, J.; Breuler, C.; Brownley, R.C.; Sung, H.H.; Chung, M.T.; Kamiya, N.; et al. Scleraxis-Lineage Cells Contribute to Ectopic Bone Formation in Muscle and Tendon. Stem Cells 2017, 35, 705–710. [Google Scholar] [CrossRef]
- Eisner, C.; Cummings, M.; Johnston, G.; Tung, L.W.; Groppa, E.; Chang, C.; Rossi, F.M. Murine Tissue-Resident PDGFRα+ Fibro-Adipogenic Progenitors Spontaneously Acquire Osteogenic Phenotype in an Altered Inflammatory Environment. J. Bone Miner. Res. 2020, 35, 1525–1534. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Yamaguchi, A.; Komaki, M.; Abe, E.; Takahashi, N.; Ikeda, T.; Rosen, V.; Wozney, J.M.; Fujisawa-Sehara, A.; Suda, T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 1994, 127, 1755–1766. [Google Scholar] [CrossRef]
- Hashimoto, N.; Kiyono, T.; Wada, M.R.; Umeda, R.; Goto, Y.; Nonaka, I.; Shimizu, S.; Yasumoto, S.; Inagawa-Ogashiwa, M. Osteogenic properties of human myogenic progenitor cells. Mech. Dev. 2008, 125, 257–269. [Google Scholar] [CrossRef]
- Kan, L.; Liu, Y.; McGuire, T.L.; Berger, D.M.; Awatramani, R.B.; Dymecki, S.M.; Kessler, J.A. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells 2009, 27, 150–156. [Google Scholar] [CrossRef]
- Matthews, B.G.; Torreggiani, E.; Roeder, E.; Matic, I.; Grcevic, D.; Kalajzic, I. Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone 2016, 84, 69–77. [Google Scholar] [CrossRef]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef]
- Winbanks, C.E.; Chen, J.L.; Qian, H.; Liu, Y.; Bernardo, B.C.; Beyer, C.; Watt, K.I.; Thomson, R.E.; Connor, T.; Turner, B.J.; et al. The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J. Cell Biol. 2013, 203, 345–357. [Google Scholar] [CrossRef]
- Liu, R.; Ginn, S.L.; Lek, M.; North, K.N.; Alexander, I.E.; Little, D.G.; Schindeler, A. Myoblast sensitivity and fibroblast insensitivity to osteogenic conversion by BMP-2 correlates with the expression of Bmpr-1a. BMC Musculoskelet. Disord. 2009, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Wright, V.; Peng, H.; Usas, A.; Young, B.; Gearhart, B.; Cummins, J.; Huard, J. BMP4-expressing muscle-derived stem cells differentiate into osteogenic lineage and improve bone healing in immunocompetent mice. Mol. Ther. 2002, 6, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rathbone, C.R. Satellite cell functional alterations following cutaneous burn in rats include an increase in their osteogenic potential. J. Surg. Res. 2013, 184, e9–e16. [Google Scholar] [CrossRef]
- Pagani, C.A.; Huber, A.K.; Hwang, C.; Marini, S.; Padmanabhan, K.; Livingston, N.; Nunez, J.; Sun, Y.; Edwards, N.; Cheng, Y.H.; et al. Novel Lineage-Tracing System to Identify Site-Specific Ectopic Bone Precursor Cells. Stem Cell Rep. 2021, 16, 626–640. [Google Scholar] [CrossRef]
- Pineault, K.M.; Song, J.Y.; Kozloff, K.M.; Lucas, D.; Wellik, D.M. Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat. Commun. 2019, 10, 3168. [Google Scholar] [CrossRef]
- Swinehart, I.T.; Schlientz, A.J.; Quintanilla, C.A.; Mortlock, D.P.; Wellik, D.M. Hox11 genes are required for regional patterning and integration of muscle, tendon and bone. Development 2013, 140, 4574–4582. [Google Scholar] [CrossRef] [PubMed]
- Rux, D.R.; Song, J.Y.; Swinehart, I.T.; Pineault, K.M.; Schlientz, A.J.; Trulik, K.G.; Goldstein, S.A.; Kozloff, K.M.; Lucas, D.; Wellik, D.M. Regionally Restricted Hox Function in Adult Bone Marrow Multipotent Mesenchymal Stem/Stromal Cells. Dev. Cell 2016, 39, 653–666. [Google Scholar] [CrossRef]
- Suda, R.K.; Billings, P.C.; Egan, K.P.; Kim, J.H.; McCarrick-Walmsley, R.; Glaser, D.L.; Porter, D.L.; Shore, E.M.; Pignolo, R.J. Circulating osteogenic precursor cells in heterotopic bone formation. Stem Cells 2009, 27, 2209–2219. [Google Scholar] [CrossRef]
- Meyers, C.; Lisiecki, J.; Miller, S.; Levin, A.; Fayad, L.; Ding, C.; Sono, T.; McCarthy, E.; Levi, B.; James, A.W. Heterotopic Ossification: A Comprehensive Review. JBMR Plus 2019, 3, e10172. [Google Scholar] [CrossRef] [PubMed]
- Grcevic, D.; Pejda, S.; Matthews, B.G.; Repic, D.; Wang, L.; Li, H.; Kronenberg, M.S.; Jiang, X.; Maye, P.; Adams, D.J.; et al. In vivo fate mapping identifies mesenchymal progenitor cells. Stem Cells 2012, 30, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Peng, C.Y.; McGuire, T.L.; Kessler, J.A. Glast-expressing progenitor cells contribute to heterotopic ossification. Bone 2013, 53, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Slezak, M.; Goritz, C.; Niemiec, A.; Frisen, J.; Chambon, P.; Metzger, D.; Pfrieger, F.W. Transgenic mice for conditional gene manipulation in astroglial cells. Glia 2007, 55, 1565–1576. [Google Scholar] [CrossRef]
- Howell, K.; Chien, C.; Bell, R.; Laudier, D.; Tufa, S.F.; Keene, D.R.; Andarawis-Puri, N.; Huang, A.H. Novel Model of Tendon Regeneration Reveals Distinct Cell Mechanisms Underlying Regenerative and Fibrotic Tendon Healing. Sci. Rep. 2017, 7, 45238. [Google Scholar] [CrossRef]
- Kan, L.; Lounev, V.Y.; Pignolo, R.J.; Duan, L.; Liu, Y.; Stock, S.R.; McGuire, T.L.; Lu, B.; Gerard, N.P.; Shore, E.M.; et al. Substance P signaling mediates BMP-dependent heterotopic ossification. J. Cell Biochem. 2011, 112, 2759–2772. [Google Scholar] [CrossRef]
- Lazard, Z.W.; Olmsted-Davis, E.A.; Salisbury, E.A.; Gugala, Z.; Sonnet, C.; Davis, E.L.; Beal, E., 2nd; Ubogu, E.E.; Davis, A.R. Osteoblasts Have a Neural Origin in Heterotopic Ossification. Clin. Orthop. Relat. Res. 2015, 473, 2790–2806. [Google Scholar] [CrossRef]
- Salisbury, E.; Rodenberg, E.; Sonnet, C.; Hipp, J.; Gannon, F.H.; Vadakkan, T.J.; Dickinson, M.E.; Olmsted-Davis, E.A.; Davis, A.R. Sensory nerve induced inflammation contributes to heterotopic ossification. J. Cell. Biochem. 2011, 112, 2748–2758. [Google Scholar] [CrossRef]
- Salisbury, E.; Sonnet, C.; Heggeness, M.; Davis, A.R.; Olmsted-Davis, E. Heterotopic ossification has some nerve. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 313–324. [Google Scholar] [CrossRef]
- Olmsted-Davis, E.; Gannon, F.H.; Ozen, M.; Ittmann, M.M.; Gugala, Z.; Hipp, J.A.; Moran, K.M.; Fouletier-Dilling, C.M.; Schumara-Martin, S.; Lindsey, R.W.; et al. Hypoxic adipocytes pattern early heterotopic bone formation. Am. J. Pathol. 2007, 170, 620–632. [Google Scholar] [CrossRef]
- Salisbury, E.A.; Dickerson, A.R.; Davis, T.A.; Forsberg, J.A.; Davis, A.R.; Olmsted-Davis, E.A. Characterization of Brown Adipose-Like Tissue in Trauma-Induced Heterotopic Ossification in Humans. Am. J. Pathol. 2017, 187, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Asharani, P.V.; Keupp, K.; Semler, O.; Wang, W.; Li, Y.; Thiele, H.; Yigit, G.; Pohl, E.; Becker, J.; Frommolt, P.; et al. Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. Am. J. Hum. Genet. 2012, 90, 661–674. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-beta/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef]
- Zhang, H.; Bradley, A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 1996, 122, 2977–2986. [Google Scholar] [CrossRef]
- Faucheux, C.; Ulysse, F.; Bareille, R.; Reddi, A.H.; Amedee, J. Opposing actions of BMP3 and TGF beta 1 in human bone marrow stromal cell growth and differentiation. Biochem. Biophys. Res. Commun. 1997, 241, 787–793. [Google Scholar] [CrossRef]
- Cole, A.E.; Murray, S.S.; Xiao, J. Bone Morphogenetic Protein 4 Signalling in Neural Stem and Progenitor Cells during Development and after Injury. Stem Cells Int. 2016, 2016, 9260592. [Google Scholar] [CrossRef]
- Lee, T.J.; Jang, J.; Kang, S.; Jin, M.; Shin, H.; Kim, D.W.; Kim, B.S. Enhancement of osteogenic and chondrogenic differentiation of human embryonic stem cells by mesodermal lineage induction with BMP-4 and FGF2 treatment. Biochem. Biophys. Res. Commun. 2013, 430, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Wolfrum, C. The dual role of BMP4 in adipogenesis and metabolism. Adipocyte 2017, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Le Douarin, N.M. A role for BMP-4 in the development of subcutaneous cartilage. Mech. Dev. 1996, 57, 69–78. [Google Scholar] [CrossRef]
- Bobinac, D.; Maric, I.; Zoricic, S.; Spanjol, J.; Dordevic, G.; Mustac, E.; Fuckar, Z. Expression of bone morphogenetic proteins in human metastatic prostate and breast cancer. Croat. Med. J. 2005, 46, 389–396. [Google Scholar] [PubMed]
- Guenther, C.A.; Wang, Z.; Li, E.; Tran, M.C.; Logan, C.Y.; Nusse, R.; Pantalena-Filho, L.; Yang, G.P.; Kingsley, D.M. A distinct regulatory region of the Bmp5 locus activates gene expression following adult bone fracture or soft tissue injury. Bone 2015, 77, 31–41. [Google Scholar] [CrossRef]
- Wordinger, R.J.; Agarwal, R.; Talati, M.; Fuller, J.; Lambert, W.; Clark, A.F. Expression of bone morphogenetic proteins (BMP), BMP receptors, and BMP associated proteins in human trabecular meshwork and optic nerve head cells and tissues. Mol. Vis. 2002, 8, 241–250. [Google Scholar]
- Camaschella, C. BMP6 orchestrates iron metabolism. Nat. Genet. 2009, 41, 386–388. [Google Scholar] [CrossRef]
- Gitelman, S.E.; Kobrin, M.S.; Ye, J.Q.; Lopez, A.R.; Lee, A.; Derynck, R. Recombinant Vgr-1/BMP-6-expressing tumors induce fibrosis and endochondral bone formation in vivo. J. Cell Biol. 1994, 126, 1595–1609. [Google Scholar] [CrossRef]
- Hahn, G.V.; Cohen, R.B.; Wozney, J.M.; Levitz, C.L.; Shore, E.M.; Zasloff, M.A.; Kaplan, F.S. A bone morphogenetic protein subfamily: Chromosomal localization of human genes for BMP5, BMP6, and BMP7. Genomics 1992, 14, 759–762. [Google Scholar] [CrossRef]
- Dudley, A.T.; Lyons, K.M.; Robertson, E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995, 9, 2795–2807. [Google Scholar] [CrossRef]
- Itoh, F.; Asao, H.; Sugamura, K.; Heldin, C.H.; ten Dijke, P.; Itoh, S. Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J. 2001, 20, 4132–4142. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- DiLeone, R.J.; King, J.A.; Storm, E.E.; Copeland, N.G.; Jenkins, N.A.; Kingsley, D.M. The Bmp8 gene is expressed in developing skeletal tissue and maps near the Achondroplasia locus on mouse chromosome 4. Genomics 1997, 40, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Liu, X.M.; Marble, A.; Lawson, K.A.; Zhao, G.Q. Requirement of Bmp8b for the generation of primordial germ cells in the mouse. Mol. Endocrinol. 2000, 14, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Q.; Deng, K.; Labosky, P.A.; Liaw, L.; Hogan, B.L. The gene encoding bone morphogenetic protein 8B is required for the initiation and maintenance of spermatogenesis in the mouse. Genes Dev. 1996, 10, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Q.; Liaw, L.; Hogan, B.L. Bone morphogenetic protein 8A plays a role in the maintenance of spermatogenesis and the integrity of the epididymis. Development 1998, 125, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Sun, M.H.; Cheng, H.; Peng, Y.; Montag, A.G.; Deyrup, A.T.; Jiang, W.; Luu, H.H.; Luo, J.; Szatkowski, J.P.; et al. Characterization of the distinct orthotopic bone-forming activity of 14 BMPs using recombinant adenovirus-mediated gene delivery. Gene Ther. 2004, 11, 1312–1320. [Google Scholar] [CrossRef]
- Levet, S.; Ciais, D.; Merdzhanova, G.; Mallet, C.; Zimmers, T.A.; Lee, S.J.; Navarro, F.P.; Texier, I.; Feige, J.J.; Bailly, S.; et al. Bone morphogenetic protein 9 (BMP9) controls lymphatic vessel maturation and valve formation. Blood 2013, 122, 598–607. [Google Scholar] [CrossRef]
- Majumdar, M.K.; Wang, E.; Morris, E.A. BMP-2 and BMP-9 promotes chondrogenic differentiation of human multipotential mesenchymal cells and overcomes the inhibitory effect of IL-1. J. Cell Physiol. 2001, 189, 275–284. [Google Scholar] [CrossRef]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef]
- Mitrofan, C.G.; Appleby, S.L.; Nash, G.B.; Mallat, Z.; Chilvers, E.R.; Upton, P.D.; Morrell, N.W. Bone morphogenetic protein 9 (BMP9) and BMP10 enhance tumor necrosis factor-alpha-induced monocyte recruitment to the vascular endothelium mainly via activin receptor-like kinase 2. J. Biol. Chem. 2017, 292, 13714–13726. [Google Scholar] [CrossRef]
- Neuhaus, H.; Rosen, V.; Thies, R.S. Heart specific expression of mouse BMP-10 a novel member of the TGF-beta superfamily. Mech. Dev. 1999, 80, 181–184. [Google Scholar] [CrossRef]
- Li, Z.; Zeng, F.; Mitchell, A.D.; Kim, Y.S.; Wu, Z.; Yang, J. Transgenic overexpression of bone morphogenetic protein 11 propeptide in skeleton enhances bone formation. Biochem. Biophys. Res. Commun. 2011, 416, 289–292. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Wei, Y.; Liu, D.; Liu, F.; Li, X.; Pan, L.; Pang, Y.; Chen, D. Role of growth differentiation factor 11 in development, physiology and disease. Oncotarget 2017, 8, 81604–81616. [Google Scholar] [CrossRef] [PubMed]
- Berasi, S.P.; Varadarajan, U.; Archambault, J.; Cain, M.; Souza, T.A.; Abouzeid, A.; Li, J.; Brown, C.T.; Dorner, A.J.; Seeherman, H.J.; et al. Divergent activities of osteogenic BMP2, and tenogenic BMP12 and BMP13 independent of receptor binding affinities. Growth Factors 2011, 29, 128–139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Settle, S.H., Jr.; Rountree, R.B.; Sinha, A.; Thacker, A.; Higgins, K.; Kingsley, D.M. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev. Biol. 2003, 254, 116–130. [Google Scholar] [CrossRef]
- Chhabra, A.; Zijerdi, D.; Zhang, J.; Kline, A.; Balian, G.; Hurwitz, S. BMP-14 deficiency inhibits long bone fracture healing: A biochemical, histologic, and radiographic assessment. J. Orthop. Trauma 2005, 19, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.M.; Scheremeta, B.H.; Boyce, A.T.; Mauck, R.L.; Tuan, R.S. Delayed fracture healing in growth differentiation factor 5-deficient mice: A pilot study. Clin. Orthop. Relat. Res. 2011, 469, 2915–2924. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Beck-Peccoz, P.; Persani, L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am. J. Hum. Genet. 2004, 75, 106–111. [Google Scholar] [CrossRef]
- Yan, C.; Wang, P.; DeMayo, J.; DeMayo, F.J.; Elvin, J.A.; Carino, C.; Prasad, S.V.; Skinner, S.S.; Dunbar, B.S.; Dube, J.L.; et al. Synergistic roles of bone morphogenetic protein 15 and growth differentiation factor 9 in ovarian function. Mol. Endocrinol. 2001, 15, 854–866. [Google Scholar] [CrossRef]
- Wang, M.; Jin, H.; Tang, D.; Huang, S.; Zuscik, M.J.; Chen, D. Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthr. Cartil. 2011, 19, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Iguchi, M.; Watanabe, K.; Hoshino, R.; Tsujimoto, M.; Kohno, M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogenetic protein-2. J. Biol. Chem. 1999, 274, 26503–26510. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Tu, Y.; Li, S.; Manske, P.R. Involvement of ERK in BMP-2 induced osteoblastic differentiation of mesenchymal progenitor cell line C3H10T1/2. Biochem. Biophys. Res. Commun. 2000, 268, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Shirai, T.; Morishita, S.; Uchida, S.; Saeki-Miura, K.; Makishima, F. p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp. Cell Res. 1999, 250, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Shirakabe, K.; Shibuya, H.; Irie, K.; Oishi, I.; Ueno, N.; Taniguchi, T.; Nishida, E.; Matsumoto, K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 1995, 270, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in Mammalian Physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef] [PubMed]
- de Caestecker, M.P.; Parks, W.T.; Frank, C.J.; Castagnino, P.; Bottaro, D.P.; Roberts, A.B.; Lechleider, R.J. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 1998, 12, 1587–1592. [Google Scholar] [CrossRef]
- Gingery, A.; Bradley, E.W.; Pederson, L.; Ruan, M.; Horwood, N.J.; Oursler, M.J. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp. Cell Res. 2008, 314, 2725–2738. [Google Scholar] [CrossRef]
- Hoffmann, A.; Preobrazhenska, O.; Wodarczyk, C.; Medler, Y.; Winkel, A.; Shahab, S.; Huylebroeck, D.; Gross, G.; Verschueren, K. Transforming growth factor-beta-activated kinase-1 (TAK1), a MAP3K, interacts with Smad proteins and interferes with osteogenesis in murine mesenchymal progenitors. J. Biol. Chem. 2005, 280, 27271–27283. [Google Scholar] [CrossRef]
- Nickel, J.; Mueller, T.D. Specification of BMP Signaling. Cells 2019, 8, 1579. [Google Scholar] [CrossRef]
- Evans, K.N.; Potter, B.K.; Brown, T.S.; Davis, T.A.; Elster, E.A.; Forsberg, J.A. Osteogenic gene expression correlates with development of heterotopic ossification in war wounds. Clin. Orthop. Relat. Res. 2014, 472, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Lv, Z.; Zhang, C.; Jiao, Y. Regulatory effect of miR-421 on humeral fracture and heterotopic ossification in elderly patients. Exp. Ther. Med. 2019, 17, 1903–1911. [Google Scholar] [CrossRef]
- Hannallah, D.; Peng, H.; Young, B.; Usas, A.; Gearhart, B.; Huard, J. Retroviral delivery of Noggin inhibits the formation of heterotopic ossification induced by BMP-4, demineralized bone matrix, and trauma in an animal model. J. Bone Jt. Surg. Am. 2004, 86, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Hu, M.; Gomes, W.A.; Kessler, J.A. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am. J. Pathol. 2004, 165, 1107–1115. [Google Scholar] [CrossRef]
- Scott, M.A.; Levi, B.; Askarinam, A.; Nguyen, A.; Rackohn, T.; Ting, K.; Soo, C.; James, A.W. Brief review of models of ectopic bone formation. Stem Cells Dev. 2012, 21, 655–667. [Google Scholar] [CrossRef]
- Ranganathan, K.; Loder, S.; Agarwal, S.; Wong, V.W.; Forsberg, J.; Davis, T.A.; Wang, S.; James, A.W.; Levi, B. Heterotopic Ossification: Basic-Science Principles and Clinical Correlates. J. Bone Jt. Surg. Am. 2015, 97, 1101–1111. [Google Scholar] [CrossRef]
- Chen, J.; Long, F. mTOR signaling in skeletal development and disease. Bone Res. 2018, 6, 1. [Google Scholar] [CrossRef]
- Phornphutkul, C.; Wu, K.Y.; Auyeung, V.; Chen, Q.; Gruppuso, P.A. mTOR signaling contributes to chondrocyte differentiation. Dev. Dyn 2008, 237, 702–712. [Google Scholar] [CrossRef]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef]
- Qureshi, A.T.; Dey, D.; Sanders, E.M.; Seavey, J.G.; Tomasino, A.M.; Moss, K.; Wheatley, B.; Cholok, D.; Loder, S.; Li, J.; et al. Inhibition of Mammalian Target of Rapamycin Signaling with Rapamycin Prevents Trauma-Induced Heterotopic Ossification. Am. J. Pathol. 2017, 187, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, Y.; Chen, G.; Tian, X.; Tang, J.; Luo, L.; Huang, M.; Yan, B.; Ao, X.; Zhou, W.; et al. Leptin accelerates the pathogenesis of heterotopic ossification in rat tendon tissues via mTORC1 signaling. J. Cell. Physiol. 2018, 233, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Frank, A.R.; Jewell, J.L. mTOR signaling in stem and progenitor cells. Development 2018, 145, dev152595. [Google Scholar] [CrossRef] [PubMed]
- Karner, C.M.; Lee, S.Y.; Long, F. Bmp Induces Osteoblast Differentiation through both Smad4 and mTORC1 Signaling. Mol. Cell. Biol. 2017, 37, e00253-16. [Google Scholar] [CrossRef]
- Chen, J.; Holguin, N.; Shi, Y.; Silva, M.J.; Long, F. mTORC2 signaling promotes skeletal growth and bone formation in mice. J. Bone Miner. Res. 2015, 30, 369–378. [Google Scholar] [CrossRef]
- Sun, W.; Shi, Y.; Lee, W.C.; Lee, S.Y.; Long, F. Rictor is required for optimal bone accrual in response to anti-sclerostin therapy in the mouse. Bone 2016, 85, 1–8. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 2007, 117, 1616–1626. [Google Scholar] [CrossRef]
- Galdones, E.; Hales, B.F. Retinoic acid receptor gamma-induced misregulation of chondrogenesis in the murine limb bud in vitro. Toxicol. Sci. 2008, 106, 223–232. [Google Scholar] [CrossRef]
- Romand, R.; Hashino, E.; Dolle, P.; Vonesch, J.L.; Chambon, P.; Ghyselinck, N.B. The retinoic acid receptors RARalpha and RARgamma are required for inner ear development. Mech. Dev. 2002, 119, 213–223. [Google Scholar] [CrossRef]
- Weston, A.D.; Chandraratna, R.A.; Torchia, J.; Underhill, T.M. Requirement for RAR-mediated gene repression in skeletal progenitor differentiation. J. Cell Biol. 2002, 158, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.D.; Rosen, V.; Chandraratna, R.A.; Underhill, T.M. Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. J. Cell Biol. 2000, 148, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Cash, D.E.; Bock, C.B.; Schughart, K.; Linney, E.; Underhill, T.M. Retinoic acid receptor alpha function in vertebrate limb skeletogenesis: A modulator of chondrogenesis. J. Cell Biol. 1997, 136, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.; Chen, L.; Hu, Y.; Ding, N.; Lu, H.; Li, Y.; Kessler, J.A.; Kan, L. Conserved signaling pathways underlying heterotopic ossification. Bone 2018, 109, 43–48. [Google Scholar] [CrossRef]
- Liu, X.; Qin, J.; Luo, Q.; Bi, Y.; Zhu, G.; Jiang, W.; Kim, S.H.; Li, M.; Su, Y.; Nan, G.; et al. Cross-talk between EGF and BMP9 signalling pathways regulates the osteogenic differentiation of mesenchymal stem cells. J. Cell. Mol. Med. 2013, 17, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef]
- Nakashima, A.; Katagiri, T.; Tamura, M. Cross-talk between Wnt and bone morphogenetic protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J. Biol. Chem. 2005, 280, 37660–37668. [Google Scholar] [CrossRef]
- Zhang, T.; Wen, F.; Wu, Y.; Goh, G.S.; Ge, Z.; Tan, L.P.; Hui, J.H.; Yang, Z. Cross-talk between TGF-beta/SMAD and integrin signaling pathways in regulating hypertrophy of mesenchymal stem cell chondrogenesis under deferral dynamic compression. Biomaterials 2015, 38, 72–85. [Google Scholar] [CrossRef]
- Pavlou, G.; Kyrkos, M.; Tsialogiannis, E.; Korres, N.; Tsiridis, E. Pharmacological treatment of heterotopic ossification following hip surgery: An update. Expert Opin. Pharm. 2012, 13, 619–622. [Google Scholar] [CrossRef]
- Joice, M.; Vasileiadis, G.I.; Amanatullah, D.F. Non-steroidal anti-inflammatory drugs for heterotopic ossification prophylaxis after total hip arthroplasty: A systematic review and meta-analysis. Bone Jt. J. 2018, 100-B, 915–922. [Google Scholar] [CrossRef]
- Kan, S.L.; Yang, B.; Ning, G.Z.; Chen, L.X.; Li, Y.L.; Gao, S.J.; Chen, X.Y.; Sun, J.C.; Feng, S.Q. Nonsteroidal Anti-inflammatory Drugs as Prophylaxis for Heterotopic Ossification after Total Hip Arthroplasty: A Systematic Review and Meta-Analysis. Medicine 2015, 94, e828. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, F.; Trivellas, A.; Eschweiler, J.; Driessen, A.; Tingart, M.; Maffulli, N. NSAIDs for Prophylaxis for Heterotopic Ossification After Total Hip Arthroplasty: A Bayesian Network Meta-analysis. Calcif. Tissue Int. 2021, 108, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.H.; Chen, W.; Sun, J.N.; Zhang, Y.; Zhang, Y.; Chen, X.Y.; Feng, S. Radiotherapy for the prophylaxis of heterotopic ossification after total hip arthroplasty: A systematic review and meta-analysis of randomized controlled trails. Med. Dosim. 2021, 46, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Popovic, M.; Agarwal, A.; Zhang, L.; Yip, C.; Kreder, H.J.; Nousiainen, M.T.; Jenkinson, R.; Tsao, M.; Lam, H.; Milakovic, M.; et al. Radiotherapy for the prophylaxis of heterotopic ossification: A systematic review and meta-analysis of published data. Radiother. Oncol. 2014, 113, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Sheybani, A.; TenNapel, M.J.; Lack, W.D.; Clerkin, P.; Hyer, D.E.; Sun, W.; Jacobson, G.M. Risk of radiation-induced malignancy with heterotopic ossification prophylaxis: A case-control analysis. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 584–589. [Google Scholar] [CrossRef]
- Balboni, T.A.; Gobezie, R.; Mamon, H.J. Heterotopic ossification: Pathophysiology, clinical features, and the role of radiotherapy for prophylaxis. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Hamid, N.; Ashraf, N.; Bosse, M.J.; Connor, P.M.; Kellam, J.F.; Sims, S.H.; Stull, D.E.; Jeray, K.J.; Hymes, R.A.; Lowe, T.J. Radiation therapy for heterotopic ossification prophylaxis acutely after elbow trauma: A prospective randomized study. J. Bone Jt. Surg. Am. 2010, 92, 2032–2038. [Google Scholar] [CrossRef]
- Pavey, G.J.; Polfer, E.M.; Nappo, K.E.; Tintle, S.M.; Forsberg, J.A.; Potter, B.K. What Risk Factors Predict Recurrence of Heterotopic Ossification After Excision in Combat-related Amputations? Clin. Orthop. Relat. Res. 2015, 473, 2814–2824. [Google Scholar] [CrossRef]
- Meiners, T.; Abel, R.; Bohm, V.; Gerner, H.J. Resection of heterotopic ossification of the hip in spinal cord injured patients. Spinal Cord 1997, 35, 443–445. [Google Scholar] [CrossRef]
- Lee, E.K.; Namdari, S.; Hosalkar, H.S.; Keenan, M.A.; Baldwin, K.D. Clinical results of the excision of heterotopic bone around the elbow: A systematic review. J. Shoulder Elb. Surg. 2013, 22, 716–722. [Google Scholar] [CrossRef]
- Thomas, B.J.; Amstutz, H.C. Results of the administration of diphosphonate for the prevention of heterotopic ossification after total hip arthroplasty. J. Bone Jt. Surg. Am. 1985, 67, 400–403. [Google Scholar] [CrossRef]
- Shafer, D.M.; Bay, C.; Caruso, D.M.; Foster, K.N. The use of eidronate disodium in the prevention of heterotopic ossification in burn patients. Burns 2008, 34, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Morrison, T.N.; Tung, W.E.; Chandraratna, R.A.; Williams, J.A.; Iwamoto, M.; Pacifici, M. Inhibition of ectopic bone formation by a selective retinoic acid receptor alpha-agonist: A new therapy for heterotopic ossification? J. Orthop. Res. 2010, 28, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Koyama, E.; Golden, E.B.; Kirsch, T.; Adams, S.L.; Chandraratna, R.A.; Michaille, J.J.; Pacifici, M. Retinoid signaling is required for chondrocyte maturation and endochondral bone formation during limb skeletogenesis. Dev. Biol. 1999, 208, 375–391. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weston, A.D.; Hoffman, L.M.; Underhill, T.M. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res. C Embryo Today 2003, 69, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice with the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Lees-Shepard, J.B.; Nicholas, S.E.; Stoessel, S.J.; Devarakonda, P.M.; Schneider, M.J.; Yamamoto, M.; Goldhamer, D.J. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. eLife 2018, 7, e40814. [Google Scholar] [CrossRef]
- Wheatley, B.M.; Cilwa, K.E.; Dey, D.; Qureshi, A.T.; Seavey, J.G.; Tomasino, A.M.; Sanders, E.M.; Bova, W.; Boehm, C.A.; Iwamoto, M.; et al. Palovarotene inhibits connective tissue progenitor cell proliferation in a rat model of combat-related heterotopic ossification. J. Orthop. Res. 2018, 36, 1135–1144. [Google Scholar] [CrossRef]
- Pavey, G.J.; Qureshi, A.T.; Tomasino, A.M.; Honnold, C.L.; Bishop, D.K.; Agarwal, S.; Loder, S.; Levi, B.; Pacifici, M.; Iwamoto, M.; et al. Targeted stimulation of retinoic acid receptor-gamma mitigates the formation of heterotopic ossification in an established blast-related traumatic injury model. Bone 2016, 90, 159–167. [Google Scholar] [CrossRef]
- Lebrun, J.J.; Takabe, K.; Chen, Y.; Vale, W. Roles of pathway-specific and inhibitory Smads in activin receptor signaling. Mol. Endocrinol. 1999, 13, 15–23. [Google Scholar] [CrossRef]
- Mueller, T.D.; Nickel, J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012, 586, 1846–1859. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jiang, W. The Role of SMAD2/3 in Human Embryonic Stem Cells. Front. Cell Dev. Biol. 2020, 8, 653. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wahdan-Alaswad, R.; Danielpour, D. Critical role of Smad2 in tumor suppression and transforming growth factor-beta-induced apoptosis of prostate epithelial cells. Cancer Res. 2009, 69, 2185–2190. [Google Scholar] [CrossRef]
- Culbert, A.L.; Chakkalakal, S.A.; Theosmy, E.G.; Brennan, T.A.; Kaplan, F.S.; Shore, E.M. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells 2014, 32, 1289–1300. [Google Scholar] [CrossRef]
- van Dinther, M.; Visser, N.; de Gorter, D.J.; Doorn, J.; Goumans, M.J.; de Boer, J.; ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef]
- Lyu, H.; Elkins, C.M.; Pierce, J.L.; Serezani, C.H.; Perrien, D.S. MyD88 Is Not Required for Muscle Injury-Induced Endochondral Heterotopic Ossification in a Mouse Model of Fibrodysplasia Ossificans Progressiva. Biomedicines 2021, 9, 630. [Google Scholar] [CrossRef]
- Lin, H.; Ying, Y.; Wang, Y.Y.; Wang, G.; Jiang, S.S.; Huang, D.; Luo, L.; Chen, Y.G.; Gerstenfeld, L.C.; Luo, Z. AMPK downregulates ALK2 via increasing the interaction between Smurf1 and Smad6, leading to inhibition of osteogenic differentiation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2369–2377. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Song, G.A.; Kim, H.J.; Woo, K.M.; Baek, J.H.; Kim, G.S.; Choi, J.Y.; Ryoo, H.M. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J. Biol. Chem. 2010, 285, 22542–22553. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Le Merrer, M.; Glaser, D.L.; Pignolo, R.J.; Goldsby, R.E.; Kitterman, J.A.; Groppe, J.; Shore, E.M. Fibrodysplasia ossificans progressiva. Best Pract. Res. Clin. Rheumatol. 2008, 22, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Shi, F.; Gao, J.; Hua, P. The role of Activin A in fibrodysplasia ossificans progressiva: A prominent mediator. Biosci. Rep. 2019, 39, BSR20190377. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.M.; Ramirez, M.R.; Reginato, A.M.; Medici, D. Molecular and cellular mechanisms of heterotopic ossification. Histol. Histopathol. 2014, 29, 1281–1285. [Google Scholar] [PubMed]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9, e54582. [Google Scholar] [CrossRef]
- Latres, E.; Mastaitis, J.; Fury, W.; Miloscio, L.; Trejos, J.; Pangilinan, J.; Okamoto, H.; Cavino, K.; Na, E.; Papatheodorou, A.; et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017, 8, 15153. [Google Scholar] [CrossRef]
- Vanhoutte, F.; Liang, S.; Ruddy, M.; Zhao, A.; Drewery, T.; Wang, Y.; DelGizzi, R.; Forleo-Neto, E.; Rajadhyaksha, M.; Herman, G.; et al. Pharmacokinetics and Pharmacodynamics of Garetosmab (Anti-Activin A): Results from a First-in-Human Phase 1 Study. J. Clin. Pharm. 2020, 60, 1424–1431. [Google Scholar] [CrossRef]
- Yamamoto, R.; Matsushita, M.; Kitoh, H.; Masuda, A.; Ito, M.; Katagiri, T.; Kawai, T.; Ishiguro, N.; Ohno, K. Clinically applicable antianginal agents suppress osteoblastic transformation of myogenic cells and heterotopic ossifications in mice. J. Bone Miner. Metab. 2013, 31, 26–33. [Google Scholar] [CrossRef]
- Kitoh, H.; Achiwa, M.; Kaneko, H.; Mishima, K.; Matsushita, M.; Kadono, I.; Horowitz, J.D.; Sallustio, B.C.; Ohno, K.; Ishiguro, N. Perhexiline maleate in the treatment of fibrodysplasia ossificans progressiva: An open-labeled clinical trial. Orphanet J. Rare Dis. 2013, 8, 163. [Google Scholar] [CrossRef]
- Molinuevo, M.S.; Schurman, L.; McCarthy, A.D.; Cortizo, A.M.; Tolosa, M.J.; Gangoiti, M.V.; Arnol, V.; Sedlinsky, C. Effect of metformin on bone marrow progenitor cell differentiation: In vivo and in vitro studies. J. Bone Miner. Res. 2010, 25, 211–221. [Google Scholar] [CrossRef]
- Jang, W.G.; Kim, E.J.; Bae, I.H.; Lee, K.N.; Kim, Y.D.; Kim, D.K.; Kim, S.H.; Lee, C.H.; Franceschi, R.T.; Choi, H.S.; et al. Metformin induces osteoblast differentiation via orphan nuclear receptor SHP-mediated transactivation of Runx2. Bone 2011, 48, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Shi, F.; Jiang, S.; Wang, Y.; Zou, J.; Ying, Y.; Huang, D.; Luo, L.; Yan, X.; Luo, Z. Metformin attenuates trauma-induced heterotopic ossification via inhibition of Bone Morphogenetic Protein signalling. J. Cell. Mol. Med. 2020, 24, 14491–14501. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Bullock, A.N. Structural basis for the potent and selective binding of LDN-212854 to the BMP receptor kinase ALK2. Bone 2018, 109, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.L.; Spreadborough, P.J.; Dey, D.; Yang, P.; Li, S.; Lee, A.; Haskins, R.M.; Grimm, P.D.; Kumar, R.; Bradley, M.J.; et al. BMP Ligand Trap ALK3-Fc Attenuates Osteogenesis and Heterotopic Ossification in Blast-Related Lower Extremity Trauma. Stem Cells Dev. 2021, 30, 91–105. [Google Scholar] [CrossRef]
- Mohedas, A.H.; Wang, Y.; Sanvitale, C.E.; Canning, P.; Choi, S.; Xing, X.; Bullock, A.N.; Cuny, G.D.; Yu, P.B. Structure-activity relationship of 3,5-diaryl-2-aminopyridine ALK2 inhibitors reveals unaltered binding affinity for fibrodysplasia ossificans progressiva causing mutants. J. Med. Chem. 2014, 57, 7900–7915. [Google Scholar] [CrossRef]
- Hao, J.; Ho, J.N.; Lewis, J.A.; Karim, K.A.; Daniels, R.N.; Gentry, P.R.; Hopkins, C.R.; Lindsley, C.W.; Hong, C.C. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem. Biol. 2010, 5, 245–253. [Google Scholar] [CrossRef]
- Tsugawa, D.; Oya, Y.; Masuzaki, R.; Ray, K.; Engers, D.W.; Dib, M.; Do, N.; Kuramitsu, K.; Ho, K.; Frist, A.; et al. Specific activin receptor-like kinase 3 inhibitors enhance liver regeneration. J. Pharm. Exp. Ther. 2014, 351, 549–558. [Google Scholar] [CrossRef]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef]
- Kitoh, H. Clinical Aspects and Current Therapeutic Approaches for FOP. Biomedicines 2020, 8, 325. [Google Scholar] [CrossRef]
- Hildebrandt, S.; Kampfrath, B.; Fischer, K.; Hildebrand, L.; Haupt, J.; Stachelscheid, H.; Knaus, P. ActivinA Induced SMAD1/5 Signaling in an iPSC Derived EC Model of Fibrodysplasia Ossificans Progressiva (FOP) Can Be Rescued by the Drug Candidate Saracatinib. Stem Cell Rev. Rep. 2021, 17, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Bagarova, J.; Kerr, G.; Xia, D.D.; Place, E.S.; Dey, D.; Shen, Y.; Bocobo, G.A.; Mohedas, A.H.; Huang, X.; et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. JCI Insight 2021, 6, e95042. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Zuo, Y.; Chen, Y.; Song, L.; Zhu, Q.; Yu, J.; Shan, C.; Cai, Z.; Hao, J.; Kaplan, F.S.; et al. ACVR1-Fc suppresses BMP signaling and chondro-osseous differentiation in an in vitro model of Fibrodysplasia ossificans progressiva. Bone 2016, 92, 29–36. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Pignolo, R.J.; Al Mukaddam, M.M.; Shore, E.M. Hard targets for a second skeleton: Therapeutic horizons for fibrodysplasia ossificans progressiva (FOP). Expert Opin. Orphan Drugs 2017, 5, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Pignolo, R.J.; Shore, E.M. From mysteries to medicines: Drug development for fibrodysplasia ossificans progressive. Expert Opin. Orphan Drugs 2013, 1, 637–649. [Google Scholar] [CrossRef]
- Cappato, S.; Tonachini, L.; Giacopelli, F.; Tirone, M.; Galietta, L.J.; Sormani, M.; Giovenzana, A.; Spinelli, A.E.; Canciani, B.; Brunelli, S.; et al. High-throughput screening for modulators of ACVR1 transcription: Discovery of potential therapeutics for fibrodysplasia ossificans progressiva. Dis. Model. Mech. 2016, 9, 685–696. [Google Scholar] [CrossRef]
- Tirone, M.; Giovenzana, A.; Vallone, A.; Zordan, P.; Sormani, M.; Nicolosi, P.A.; Meneveri, R.; Gigliotti, C.R.; Spinelli, A.E.; Bocciardi, R.; et al. Severe Heterotopic Ossification in the Skeletal Muscle and Endothelial Cells Recruitment to Chondrogenesis Are Enhanced by Monocyte/Macrophage Depletion. Front. Immunol. 2019, 10, 1640. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Lu, Y.Q.; Han, J.X. MicroRNAs: Important mediators of ossification. Chin. Med. J. 2012, 125, 4111–4116. [Google Scholar]
- Mura, M.; Cappato, S.; Giacopelli, F.; Ravazzolo, R.; Bocciardi, R. The role of the 3′UTR region in the regulation of the ACVR1/Alk-2 gene expression. PLoS ONE 2012, 7, e50958. [Google Scholar] [CrossRef]
- Oishi, T.; Uezumi, A.; Kanaji, A.; Yamamoto, N.; Yamaguchi, A.; Yamada, H.; Tsuchida, K. Osteogenic differentiation capacity of human skeletal muscle-derived progenitor cells. PLoS ONE 2013, 8, e56641. [Google Scholar] [CrossRef]
- Guerit, D.; Philipot, D.; Chuchana, P.; Toupet, K.; Brondello, J.M.; Mathieu, M.; Jorgensen, C.; Noel, D. Sox9-regulated miRNA-574-3p inhibits chondrogenic differentiation of mesenchymal stem cells. PLoS ONE 2013, 8, e62582. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cai, J.; Yu, S.; Chen, S.; Li, F.; Fan, C. MiR-630 Inhibits Endothelial-Mesenchymal Transition by Targeting Slug in Traumatic Heterotopic Ossification. Sci. Rep. 2016, 6, 22729. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhu, B.; Jiang, T.; Tan, J.; Wu, Z.; Yuan, Z.; Zheng, L.; Zhao, J. miR-17-5p Regulates Heterotopic Ossification by Targeting ANKH in Ankylosing Spondylitis. Mol. Nucleic Acids 2019, 18, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, Z.; Li, L.; Shen, X.; Sun, B.; Wang, R.; Zhong, H.; Shi, Q.; Wei, L.; Zhang, Y.; et al. MicroRNA-181 regulates the development of Ossification of Posterior longitudinal ligament via Epigenetic Modulation by targeting PBX1. Theranostics 2020, 10, 7492–7509. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- Shi, S.; Cai, J.; de Gorter, D.J.; Sanchez-Duffhues, G.; Kemaladewi, D.U.; Hoogaars, W.M.; Aartsma-Rus, A.; ’t Hoen, P.A.; ten Dijke, P. Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: Inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PLoS ONE 2013, 8, e69096. [Google Scholar] [CrossRef]
- Shi, F.; Gao, J.; Zou, J.; Ying, Y.; Lin, H. Targeting heterotopic ossification by inhibiting activin receptorlike kinase 2 function (Review). Mol. Med. Rep. 2019, 20, 2979–2989. [Google Scholar]
- Kaplan, J.; Kaplan, F.S.; Shore, E.M. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther. 2012, 19, 786–790. [Google Scholar] [CrossRef]
- Shrivats, A.R.; Hsu, E.; Averick, S.; Klimak, M.; Watt, A.C.; DeMaio, M.; Matyjaszewski, K.; Hollinger, J.O. Cationic Nanogel-mediated Runx2 and Osterix siRNA Delivery Decreases Mineralization in MC3T3 Cells. Clin. Orthop. Relat. Res. 2015, 473, 2139–2149. [Google Scholar] [CrossRef]
- Patil, S.; Dang, K.; Zhao, X.; Gao, Y.; Qian, A. Role of LncRNAs and CircRNAs in Bone Metabolism and Osteoporosis. Front. Genet. 2020, 11, 584118. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Avval, S.T.; Rahmani, S.; Shoorei, H.; Taheri, M.; Samadian, M. Contribution of miRNAs and lncRNAs in osteogenesis and related disorders. Biomed. Pharmacother. 2021, 142, 111942. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Z.; Chen, S.; Cui, H.; Li, X.; Dai, G.; Zhong, F.; Hao, W.; Zhang, K.; Liu, H. BRD4 promotes heterotopic ossification through upregulation of LncRNA MANCR. Bone Jt. Res. 2021, 10, 668–676. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felix-Ilemhenbhio, F.; Pickering, G.A.E.; Kiss-Toth, E.; Wilkinson, J.M. Pathophysiology and Emerging Molecular Therapeutic Targets in Heterotopic Ossification. Int. J. Mol. Sci. 2022, 23, 6983. https://doi.org/10.3390/ijms23136983
Felix-Ilemhenbhio F, Pickering GAE, Kiss-Toth E, Wilkinson JM. Pathophysiology and Emerging Molecular Therapeutic Targets in Heterotopic Ossification. International Journal of Molecular Sciences. 2022; 23(13):6983. https://doi.org/10.3390/ijms23136983
Chicago/Turabian StyleFelix-Ilemhenbhio, Favour, George A. E. Pickering, Endre Kiss-Toth, and Jeremy Mark Wilkinson. 2022. "Pathophysiology and Emerging Molecular Therapeutic Targets in Heterotopic Ossification" International Journal of Molecular Sciences 23, no. 13: 6983. https://doi.org/10.3390/ijms23136983
APA StyleFelix-Ilemhenbhio, F., Pickering, G. A. E., Kiss-Toth, E., & Wilkinson, J. M. (2022). Pathophysiology and Emerging Molecular Therapeutic Targets in Heterotopic Ossification. International Journal of Molecular Sciences, 23(13), 6983. https://doi.org/10.3390/ijms23136983