
Interplay between Zn2+ Homeostasis and Mitochondrial Functions in Cardiovascular Diseases and Heart Ageing

 ,
,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Zn2+ in Mitochondrial Homeostasis
3. Zn2+ in Cardiac Function and Pathology
3.1. Zn2+ in Heart Development
3.2. Zn2+ in Excitation–Contraction Coupling
3.3. Zn2+ in Heart Failure
3.4. Zn2+ in Ischemia/Reperfusion Injury
3.4.1. Mitochondria as the Main Source of Chemically Reactive Species in I/R Injury
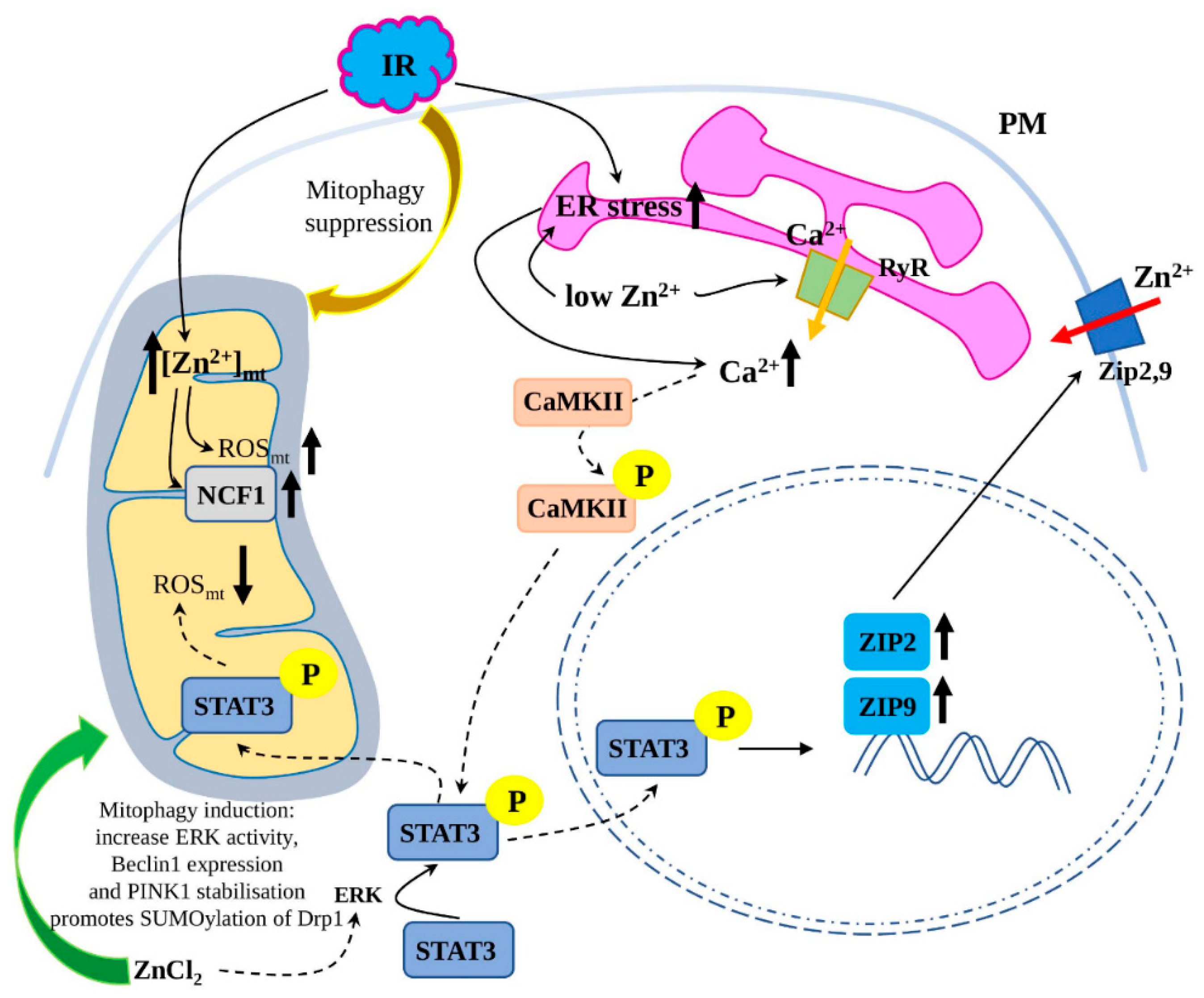
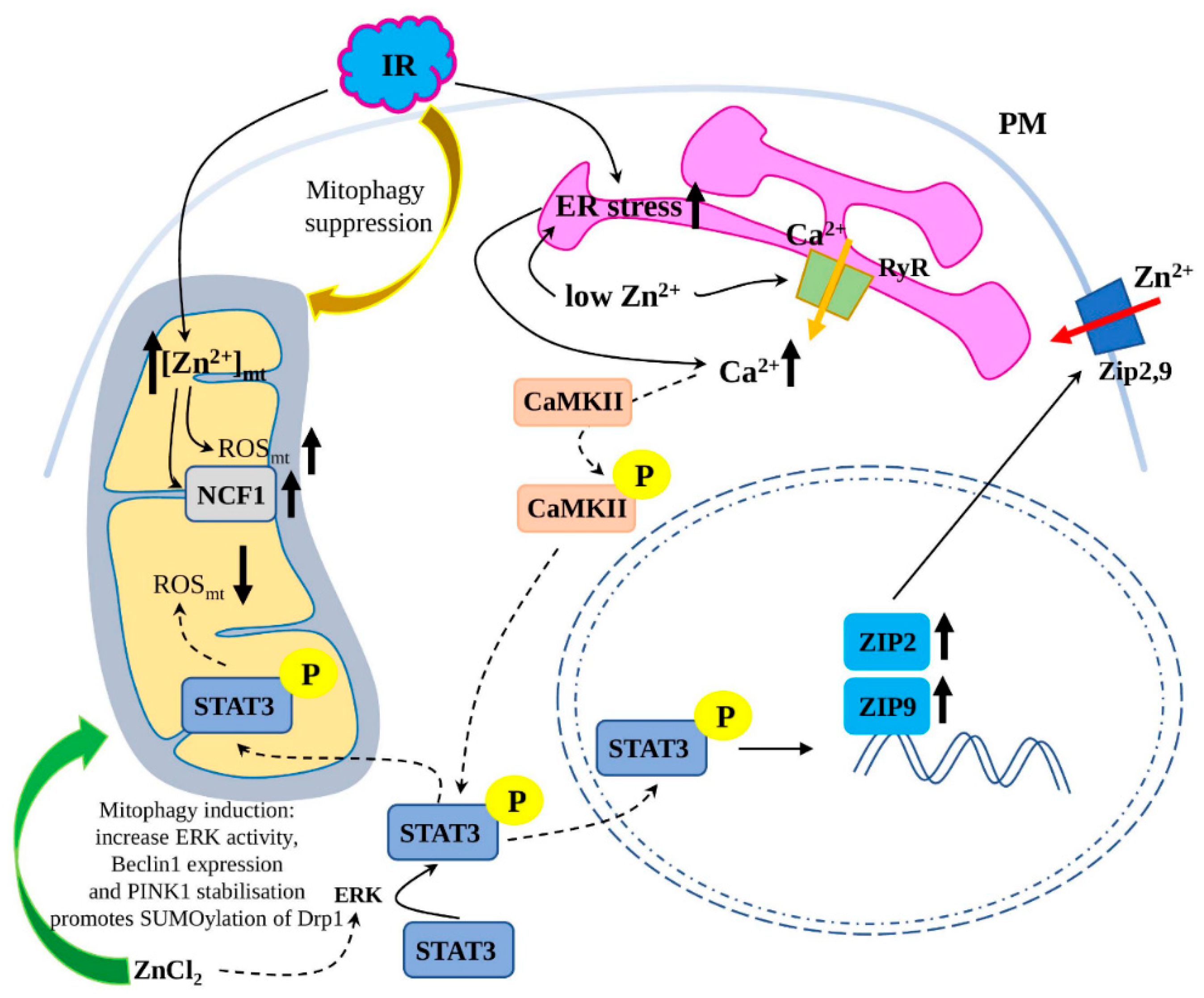
3.4.2. Zn2+ Regulation of Mitophagy in I/R Injury
3.4.3. Zn2+-Mediated Regulation of ER Stress in I/R Injury
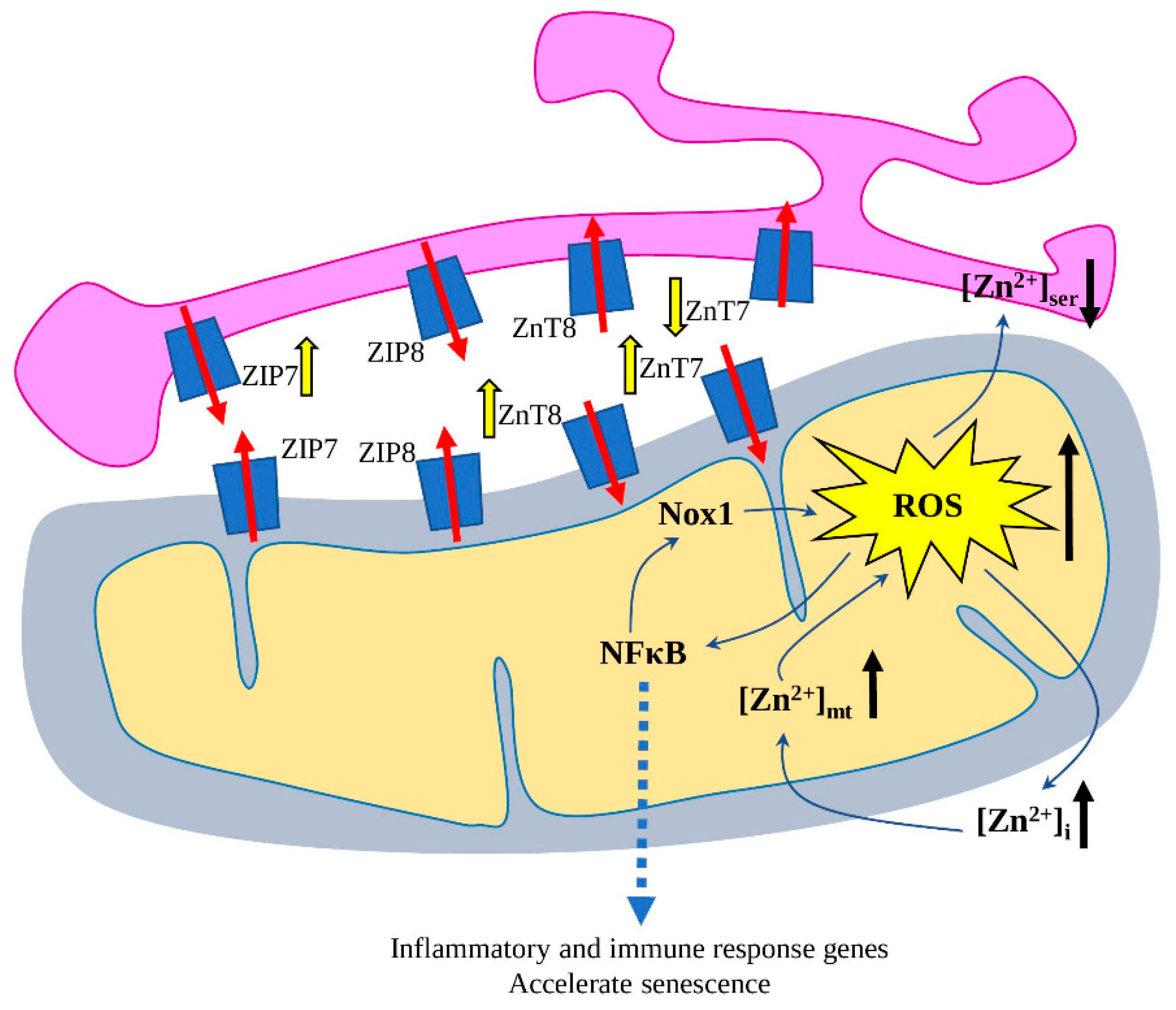
4. Aged Heart
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| [Zn2+]i | intracellular labile Zn2+ |
| [Zn2+]mt | mitochondrial Zn2+ |
| [Zn2+]ser | sarco/endoplasmic reticulum Zn2+ |
| ADAMTS | A Disintegrin And Metalloproteinase With Thrombospondin Motifs 1 |
| BAX | B-cell lymphoma 2–associated X protein |
| BBC3 | BCL2 Binding Component 3 |
| Beclin1 | ATG6, Autophagy Related |
| calregulin | Endoplasmic Reticulum Resident Protein 60 |
| CaMKII | Ca2+-calmodulin-dependent protein kinase |
| CASP | caspase 9 |
| CDKN1A | cyclin-dependent kinase inhibitor 1A |
| CHD | congenital heart disease |
| DRP1 | DNM1L or dynamin 1 like |
| ECM | extracellular matrix |
| EI24 | etoposide-induced 2.4 |
| ER | endoplasmic reticulum |
| ErbB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| ETC | electron transport chain |
| FAS | Fas cell-surface death receptor |
| GRP78 | Heat Shock Protein 70 Family Protein 5 |
| GSK3B | Glycogen Synthase Kinase-3 Beta |
| H/R | hypoxia-oxygenation |
| HF | Heart failure |
| I/R | ischemia/reperfusion injury |
| IκBα | NF-Kappa-B Inhibitor Alpha |
| LVNC | left ventricular noncompaction |
| MAPK1 | Mitogen-Activated Protein Kinase 1, or ERK |
| MG23 | transmembrane protein mitsugumin 23 |
| MMP | mitochondrial membrane potential |
| mPTP | mitochondrial permeability transition pore |
| MTF1 | metal regulatory transcription factor 1 |
| NOX2 | NADPH oxidase 2 |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SENP5 | SUMO1/Sentrin Specific Peptidase 5 |
| SR | sarcoplasmic reticulum |
| ZIP | Zrt-/Irt-like protein |
| ZnT | zinc transporters |
| Znt9 | SLC-30A9, Solute Carrier Family 30 Member 9 |
References
- Hübner, C.; Haase, H. Interactions of Zinc- and Redox-Signaling Pathways. Redox. Biol. 2021, 41, 101916. [Google Scholar] [CrossRef] [PubMed]
- Skalny, A.V.; Aschner, M.; Tinkov, A.A. Zinc. Adv. Food Nutr. Res. 2021, 96, 251–310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Chen, H. Aberrance of Zinc Metalloenzymes-Induced Human Diseases and Its Potential Mechanisms. Nutrients 2021, 13, 4456. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Turan, B. Intracellular Zn(2+) Increase in Cardiomyocytes Induces Both Electrical and Mechanical Dysfunction in Heart via Endogenous Generation of Reactive Nitrogen Species. Biol. Trace Elem. Res. 2016, 169, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Taylor, K.M.; Fu, D. Zinc Transporters and Their Functional Integration in Mammalian Cells. J. Biol. Chem. 2021, 296, 100320. [Google Scholar] [CrossRef] [PubMed]
- Asghari, K.; Shargh, Z.; Fatehfar, S.; Chodari, L.; Sameei, P. The Impact of Zinc on the Molecular Signaling Pathways in the Diabetes Disease. J. Trace Elem. Med. Biol. 2022, 72, 126985. [Google Scholar] [CrossRef] [PubMed]
- Gaburjakova, J.; Gaburjakova, M. The Cardiac Ryanodine Receptor Provides a Suitable Pathway for the Rapid Transport of Zinc (Zn2+). Cells 2022, 11, 868. [Google Scholar] [CrossRef]
- Di Filippo, E.S.; Checcaglini, F.; Fanò-Illic, G.; Fulle, S. H2O2/Ca2+/Zn2+ Complex Can Be Considered a “Collaborative Sensor” of the Mitochondrial Capacity? Antioxidants 2022, 11, 342. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, L. Zinc Homeostasis Plays an Important Role in the Prevention of Obesity-Induced Cardiac Inflammation, Remodeling and Dysfunction. J. Trace Elem. Med. Biol. 2020, 62, 126615. [Google Scholar] [CrossRef]
- Abregú, F.M.G.; Caniffi, C.; Arranz, C.T.; Tomat, A.L. Impact of Zinc Deficiency During Prenatal and/or Postnatal Life on Cardiovascular and Metabolic Diseases: Experimental and Clinical Evidence. Adv. Nutr. 2022, 13, nmac012. [Google Scholar] [CrossRef]
- Thokala, S.; Bodiga, V.L.; Kudle, M.R.; Bodiga, S. Comparative Response of Cardiomyocyte ZIPs and ZnTs to Extracellular Zinc and TPEN. Biol. Trace Elem. Res. 2019, 192, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Brugger, D.; Windisch, W.M. Short-Term Subclinical Zinc Deficiency in Weaned Piglets Affects Cardiac Redox Metabolism and Zinc Concentration. J. Nutr. 2017, 147, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabosseau, P.; Tuncay, E.; Meur, G.; Bellomo, E.A.; Hessels, A.; Hughes, S.; Johnson, P.R.V.; Bugliani, M.; Marchetti, P.; Turan, B.; et al. Mitochondrial and ER-Targeted ECALWY Probes Reveal High Levels of Free Zn2+. ACS Chem. Biol. 2014, 9, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Bitirim, V.C.; Durak, A.; Carrat, G.R.J.; Taylor, K.M.; Rutter, G.A.; Turan, B. Hyperglycemia-Induced Changes in ZIP7 and ZnT7 Expression Cause Zn2+ Release From the Sarco(Endo)Plasmic Reticulum and Mediate ER Stress in the Heart. Diabetes 2017, 66, 1346–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, Y. The Role of Zinc Homeostasis in the Prevention of Diabetes Mellitus and Cardiovascular Diseases. JAT 2021, 28, 1109–1122. [Google Scholar] [CrossRef]
- MacKenzie, S.; Bergdahl, A. Zinc Homeostasis in Diabetes Mellitus and Vascular Complications. Biomedicines 2022, 10, 139. [Google Scholar] [CrossRef]
- Turan, B.; Tuncay, E. The Role of Labile Zn2+ and Zn2+-Transporters in the Pathophysiology of Mitochondria Dysfunction in Cardiomyocytes. Mol. Cell. Biochem. 2021, 476, 971–989. [Google Scholar] [CrossRef]
- Knies, K.A.; Li, Y.V. Zinc Cytotoxicity Induces Mitochondrial Morphology Changes in Hela Cell Line. Int. J. Physiol. Pathophysiol. Pharmacol. 2021, 13, 43–51. [Google Scholar]
- Billur, D.; Tuncay, E.; Okatan, E.N.; Olgar, Y.; Durak, A.T.; Degirmenci, S.; Can, B.; Turan, B. Interplay Between Cytosolic Free Zn2+ and Mitochondrion Morphological Changes in Rat Ventricular Cardiomyocytes. Biol. Trace Elem. Res. 2016, 174, 177–188. [Google Scholar] [CrossRef]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The Role of Mitochondrial Dynamics in Cardiovascular Diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef]
- Onishi, M.; Okamoto, K. Mitochondrial Clearance: Mechanisms and Roles in Cellular Fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.A.; Postnov, A.Y.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative Assessment of Heteroplasmy of Mitochondrial Genome: Perspectives in Diagnostics and Methodological Pitfalls. BioMed Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular Mechanisms and Physiological Functions of Mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Myeloid Dendritic Cells: Development, Functions, and Role in Atherosclerotic Inflammation. Immunobiology 2015, 220, 833–844. [Google Scholar] [CrossRef]
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Li, J.; Sivaprasadarao, A. High Glucose-Induced ROS Activates TRPM2 to Trigger Lysosomal Membrane Permeabilization and Zn2+-Mediated Mitochondrial Fission. Sci. Signal 2017, 10, eaal4161. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.-W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Bian, X.; Teng, T.; Zhao, H.; Qin, J.; Qiao, Z.; Sun, Y.; Liun, Z.; Xu, Z. Zinc Prevents Mitochondrial Superoxide Generation by Inducing Mitophagy in the Setting of Hypoxia/Reoxygenation in Cardiac Cells. Free Radic. Res. 2018, 52, 80–91. [Google Scholar] [CrossRef]
- Ma, T.; Zhao, L.; Zhang, J.; Tang, R.; Wang, X.; Liu, N.; Zhang, Q.; Wang, F.; Li, M.; Shan, Q.; et al. A Pair of Transporters Controls Mitochondrial Zn2+ Levels to Maintain Mitochondrial Homeostasis. Protein Cell 2022, 13, 180–202. [Google Scholar] [CrossRef]
- Perez, Y.; Shorer, Z.; Liani-Leibson, K.; Chabosseau, P.; Kadir, R.; Volodarsky, M.; Halperin, D.; Barber-Zucker, S.; Shalev, H.; Schreiber, R.; et al. SLC30A9 Mutation Affecting Intracellular Zinc Homeostasis Causes a Novel Cerebro-Renal Syndrome. Brain 2017, 140, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Qiao, X.; Xie, T.; Fu, W.; Li, H.; Zhao, Y.; Guo, M.; Feng, Y.; Chen, L.; Zhao, Y.; et al. SLC-30A9 Is Required for Zn2+ Homeostasis, Zn2+ Mobilization, and Mitochondrial Health. Proc. Natl. Acad. Sci. USA 2021, 118, e2023909118. [Google Scholar] [CrossRef]
- Turan, B.; Tuncay, E. Impact of Labile Zinc on Heart Function: From Physiology to Pathophysiology. Int. J. Mol. Sci. 2017, 18, 2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poelmann, R.E.; Gittenberger-de Groot, A.C. Development and Evolution of the Metazoan Heart. Dev. Dyn. 2019, 248, 634–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Li, D. Zinc and Zinc Transporters: Novel Regulators of Ventricular Myocardial Development. Pediatr. Cardiol. 2018, 39, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Zhao, D.; Chen, X.; Zhang, C.; Zheng, J.; Liu, X. Zinc Deficiency Induces Abnormal Development of the Myocardium by Promoting SENP5 Overexpression. PLoS ONE 2020, 15, e0242606. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 2018, 71, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Li, D.; Cheng, L.; Li, L.; Liu, F.; Hand, N.J.; Epstein, J.A.; Rader, D.J. Zinc Transporter Slc39a8 Is Essential for Cardiac Ventricular Compaction. J. Clin. Investig. 2018, 128, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Rohde, S.; Muslem, R.; Kaya, E.; Dalinghaus, M.; van Waning, J.I.; Majoor-Krakauer, D.; Towbin, J.; Caliskan, K. State-of-the Art Review: Noncompaction Cardiomyopathy in Pediatric Patients. Heart Fail. Rev. 2022, 27, 15–28. [Google Scholar] [CrossRef]
- Soldatov, V.O.; Malorodova, T.N.; Balamutova, T.I.; Ksenofontov, A.O.; Dovgan, A.P.; Urozhevskaya, Z.S. Endothelial Dysfunction: Comparative Evaluation of Ultrasound Dopplerography, Laser Dopplerflowmetry and Direct Monitoring of Arterial Pressure for Conducting Pharmacological Tests in Rats. Res. Results Pharmacol. 2018, 4, 73–80. [Google Scholar] [CrossRef]
- Peralta, F.A.; Huidobro-Toro, J.P. Zinc as Allosteric Ion Channel Modulator: Ionotropic Receptors as Metalloproteins. Int. J. Mol. Sci. 2016, 17, 1059. [Google Scholar] [CrossRef] [Green Version]
- Degirmenci, S.; Olgar, Y.; Durak, A.; Tuncay, E.; Turan, B. Cytosolic Increased Labile Zn2+ Contributes to Arrhythmogenic Action Potentials in Left Ventricular Cardiomyocytes through Protein Thiol Oxidation and Cellular ATP Depletion. J. Trace Elem. Med. Biol. 2018, 48, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Sanchis-Gomar, F. Global Epidemiology and Future Trends of Heart Failure. AME Med. J. 2020, 5, 15. [Google Scholar] [CrossRef]
- Puchenkova, O.A.; Nadezhdin, S.V.; Soldatov, V.O.; Zhuchenko, M.A.; Korshunova, D.S.; Kubekina, M.V.; Korshunov, E.N.; Korokina, L.V.; Golubinskaya, P.A.; Kulikov, A.L.; et al. Study of antiatherosclerotic and endothelioprotective activity of peptide agonists of epor/cd131 heteroreceptor. Farm. Farmakol. 2020, 8, 100–111. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial Dysfunction and Mitochondrial Therapies in Heart Failure. Pharmacol. Res. 2022, 175, 106038. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Lemos, F.O.; Bultynck, G.; Parys, J.B. A Comprehensive Overview of the Complex World of the Endo- and Sarcoplasmic Reticulum Ca2+-Leak Channels. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 119020. [Google Scholar] [CrossRef]
- Reilly-O’Donnell, B.; Robertson, G.B.; Karumbi, A.; McIntyre, C.; Bal, W.; Nishi, M.; Takeshima, H.; Stewart, A.J.; Pitt, S.J. Dysregulated Zn2+ Homeostasis Impairs Cardiac Type-2 Ryanodine Receptor and Mitsugumin 23 Functions, Leading to Sarcoplasmic Reticulum Ca2+ Leakage. J. Biol. Chem. 2017, 292, 13361–13373. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, H.; Venturi, E.; Sitsapesan, R. New and Notable Ion-Channels in the Sarcoplasmic/Endoplasmic Reticulum: Do They Support the Process of Intracellular Ca2+ Release? J. Physiol. 2015, 593, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- Olgar, Y.; Durak, A.; Tuncay, E.; Bitirim, C.V.; Ozcinar, E.; Inan, M.B.; Tokcaer-Keskin, Z.; Akcali, K.C.; Akar, A.R.; Turan, B. Increased Free Zn2+ Correlates Induction of Sarco(Endo)Plasmic Reticulum Stress via Altered Expression Levels of Zn2+ -Transporters in Heart Failure. J. Cell. Mol. Med. 2018, 22, 1944–1956. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, S.; El Khoury, N.; Rivard, K.; Gélinas, R.; Goyette, P.; Paradis, P.; Nemer, M.; Fiset, C. Reduction in Na(+) Current by Angiotensin II Is Mediated by PKCα in Mouse and Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Heart Rhythm. 2016, 13, 1346–1354. [Google Scholar] [CrossRef]
- Guo, W.-H.; Wang, X.; Shang, M.-S.; Chen, Z.; Guo, Q.; Li, L.; Wang, H.-Y.; Yu, R.-H.; Ma, C.-S. Crosstalk between PKC and MAPK Pathway Activation in Cardiac Fibroblasts in a Rat Model of Atrial Fibrillation. Biotechnol. Lett. 2020, 42, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Olgar, Y.; Ozdemir, S.; Turan, B. Induction of Endoplasmic Reticulum Stress and Changes in Expression Levels of Zn2+-Transporters in Hypertrophic Rat Heart. Mol. Cell. Biochem. 2018, 440, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wang, S.; Lv, J.; Zhao, Z.; Guo, N.; Wu, G.; Tong, J.; Wang, Z. Slc39a2-Mediated Zinc Homeostasis Modulates Innate Immune Signaling in Phenylephrine-Induced Cardiomyocyte Hypertrophy. Front. Cardiovasc. Med. 2021, 8, 736911. [Google Scholar] [CrossRef] [PubMed]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Heim, V.J.; Stafford, C.A.; Nachbur, U. NOD Signaling and Cell Death. Front. Cell. Dev. Biol. 2019, 7, 208. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Salonen, J.T.; Zhelankin, A.V.; Melnichenko, A.A.; Kaikkonen, J.; Bobryshev, Y.V.; Orekhov, A.N. Low Density Lipoprotein-Containing Circulating Immune Complexes: Role in Atherosclerosis and Diagnostic Value. BioMed Res. Int. 2014, 2014, 205697. [Google Scholar] [CrossRef]
- Yu, X.; Huang, L.; Zhao, J.; Wang, Z.; Yao, W.; Wu, X.; Huang, J.; Bian, B. The Relationship between Serum Zinc Level and Heart Failure: A Meta-Analysis. Biomed. Res. Int. 2018, 2018, 2739014. [Google Scholar] [CrossRef]
- Rosenblum, H.; Wessler, J.D.; Gupta, A.; Maurer, M.S.; Bikdeli, B. Zinc Deficiency and Heart Failure: A Systematic Review of the Current Literature. J. Card. Fail. 2020, 26, 180–189. [Google Scholar] [CrossRef]
- Rosenblum, H.; Bikdeli, B.; Wessler, J.; Gupta, A.; Jacoby, D.L. Zinc Deficiency as a Reversible Cause of Heart Failure. Tex. Heart Inst. J. 2020, 47, 152–154. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial Ischaemia–Reperfusion Injury and Cardioprotection in Perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Chistiakov, D.; Revin, V.; Sobenin, I.; Orekhov, A.; Bobryshev, Y. Vascular Endothelium: Functioning in Norm, Changes in Atherosclerosis and Current Dietary Approaches to Improve Endothelial Function. Mini Rev. Med. Chem. 2015, 15, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Akbari, G. Role of Zinc Supplementation on Ischemia/Reperfusion Injury in Various Organs. Biol. Trace Elem. Res. 2020, 196, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The Role of Mitochondrial Reactive Oxygen Species, NO and H2S in Ischaemia/Reperfusion Injury and Cardioprotection. J. Cell. Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” Target of Myocardial Conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Cross Talk between Increased Intracellular Zinc (Zn2+) and Accumulation of Reactive Oxygen Species in Chemical Ischemia. Am. J. Physiol. Cell Physiol. 2017, 313, C448–C459. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ma, J.; Deng, Y.; Kelly, J.A.; Kim, K.; Bang, S.-Y.; Lee, H.-S.; Li, Q.-Z.; Wakeland, E.K.; Qiu, R.; et al. A Missense Variant in NCF1 Is Associated with Susceptibility to Multiple Autoimmune Diseases. Nat. Genet. 2017, 49, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-Induced Dynamic Regulation of Mitochondrial STAT3 and Its Association with Cyclophilin D Reduce Mitochondrial ROS Production. Sci. Signal. 2017, 10, eaag2588. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, K.; Keasey, M.P.; Razskazovskiy, V.; Visavadiya, N.P.; Jia, C.; Hagg, T. Reduced FAK-STAT3 Signaling Contributes to ER Stress-Induced Mitochondrial Dysfunction and Death in Endothelial Cells. Cell. Signal. 2017, 36, 154–162. [Google Scholar] [CrossRef]
- Zhang, G.; Sheng, M.; Wang, J.; Teng, T.; Sun, Y.; Yang, Q.; Xu, Z. Zinc Improves Mitochondrial Respiratory Function and Prevents Mitochondrial ROS Generation at Reperfusion by Phosphorylating STAT3 at Ser727. J. Mol. Cell. Cardiol. 2018, 118, 169–182. [Google Scholar] [CrossRef]
- Du, L.; Zhang, H.; Zhao, H.; Cheng, X.; Qin, J.; Teng, T.; Yang, Q.; Xu, Z. The Critical Role of the Zinc Transporter Zip2 (SLC39A2) in Ischemia/Reperfusion Injury in Mouse Hearts. J. Mol. Cell. Cardiol. 2019, 132, 136–145. [Google Scholar] [CrossRef]
- Zech, A.T.L.; Singh, S.R.; Schlossarek, S.; Carrier, L. Autophagy in Cardiomyopathies. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118432. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Pazos, R. Interplay Between Autophagy and Zinc. J. Trace Elem. Med. Biol. 2020, 62, 126636. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Zeng, Y.; Sun, R.-R.; Liu, M.; Chen, S.; Zhang, P.-Y. SUMOylation in Cardiac Disorders—A Review. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1583–1587. [Google Scholar] [PubMed]
- Bian, X.; Xu, J.; Zhao, H.; Zheng, Q.; Xiao, X.; Ma, X.; Li, Y.; Du, X.; Liu, X. Zinc-Induced SUMOylation of Dynamin-Related Protein 1 Protects the Heart against Ischemia-Reperfusion Injury. Oxidative Med. Cell. Longev. 2019, 2019, 1232146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, N.; He, H.; Chai, J.; Cheng, X.; Zhao, H.; Zhou, D.; Teng, T.; Kong, X.; Yang, Q.; et al. The Zinc Transporter ZIP7 (Slc39a7) Controls Myocardial Reperfusion Injury by Regulating Mitophagy. Basic Res. Cardiol. 2021, 116, 54. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Nikiforov, N.G.; Zhuravlev, A.D.; Orekhov, N.A.; Grechko, A.V.; Orekhov, A.N. Role of the MtDNA Mutations and Mitophagy in Inflammaging. Int. J. Mol. Sci. 2022, 23, 1323. [Google Scholar] [CrossRef]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.-F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, L.; Gillette, T.G.; Jiang, X.; Wang, Z.V. The Unfolded Protein Response in Ischemic Heart Disease. J. Mol. Cell Cardiol 2018, 117, 19–25. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Q.; Gao, A.; Chen, L.; Li, L. Endoplasmic Reticulum Stress and Focused Drug Discovery in Cardiovascular Disease. Clin. Chim Acta 2020, 504, 125–137. [Google Scholar] [CrossRef]
- Wang, G.; Huang, H.; Zheng, H.; He, Y.; Zhang, Y.; Xu, Z.; Zhang, L.; Xi, J. Zn2+ and MPTP Mediate Endoplasmic Reticulum Stress Inhibition-Induced Cardioprotection Against Myocardial Ischemia/Reperfusion Injury. Biol. Trace Elem. Res. 2016, 174, 189–197. [Google Scholar] [CrossRef]
- Bodiga, V.L.; Vemuri, P.K.; Nimmagadda, G.; Bodiga, S. Zinc-Dependent Changes in Oxidative and Endoplasmic Reticulum Stress during Cardiomyocyte Hypoxia/Reoxygenation. Biol. Chem. 2020, 401, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, Z.; Segers, V.F.M.; De Keulenaer, G.W. ErbB2 Signaling at the Crossing between Heart Failure and Cancer. Basic Res. Cardiol 2016, 111, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Liu, D.; Yan, Q.; Bian, X.; Yu, J.; Wang, J.; Cheng, X.; Xu, Z. Endoplasmic Reticulum Stress/Ca2+-Calmodulin-Dependent Protein Kinase/Signal Transducer and Activator of Transcription 3 Pathway Plays a Role in the Regulation of Cellular Zinc Deficiency in Myocardial Ischemia/Reperfusion Injury. Front. Physiol. 2022, 12, 736920. [Google Scholar] [CrossRef]
- Zhao, N.; Li, Q.; Sui, H.; Zhang, H. Role of Oxidation-Dependent CaMKII Activation in the Genesis of Abnormal Action Potentials in Atrial Cardiomyocytes: A Simulation Study. Biomed. Res. Int. 2020, 2020, 1597012. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Zhang, G.; Wang, J.; Yang, Q.; Zhao, H.; Cheng, X.; Xu, Z. Remifentanil Induces Cardio Protection Against Ischemia/Reperfusion Injury by Inhibiting Endoplasmic Reticulum Stress Through the Maintenance of Zinc Homeostasis. Anesth. Analg. 2018, 127, 267–276. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, X.; Zhao, H.; Yang, Q.; Xu, Z. Downregulation of the Zinc Transporter SLC39A13 (ZIP13) Is Responsible for the Activation of CaMKII at Reperfusion and Leads to Myocardial Ischemia/Reperfusion Injury in Mouse Hearts. J. Mol. Cell. Cardiol. 2021, 152, 69–79. [Google Scholar] [CrossRef]
- Bou-Teen, D.; Kaludercic, N.; Weissman, D.; Turan, B.; Maack, C.; Di Lisa, F.; Ruiz-Meana, M. Mitochondrial ROS and Mitochondria-Targeted Antioxidants in the Aged Heart. Free. Radic. Biol. Med. 2021, 167, 109–124. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Bou-Teen, D.; Ferdinandy, P.; Gyongyosi, M.; Pesce, M.; Perrino, C.; Schulz, R.; Sluijter, J.P.G.; Tocchetti, C.G.; Thum, T.; et al. Cardiomyocyte Ageing and Cardioprotection: Consensus Document from the ESC Working Groups Cell Biology of the Heart and Myocardial Function. Cardiovasc. Res. 2020, 116, 1835–1849. [Google Scholar] [CrossRef]
- Olgar, Y.; Degirmenci, S.; Durak, A.; Billur, D.; Can, B.; Kayki-Mutlu, G.; Arioglu-Inan, E.E.; Turan, B. Aging Related Functional and Structural Changes in the Heart and Aorta: MitoTEMPO Improves Aged-Cardiovascular Performance. Exp. Gerontol. 2018, 110, 172–181. [Google Scholar] [CrossRef]
- Olgar, Y.; Billur, D.; Tuncay, E.; Turan, B. MitoTEMPO Provides an Antiarrhythmic Effect in Aged-Rats through Attenuation of Mitochondrial Reactive Oxygen Species. Exp. Gerontol. 2020, 136, 110961. [Google Scholar] [CrossRef]
- Novakovic, S.; Molesworth, L.W.; Gourley, T.E.; Boag, P.R.; Davis, G.M. Zinc Transporters Maintain Longevity by Influencing Insulin/IGF-1 Activity in Caenorhabditis Elegans. FEBS Lett. 2020, 594, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Magnusson, K.R.; Sharpton, T.J.; Ho, E. Effects of Zinc Status on Age-Related T Cell Dysfunction and Chronic Inflammation. Biometals 2021, 34, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Troche, C.; Kim, J.; Kim, M.-H.; Teran, O.Y.; Leeuwenburgh, C.; Cousins, R.J. Aging Amplifies Multiple Phenotypic Defects in Mice with Zinc Transporter Zip14 (Slc39a14) Deletion. Exp. Gerontol. 2016, 85, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Olgar, Y.; Tuncay, E.; Turan, B. Mitochondria-Targeting Antioxidant Provides Cardioprotection through Regulation of Cytosolic and Mitochondrial Zn2+ Levels with Re-Distribution of Zn2+-Transporters in Aged Rat Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 3783. [Google Scholar] [CrossRef] [Green Version]
- Salazar, G.; Huang, J.; Feresin, R.G.; Zhao, Y.; Griendling, K.K. Zinc Regulates Nox1 Expression through a NF-ΚB and Mitochondrial ROS Dependent Mechanism to Induce Senescence of Vascular Smooth Muscle Cells. Free. Radic. Biol. Med. 2017, 108, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The Non-Canonical NF-ΚB Pathway in Immunity and Inflammation. Nat. Rev. Immunol 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Barnabei, L.; Laplantine, E.; Mbongo, W.; Rieux-Laucat, F.; Weil, R. NF-ΚB: At the Borders of Autoimmunity and Inflammation. Front. Immunol. 2021, 12, 716469. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabravolski, S.A.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Interplay between Zn2+ Homeostasis and Mitochondrial Functions in Cardiovascular Diseases and Heart Ageing. Int. J. Mol. Sci. 2022, 23, 6890. https://doi.org/10.3390/ijms23136890
Dabravolski SA, Sadykhov NK, Kartuesov AG, Borisov EE, Sukhorukov VN, Orekhov AN. Interplay between Zn2+ Homeostasis and Mitochondrial Functions in Cardiovascular Diseases and Heart Ageing. International Journal of Molecular Sciences. 2022; 23(13):6890. https://doi.org/10.3390/ijms23136890
Chicago/Turabian StyleDabravolski, Siarhei A., Nikolay K. Sadykhov, Andrey G. Kartuesov, Evgeny E. Borisov, Vasily N. Sukhorukov, and Alexander N. Orekhov. 2022. "Interplay between Zn2+ Homeostasis and Mitochondrial Functions in Cardiovascular Diseases and Heart Ageing" International Journal of Molecular Sciences 23, no. 13: 6890. https://doi.org/10.3390/ijms23136890
APA StyleDabravolski, S. A., Sadykhov, N. K., Kartuesov, A. G., Borisov, E. E., Sukhorukov, V. N., & Orekhov, A. N. (2022). Interplay between Zn2+ Homeostasis and Mitochondrial Functions in Cardiovascular Diseases and Heart Ageing. International Journal of Molecular Sciences, 23(13), 6890. https://doi.org/10.3390/ijms23136890