
Neuroimaging Methods to Map In Vivo Changes of OXPHOS and Oxidative Stress in Neurodegenerative Disorders


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. What Is Mitochondrial Impairment? The Molecular Complexity of a Fundamental Cell Organelle
1.2. Why Could It Be Helpful to Identify Patients with Predominant Mitochondrial Dysfunction? On the Leap to Individualized Treatment Decisions
1.3. The Unbundling of Metabolic Pathways: Mitochondria at the Convergence of Human Metabolism
1.4. Neuroimaging for Patient Stratification and Therapy Monitoring in Patients with Suspected Mitochondrial Dysfunction?
1.5. What Can We Measure? Mitochondrial Bioenergetics and Oxidative Stress as Promising Neuroimaging Markers of Mitochondrial Dysfunction
- Nuclear magnetic resonance (NMR). Atomic nuclei with a non-zero nuclear spin can be considered NMR-active. MRI and magnetic resonance spectroscopy (MRSI) utilize this phenomenon to generate image contrast or simultaneously measure multiple metabolites. In most MRI or MRSI studies, the NMR-phenomenon of protons (1H) is used to derive imaging data. However, these methods can also be applied to other NMR-active nuclei (e.g., 13C-, 15N-, 17O-, or 31P-nuclei). The use of non-1H nuclei in neuroimaging is often referred to as hetero-, multi-, or X-nuclear MR(S)I [38,45]. The acquisition of adequate X-nuclear MRSI signals critically depends on the natural abundance, the relative sensitivity (defined by the gyromagnetic ratio and the nuclear spin), and T1/T2 relaxation times (using quadrupolar relaxation) of the NMR-active isotope [38,46].
- Near-infrared spectroscopy (NIRS). NIRS is a physical analysis technique based on the absorption and emission of short-waved (within the near-infrared region of the electromagnetic spectrum) light to detect chromophoric metabolites (e.g., hemoglobin) [47].
- Radioactive decay. PET imaging is based on the simultaneous detection of two gamma-ray photons produced after a positron-emitting radionuclide (β+ decay) decay. The target-specific generation of radiotracers and the distribution of these weakly radioactively labeled substances can be used to image biochemical and physiological functions of the brain [48].
1.6. The Scope of this Review
2. Neuroimaging-Based Assessments of OXPHOS-Related Complexes and Metabolites
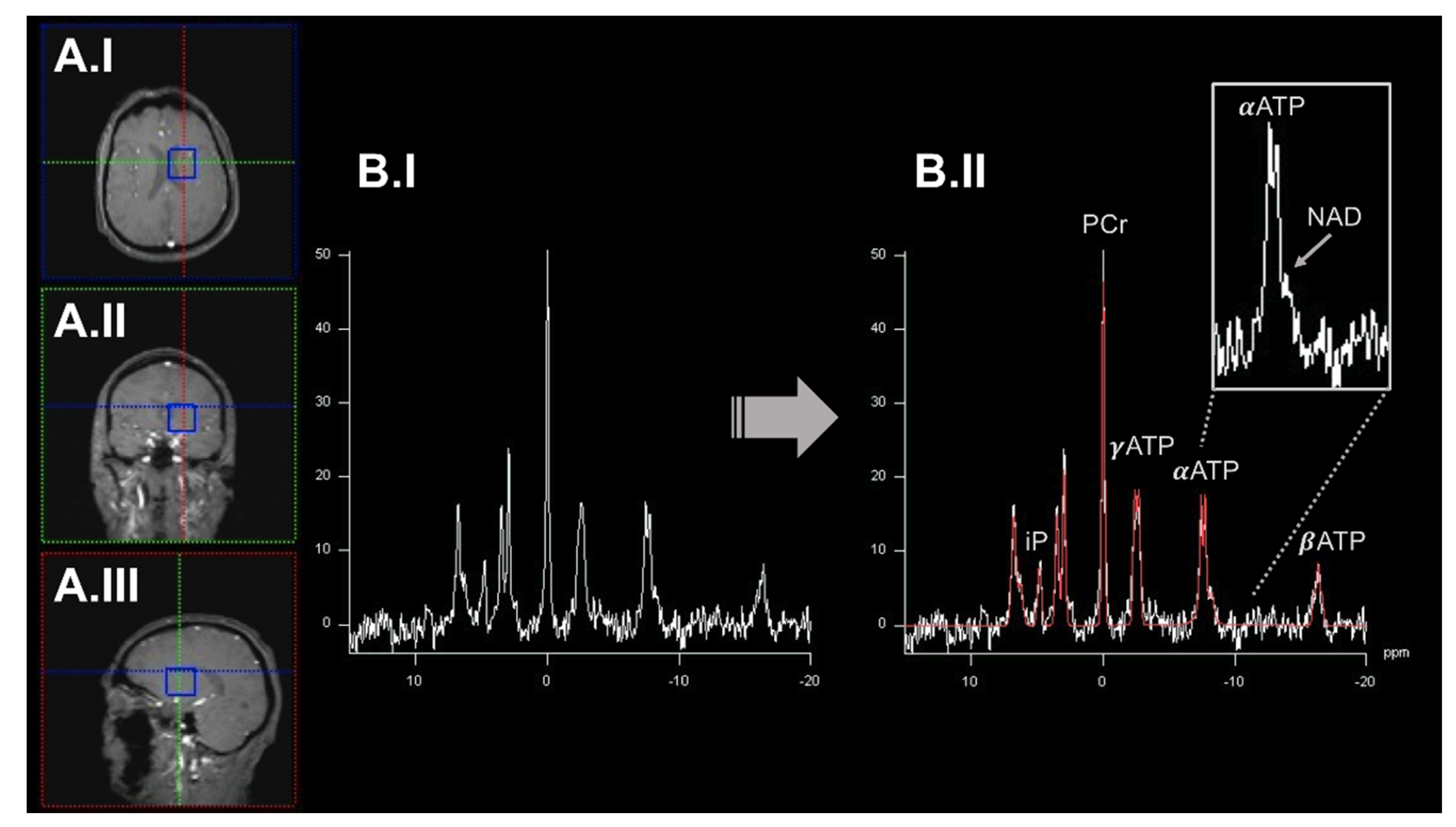
2.1. 31Phosphorus-Magnetic Resonance Spectroscopy Imaging to Quantify OXPHOS-Related Metabolite Levels In Vivo
2.2. Dynamic Measurements of OXPHOS Reaction Kinetics by 31phosphorus Magnetization Transfer Magnetic Resonance Spectroscopy Imaging
2.3. Quantitative Assessment of Mitochondrial Complex I by Positron Emission Tomography Imaging Radiotracer
2.4. Broadband Near-Infrared Spectroscopy to Dynamically Map Cytochrome c Oxidase Activity
3. Neuroimaging Assessments of In Vivo Oxygen Consumption
3.1. 15Oxygen-Positron Emission Tomography Imaging of In Vivo Oxygen Metabolism
3.2. 17Oxygen-Magnetic Resonance Spectroscopy Imaging to Assess the Cerebral Metabolic Rate of Oxygen Consumption
- oxygen consumption in the mitochondria via complex IV,
- leaching of H217O from the brain via blood perfusion, and
- recirculation of H217O-containing blood. Here, H217O is generated throughout the whole body of a given organism and returns to the brain via the bloodstream [38].
3.3. Estimating Brain Oxygen Metabolism by Conventional Magnetic Resonance Imaging Methods
- 1.
- BOLD functional magnetic resonance imaging (fMRI). Here, physiological challenges (using interleaved hypercapnic and hyperoxic states) are demanded to calibrate the fMRI signal [115].
- 2.
- 3.
4. Neuroimaging-Based In Vivo Assessment of Oxidative Stress
- 1.
- antioxidant enzyme systems (such as glutathione reductase, glutathione peroxidase, and catalases, among others) [127] and
- 2.
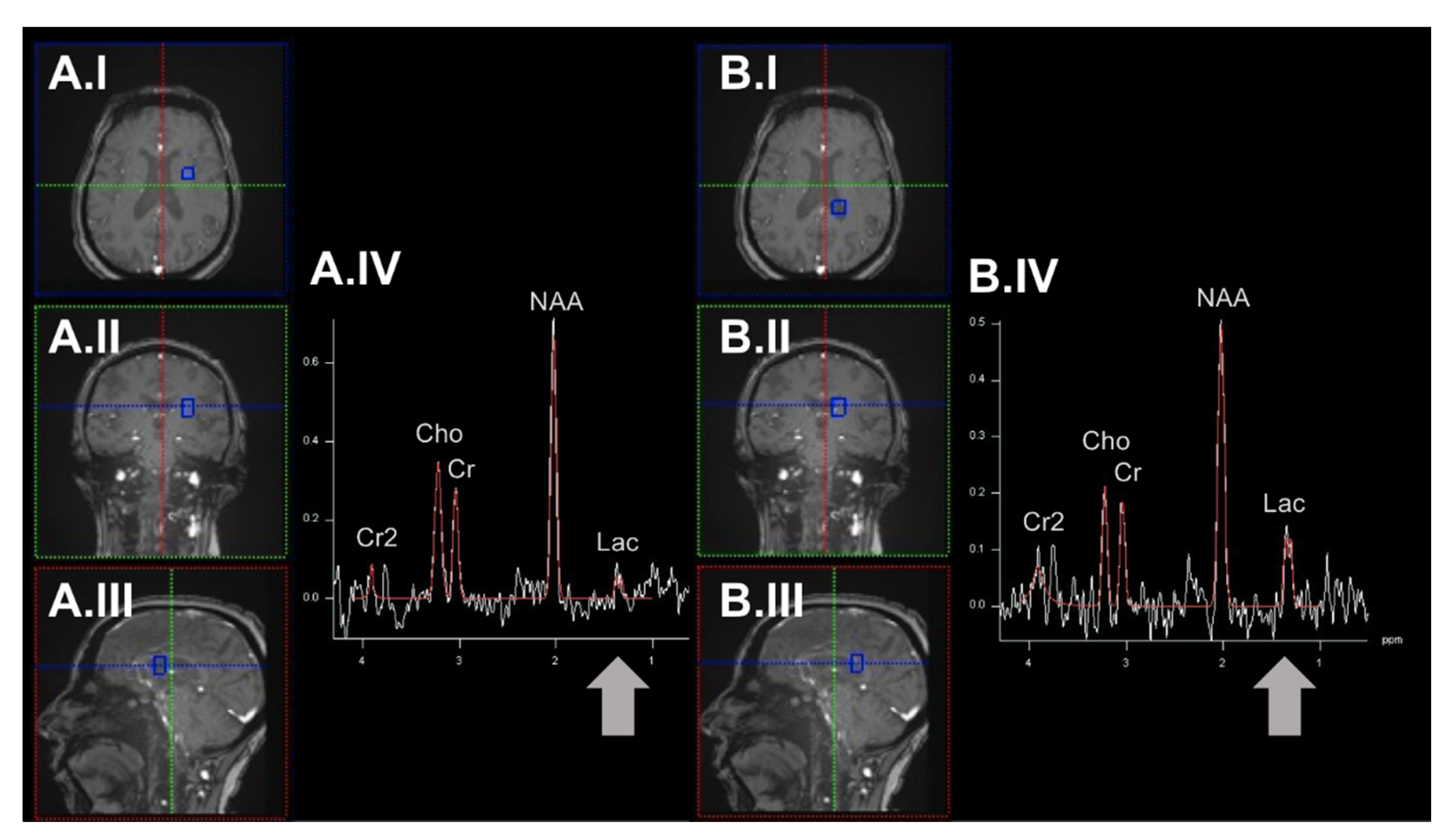
4.1. Proton-Magnetic Resonance Spectroscopy Imaging to Quantify Low-Molecular-Weight Antioxidants
4.2. Probing Oxidative Stress by Over-Reductive Tissue State-Specific Radiotracer
4.3. Iron-Sensitive Magnetic Resonance Imaging to Unravel the Generation of Oxidative Stress
4.4. Assessments of the Short-Term Response to Antioxidant Treatments by QUEnch-assiSTed MRI
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef] [PubMed]
- Ries, V.; Oertel, W.H.; Hoglinger, G.U. Mitochondrial dysfunction as a therapeutic target in progressive supranuclear palsy. J. Mol. Neurosci. 2011, 45, 684–689. [Google Scholar] [CrossRef]
- Rummel, N.G.; Butterfield, D.A. Altered Metabolism in Alzheimer Disease Brain: Role of Oxidative Stress. Antioxid. Redox. Signal 2021, 36, 1289–1305. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Behl, T.; Sharma, L.; Aelya, L.; Bungau, S. Mitochondrial Dysfunction in Huntington’s disease: Pathogenesis and Therapeutic Opportunities. Curr. Drug Targets 2021, 22, 1637–1667. [Google Scholar] [CrossRef]
- Flones, I.H.; Ricken, G.; Klotz, S.; Lang, A.; Strobel, T.; Dolle, C.; Kovacs, G.G.; Tzoulis, C. Mitochondrial respiratory chain deficiency correlates with the severity of neuropathology in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. Commun. 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Doni, D.; Rigoni, G.; Palumbo, E.; Baschiera, E.; Peruzzo, R.; De Rosa, E.; Caicci, F.; Passerini, L.; Bettio, D.; Russo, A.; et al. The displacement of frataxin from the mitochondrial cristae correlates with abnormal respiratory supercomplexes formation and bioenergetic defects in cells of Friedreich ataxia patients. FASEB J. 2021, 35, e21362. [Google Scholar] [CrossRef]
- Rey, F.; Ottolenghi, S.; Zuccotti, G.V.; Samaja, M.; Carelli, S. Mitochondrial dysfunctions in neurodegenerative diseases: Role in disease pathogenesis, strategies for analysis and therapeutic prospects. Neural Regen. Res. 2022, 17, 754–758. [Google Scholar] [CrossRef]
- Lashuel, H.A. Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity? Curr. Opin. Chem. Biol. 2021, 64, 67–75. [Google Scholar] [CrossRef]
- Morais, V.A.; De Strooper, B. Mitochondria dysfunction and neurodegenerative disorders: Cause or consequence. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S255–S263. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Cummings, J. Disease modification and Neuroprotection in neurodegenerative disorders. Transl. Neurodegener. 2017, 6, 25. [Google Scholar] [CrossRef]
- de Aquino, C.H. Methodological Issues in Randomized Clinical Trials for Prodromal Alzheimer’s and Parkinson’s Disease. Front. Neurol. 2021, 12, 694329. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef]
- Gong, P.; Fanos, J.H.; Korty, L.; Siskind, C.E.; Hanson-Kahn, A.K. Impact of Huntington Disease Gene-Positive Status on Pre-Symptomatic Young Adults and Recommendations for Genetic Counselors. J. Genet. Couns. 2016, 25, 1188–1197. [Google Scholar] [CrossRef]
- Prasuhn, J.; Bruggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; Konig, I.R.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 31. [Google Scholar] [CrossRef]
- Dansson, H.V.; Stempfle, L.; Egilsdottir, H.; Schliep, A.; Portelius, E.; Blennow, K.; Zetterberg, H.; Johansson, F.D.; the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Predicting progression and cognitive decline in amyloid-positive patients with Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 151. [Google Scholar] [CrossRef]
- Mitchell, T.; Lehericy, S.; Chiu, S.Y.; Strafella, A.P.; Stoessl, A.J.; Vaillancourt, D.E. Emerging Neuroimaging Biomarkers Across Disease Stage in Parkinson Disease: A Review. JAMA Neurol. 2021, 78, 1262–1272. [Google Scholar] [CrossRef]
- Abeyasinghe, P.M.; Long, J.D.; Razi, A.; Pustina, D.; Paulsen, J.S.; Tabrizi, S.J.; Poudel, G.R.; Georgiou-Karistianis, N. Tracking Huntington’s Disease Progression Using Motor, Functional, Cognitive, and Imaging Markers. Mov. Disord. 2021, 36, 2282–2292. [Google Scholar] [CrossRef]
- Gotovac, K.; Hajnsek, S.; Pasic, M.B.; Pivac, N.; Borovecki, F. Personalized medicine in neurodegenerative diseases: How far away? Mol. Diagn. Ther. 2014, 18, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front. Aging Neurosci. 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Bruggemann, N. Gene Therapeutic Approaches for the Treatment of Mitochondrial Dysfunction in Parkinson’s Disease. Genes 2021, 12, 1840. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Bruggemann, N. Genotype-driven therapeutic developments in Parkinson’s disease. Mol. Med. 2021, 27, 42. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The multi-faceted role of mitochondria in the pathology of Parkinson’s disease. J. Neurochem. 2021, 156, 715–752. [Google Scholar] [CrossRef]
- Wiggins, R.; Feigin, A. Emerging therapeutics in Huntington’s disease. Expert Opin. Emerg. Drugs 2021, 26, 295–302. [Google Scholar] [CrossRef]
- Park, S.S.; Jeong, H.; Andreazza, A.C. Circulating cell-free mitochondrial DNA in brain health and disease: A systematic review and meta-analysis. World J. Biol. Psychiatry 2021, 23, 87–102. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Beal, M.F.; Nirenberg, M.J.; Shungu, D.C.; Henchcliffe, C. Usefulness of proton and phosphorus MR spectroscopic imaging for early diagnosis of Parkinson’s disease. J. Neuroimaging 2015, 25, 105–110. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. How the brain fights fatty acids’ toxicity. Neurochem. Int. 2021, 148, 105050. [Google Scholar] [CrossRef]
- Reiten, O.K.; Wilvang, M.A.; Mitchell, S.J.; Hu, Z.; Fang, E.F. Preclinical and clinical evidence of NAD(+) precursors in health, disease, and ageing. Mech. Ageing Dev. 2021, 199, 111567. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Faitg, J.; Lacefield, C.; Davey, T.; White, K.; Laws, R.; Kosmidis, S.; Reeve, A.K.; Kandel, E.R.; Vincent, A.E.; Picard, M. 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 2021, 36, 109509. [Google Scholar] [CrossRef] [PubMed]
- Polimeni, J.R.; Lewis, L.D. Imaging faster neural dynamics with fast fMRI: A need for updated models of the hemodynamic response. Prog. Neurobiol. 2021, 207, 102174. [Google Scholar] [CrossRef]
- Davis, T.L.; Kwong, K.K.; Weisskoff, R.M.; Rosen, B.R. Calibrated functional MRI: Mapping the dynamics of oxidative metabolism. Proc. Natl. Acad. Sci. USA 1998, 95, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial respiratory supercomplexes in mammalian cells: Structural versus functional role. J. Mol. Med. 2021, 99, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, K.; Nowicka, U.; Chacinska, A. Mitochondrial control of cellular protein homeostasis. Biochem. J. 2020, 477, 3033–3054. [Google Scholar] [CrossRef]
- Zhu, X.H.; Lu, M.; Chen, W. Quantitative imaging of brain energy metabolisms and neuroenergetics using in vivo X-nuclear 2H, 17O and 31P MRS at ultra-high field. J. Magn. Reson. 2018, 292, 155–170. [Google Scholar] [CrossRef]
- Saito, S.; Takahashi, Y.; Ohki, A.; Shintani, Y.; Higuchi, T. Early detection of elevated lactate levels in a mitochondrial disease model using chemical exchange saturation transfer (CEST) and magnetic resonance spectroscopy (MRS) at 7T-MRI. Radiol. Phys. Technol. 2019, 12, 46–54. [Google Scholar] [CrossRef]
- Bagwe-Parab, S.; Kaur, G. Molecular targets and therapeutic interventions for iron induced neurodegeneration. Brain. Res. Bull. 2020, 156, 1–9. [Google Scholar] [CrossRef]
- Sotoudeh, H.; Sarrami, A.H.; Wang, J.X.; Saadatpour, Z.; Razaei, A.; Gaddamanugu, S.; Choudhary, G.; Shafaat, O.; Singhal, A. Susceptibility-Weighted Imaging in Neurodegenerative Disorders: A Review. J. Neuroimaging 2021, 31, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Gauberti, M.; Martinez de Lizarrondo, S. Molecular MRI of Neuroinflammation: Time to Overcome the Translational Roadblock. Neuroscience 2021, 474, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Beaino, W.; Janssen, B.; Vugts, D.J.; de Vries, H.E.; Windhorst, A.D. Towards PET imaging of the dynamic phenotypes of microglia. Clin. Exp. Immunol. 2021, 206, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Chen, W. In vivo X-Nuclear MRS Imaging Methods for Quantitative Assessment of Neuroenergetic Biomarkers in Studying Brain Function and Aging. Front. Aging Neurosci. 2018, 10, 394. [Google Scholar] [CrossRef] [PubMed]
- Henning, A. Proton and multinuclear magnetic resonance spectroscopy in the human brain at ultra-high field strength: A review. Neuroimage 2018, 168, 181–198. [Google Scholar] [CrossRef]
- Bale, G.; Elwell, C.E.; Tachtsidis, I. From Jobsis to the present day: A review of clinical near-infrared spectroscopy measurements of cerebral cytochrome-c-oxidase. J. Biomed. Opt. 2016, 21, 091307. [Google Scholar] [CrossRef]
- Nasrallah, I.; Dubroff, J. An overview of PET neuroimaging. Semin. Nucl. Med. 2013, 43, 449–461. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Subrahmanian, N.; LaVoie, M.J. Is there a special relationship between complex I activity and nigral neuronal loss in Parkinson’s disease? A critical reappraisal. Brain Res. 2021, 1767, 147434. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed]
- Ranganayaki, S.; Jamshidi, N.; Aiyaz, M.; Rashmi, S.K.; Gayathri, N.; Harsha, P.K.; Padmanabhan, B.; Srinivas Bharath, M.M. Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration. Sci. Rep. 2021, 11, 1483. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, N.; Boroujeni, M.E.; Abdollahifar, M.A.; Piryaei, A.; Khodagholi, F.; Mirbehbahani, S.H.; Siroosi, S.; Moghaddam, M.H.; Aliaghaei, A.; Sadeghi, Y. Transplantation of human dental pulp stem cells compensates for striatal atrophy and modulates neuro-inflammation in 3-nitropropionic acid rat model of Huntington’s disease. Neurosci. Res. 2021, 170, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Morais, F.M.; Ribeiro, A.M.; Moreira, F.A.; Silva, P.V.G. Systematic review and meta-analysis on the role of mitochondrial cytochrome c oxidase in Alzheimer’s disease. Acta Neuropsychiatr. 2021, 33, 55–64. [Google Scholar] [CrossRef]
- Buonocore, M.H.; Maddock, R.J. Magnetic resonance spectroscopy of the brain: A review of physical principles and technical methods. Rev. Neurosci. 2015, 26, 609–632. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Yu, X. Assessing tissue metabolism by phosphorous-31 magnetic resonance spectroscopy and imaging: A methodology review. Quant. Imaging Med. Surg. 2017, 7, 707–726. [Google Scholar] [CrossRef]
- Das, N.; Ren, J.; Spence, J.; Chapman, S.B. Phosphate Brain Energy Metabolism and Cognition in Alzheimer’s Disease: A Spectroscopy Study Using Whole-Brain Volume-Coil 31Phosphorus Magnetic Resonance Spectroscopy at 7Tesla. Front. Neurosci. 2021, 15, 641739. [Google Scholar] [CrossRef]
- Rango, M.; Dossi, G.; Squarcina, L.; Bonifati, C. Brain mitochondrial impairment in early-onset Parkinson’s disease with or without PINK1 mutation. Mov. Disord. 2020, 35, 504–507. [Google Scholar] [CrossRef]
- Stamelou, M.; Pilatus, U.; Reuss, A.; Magerkurth, J.; Eggert, K.M.; Knake, S.; Ruberg, M.; Schade-Brittinger, C.; Oertel, W.H.; Hoglinger, G.U. In vivo evidence for cerebral depletion in high-energy phosphates in progressive supranuclear palsy. J. Cereb. Blood Flow Metab. 2009, 29, 861–870. [Google Scholar] [CrossRef]
- Mochel, F.; N’Guyen, T.M.; Deelchand, D.; Rinaldi, D.; Valabregue, R.; Wary, C.; Carlier, P.G.; Durr, A.; Henry, P.G. Abnormal response to cortical activation in early stages of Huntington disease. Mov. Disord. 2012, 27, 907–910. [Google Scholar] [CrossRef]
- Lu, M.; Zhu, X.H.; Zhang, Y.; Chen, W. Intracellular redox state revealed by in vivo (31) P MRS measurement of NAD(+) and NADH contents in brains. Magn. Reson. Med. 2014, 71, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhu, X.H.; Chen, W. In vivo (31) P MRS assessment of intracellular NAD metabolites and NAD(+) /NADH redox state in human brain at 4 T. NMR Biomed. 2016, 29, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Chong, R.; Wakade, C.; Seamon, M.; Giri, B.; Morgan, J.; Purohit, S. Niacin Enhancement for Parkinson’s Disease: An Effectiveness Trial. Front. Aging Neurosci. 2021, 13, 667032. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Zhu, X.H.; Qiao, H.; Zhang, X.; Chen, W. Efficient in vivo 31P magnetization transfer approach for noninvasively determining multiple kinetic parameters and metabolic fluxes of ATP metabolism in the human brain. Magn. Reson. Med. 2007, 57, 103–114. [Google Scholar] [CrossRef]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaard, S.J.; Dumas, E.M.; Teeuwisse, W.M.; Kan, H.E.; Webb, A.; van Buchem, M.A.; Roos, R.A.; van der Grond, J. Longitudinal metabolite changes in Huntington’s disease during disease onset. J. Huntingtons Dis. 2014, 3, 377–386. [Google Scholar] [CrossRef]
- Harada, N.; Nishiyama, S.; Kanazawa, M.; Tsukada, H. Development of novel PET probes, [18F]BCPP-EF, [18F]BCPP-BF, and [11C]BCPP-EM for mitochondrial complex 1 imaging in the living brain. J. Labelled Comp. Radiopharm. 2013, 56, 553–561. [Google Scholar] [CrossRef]
- Tsukada, H.; Nishiyama, S.; Fukumoto, D.; Kanazawa, M.; Harada, N. Novel PET probes 18F-BCPP-EF and 18F-BCPP-BF for mitochondrial complex I: A PET study in comparison with 18F-BMS-747158-02 in rat brain. J. Nucl. Med. 2014, 55, 473–480. [Google Scholar] [CrossRef]
- Tsukada, H.; Ohba, H.; Kanazawa, M.; Kakiuchi, T.; Harada, N. Evaluation of 18F-BCPP-EF for mitochondrial complex 1 imaging in the brain of conscious monkeys using PET. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 755–763. [Google Scholar] [CrossRef]
- Tsukada, H.; Kanazawa, M.; Ohba, H.; Nishiyama, S.; Harada, N.; Kakiuchi, T. PET Imaging of Mitochondrial Complex I with 18F-BCPP-EF in the Brains of MPTP-Treated Monkeys. J. Nucl. Med. 2016, 57, 950–953. [Google Scholar] [CrossRef]
- Wilson, H.; Pagano, G.; de Natale, E.R.; Mansur, A.; Caminiti, S.P.; Polychronis, S.; Middleton, L.T.; Price, G.; Schmidt, K.F.; Gunn, R.N.; et al. Mitochondrial Complex 1, Sigma 1, and Synaptic Vesicle 2A in Early Drug-Naive Parkinson’s Disease. Mov. Disord. 2020, 35, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Therriault, J.; Kang, M.S.P.; Savard, M.; Pascoal, T.A.; Lussier, F.; Tissot, C.; Wang, Y.T.; Benedet, A.; Matsudaira, T.; et al. Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Mansur, A.; Rabiner, E.A.; Tsukada, H.; Comley, R.A.; Lewis, Y.; Huiban, M.; Passchier, J.; Gunn, R.N. Test-retest variability and reference region-based quantification of (18)F-BCPP-EF for imaging mitochondrial complex I in the human brain. J. Cereb. Blood Flow Metab. 2021, 41, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Lange, F.; Dunne, L.; Hale, L.; Tachtsidis, I. MAESTROS: A Multiwavelength Time-Domain NIRS System to Monitor Changes in Oxygenation and Oxidation State of Cytochrome-C-Oxidase. IEEE J. Sel. Top. Quantum Electron. 2019, 25, 7100312. [Google Scholar] [CrossRef]
- Kovacsova, Z.; Bale, G.; Mitra, S.; de Roever, I.; Meek, J.; Robertson, N.; Tachtsidis, I. Investigation of Confounding Factors in Measuring Tissue Saturation with NIRS Spatially Resolved Spectroscopy. Adv. Exp. Med. Biol. 2018, 1072, 307–312. [Google Scholar] [CrossRef]
- Caldwell, M.; Scholkmann, F.; Wolf, U.; Wolf, M.; Elwell, C.; Tachtsidis, I. Modelling confounding effects from extracerebral contamination and systemic factors on functional near-infrared spectroscopy. Neuroimage 2016, 143, 91–105. [Google Scholar] [CrossRef]
- Russell-Buckland, J.; Kaynezhad, P.; Mitra, S.; Bale, G.; Bauer, C.; Lingam, I.; Meehan, C.; Avdic-Belltheus, A.; Martinello, K.; Bainbridge, A.; et al. Systems Biology Model of Cerebral Oxygen Delivery and Metabolism During Therapeutic Hypothermia: Application to the Piglet Model. Adv. Exp. Med. Biol. 2021, 1269, 31–38. [Google Scholar] [CrossRef]
- Mintun, M.A.; Raichle, M.E.; Martin, W.R.; Herscovitch, P. Brain oxygen utilization measured with O-15 radiotracers and positron emission tomography. J. Nucl. Med. 1984, 25, 177–187. [Google Scholar]
- Subramanyam, R.; Alpert, N.M.; Hoop, B., Jr.; Brownell, G.L.; Taveras, J.M. A model for regional cerebral oxygen distribution during continuous inhalation of 15O2, C15O, and C15O2. J. Nucl. Med. 1978, 19, 48–53. [Google Scholar] [CrossRef]
- Fan, A.P.; An, H.; Moradi, F.; Rosenberg, J.; Ishii, Y.; Nariai, T.; Okazawa, H.; Zaharchuk, G. Quantification of brain oxygen extraction and metabolism with [15O]-gas PET: A technical review in the era of PET/MRI. Neuroimage 2020, 220, 117136. [Google Scholar] [CrossRef]
- Kudomi, N.; Hirano, Y.; Koshino, K.; Hayashi, T.; Watabe, H.; Fukushima, K.; Moriwaki, H.; Teramoto, N.; Iihara, K.; Iida, H. Rapid quantitative CBF and CMRO2 measurements from a single PET scan with sequential administration of dual 15O-labeled tracers. J. Cereb. Blood Flow Metab. 2013, 33, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.C.; Jones, T. Oxygen metabolism, oxygen extraction and positron emission tomography: Historical perspective and impact on basic and clinical neuroscience. Neuroimage 2012, 61, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Kudomi, N.; Sasakawa, Y.; Monden, T.; Kato, K.; Yamamoto, Y.; Kawai, N.; Nishiyama, Y. Applicability of emission-based attenuation map for rapid CBF, OEF, and CMRO2 measurements using gaseous 15O-labeled compounds. EJNMMI Phys. 2015, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.U.; Mikkelsen, I.K.; Aanerud, J.; Espelund, U.S.; Tietze, A.; Oettingen, G.V.; Juul, N.; Nikolajsen, L.; Ostergaard, L.; Rasmussen, M. Ephedrine versus Phenylephrine Effect on Cerebral Blood Flow and Oxygen Consumption in Anesthetized Brain Tumor Patients: A Randomized Clinical Trial. Anesthesiology 2020, 133, 304–317. [Google Scholar] [CrossRef]
- Lin, W.; Powers, W.J. Oxygen metabolism in acute ischemic stroke. J. Cereb. Blood Flow Metab. 2018, 38, 1481–1499. [Google Scholar] [CrossRef]
- Frackowiak, R.S.; Lenzi, G.L.; Jones, T.; Heather, J.D. Quantitative measurement of regional cerebral blood flow and oxygen metabolism in man using 15O and positron emission tomography: Theory, procedure, and normal values. J. Comput. Assist. Tomogr. 1980, 4, 727–736. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kanno, I.; Uemura, K.; Shishido, F.; Inugami, A.; Ogawa, T.; Murakami, M.; Suzuki, K. Reduction in regional cerebral metabolic rate of oxygen during human aging. Stroke 1986, 17, 1220–1228. [Google Scholar] [CrossRef]
- Zhu, X.H.; Chen, W. In vivo17O MRS imaging—Quantitative assessment of regional oxygen consumption and perfusion rates in living brain. Anal. Biochem. 2017, 529, 171–178. [Google Scholar] [CrossRef]
- Iguchi, S.; Moriguchi, T.; Yamazaki, M.; Hori, Y.; Koshino, K.; Toyoda, K.; Teuho, J.; Shimochi, S.; Terakawa, Y.; Fukuda, T.; et al. System evaluation of automated production and inhalation of 15O-labeled gaseous radiopharmaceuticals for the rapid 15O-oxygen PET examinations. EJNMMI Phys. 2018, 5, 37. [Google Scholar] [CrossRef]
- Zhu, X.H.; Zhang, N.; Zhang, Y.; Zhang, X.; Ugurbil, K.; Chen, W. In vivo 17O NMR approaches for brain study at high field. NMR Biomed. 2005, 18, 83–103. [Google Scholar] [CrossRef]
- Ishii, K.; Kitagaki, H.; Kono, M.; Mori, E. Decreased medial temporal oxygen metabolism in Alzheimer’s disease shown by PET. J. Nucl. Med. 1996, 37, 1159–1165. [Google Scholar] [PubMed]
- Anderson, K.E.; Brickman, A.M.; Flynn, J.; Scarmeas, N.; Van Heertum, R.; Sackeim, H.; Marder, K.S.; Bell, K.; Moeller, J.R.; Stern, Y. Impairment of nonverbal recognition in Alzheimer disease: A PET O-15 study. Neurology 2007, 69, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Albani, G.; Kunig, G.; Soelch, C.M.; Mauro, A.; Priano, L.; Martignoni, E.; Leenders, K.L. The role of language areas in motor control dysfunction in Parkinson’s disease. Neurol. Sci. 2001, 22, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, H.; Kudo, T. Clinical impact of hemodynamic parameter measurement for cerebrovascular disease using positron emission tomography and 15O-labeled tracers. Ann. Nucl. Med. 2009, 23, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Zhang, Y.; Tian, R.X.; Lei, H.; Zhang, N.; Zhang, X.; Merkle, H.; Ugurbil, K.; Chen, W. Development of 17O NMR approach for fast imaging of cerebral metabolic rate of oxygen in rat brain at high field. Proc. Natl. Acad. Sci. USA 2002, 99, 13194–13199. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Y.; Ugurbil, K.; Chen, W.; Zhu, X.H. In vitro and in vivo studies of 17O NMR sensitivity at 9.4 and 16.4 T. Magn. Reson. Med. 2013, 69, 1523–1527. [Google Scholar] [CrossRef]
- Zhu, X.H.; Chen, J.M.; Tu, T.W.; Chen, W.; Song, S.K. Simultaneous and noninvasive imaging of cerebral oxygen metabolic rate, blood flow and oxygen extraction fraction in stroke mice. Neuroimage 2013, 64, 437–447. [Google Scholar] [CrossRef]
- Zhu, X.H.; Zhang, Y.; Zhang, N.; Ugurbil, K.; Chen, W. Noninvasive and three-dimensional imaging of CMRO2 in rats at 9.4 T: Reproducibility test and normothermia/hypothermia comparison study. J. Cereb. Blood Flow Metab. 2007, 27, 1225–1234. [Google Scholar] [CrossRef]
- Zhang, N.; Zhu, X.H.; Lei, H.; Ugurbil, K.; Chen, W. Simplified methods for calculating cerebral metabolic rate of oxygen based on 17O magnetic resonance spectroscopic imaging measurement during a short 17O2 inhalation. J. Cereb. Blood Flow Metab. 2004, 24, 840–848. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, Y.; Ugurbil, K.; Chen, W. 3D imaging of CMRO2 in rat brain at different temperature using high-field 17O NMR approach. In Proceedings of the International Society of Magnetic Resonance Medicine, Toronto, ON, Canada, 10–16 July 2003; p. 569. [Google Scholar]
- Zhu, X.H.; Zhang, N.; Zhang, Y.; Ugurbil, K.; Chen, W. New insights into central roles of cerebral oxygen metabolism in the resting and stimulus-evoked brain. J. Cereb. Blood Flow Metab. 2009, 29, 10–18. [Google Scholar] [CrossRef]
- Atkinson, I.C.; Thulborn, K.R. Feasibility of mapping the tissue mass corrected bioscale of cerebral metabolic rate of oxygen consumption using 17-oxygen and 23-sodium MR imaging in a human brain at 9.4 T. Neuroimage 2010, 51, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.H.; Begovatz, P.; Nagel, A.M.; Umathum, R.; Schommer, K.; Bachert, P.; Bock, M. A measurement setup for direct 17O MRI at 7 T. Magn. Reson. Med. 2011, 66, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Niesporek, S.C.; Umathum, R.; Lommen, J.M.; Behl, N.G.R.; Paech, D.; Bachert, P.; Ladd, M.E.; Nagel, A.M. Reproducibility of CMRO2 determination using dynamic 17O MRI. Magn. Reson. Med. 2018, 79, 2923–2934. [Google Scholar] [CrossRef]
- Zhu, X.H.; Chen, W. In vivo oxygen-17 NMR for imaging brain oxygen metabolism at high field. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 319–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, W.; Zhu, X.H.; Vollmers, M.L.; Colonna, E.T.; Adriany, G.; Tramm, B.; Dubinsky, J.M.; Oz, G. Non-invasive measurement of cerebral oxygen metabolism in the mouse brain by ultra-high field 17O MR spectroscopy. J. Cereb. Blood Flow Metab. 2013, 33, 1846–1849. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Lepak, V.C.; Eberly, L.E.; Roth, B.; Cui, W.; Zhu, X.H.; Oz, G.; Dubinsky, J.M. Oxygen consumption deficit in Huntington disease mouse brain under metabolic stress. Hum. Mol. Genet. 2016, 25, 2813–2826. [Google Scholar] [CrossRef]
- Mellon, E.A.; Beesam, R.S.; Baumgardner, J.E.; Borthakur, A.; Witschey, W.R., 2nd; Reddy, R. Estimation of the regional cerebral metabolic rate of oxygen consumption with proton detected 17O MRI during precision 17O2 inhalation in swine. J. Neurosci. Methods 2009, 179, 29–39. [Google Scholar] [CrossRef]
- Buxton, R.B.; Frank, L.R. A model for the coupling between cerebral blood flow and oxygen metabolism during neural stimulation. J. Cereb. Blood Flow Metab. 1997, 17, 64–72. [Google Scholar] [CrossRef]
- Hayashi, T.; Watabe, H.; Kudomi, N.; Kim, K.M.; Enmi, J.; Hayashida, K.; Iida, H. A theoretical model of oxygen delivery and metabolism for physiologic interpretation of quantitative cerebral blood flow and metabolic rate of oxygen. J. Cereb. Blood Flow Metab. 2003, 23, 1314–1323. [Google Scholar] [CrossRef]
- Hyder, F.; Shulman, R.G.; Rothman, D.L. A model for the regulation of cerebral oxygen delivery. J. Appl. Physiol. 1998, 85, 554–564. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lundstrom, B.N.; Snyder, A.Z.; Vlassenko, A.G.; Shulman, G.L.; Raichle, M.E. Blood flow and oxygen delivery to human brain during functional activity: Theoretical modeling and experimental data. Proc. Natl. Acad. Sci. USA 2001, 98, 6859–6864. [Google Scholar] [CrossRef] [PubMed]
- Vafaee, M.S.; Gjedde, A. Model of blood-brain transfer of oxygen explains nonlinear flow-metabolism coupling during stimulation of visual cortex. J. Cereb. Blood Flow Metab. 2000, 20, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Valabregue, R.; Aubert, A.; Burger, J.; Bittoun, J.; Costalat, R. Relation between cerebral blood flow and metabolism explained by a model of oxygen exchange. J. Cereb. Blood Flow Metab. 2003, 23, 536–545. [Google Scholar] [CrossRef]
- Bulte, D.P.; Kelly, M.; Germuska, M.; Xie, J.; Chappell, M.A.; Okell, T.W.; Bright, M.G.; Jezzard, P. Quantitative measurement of cerebral physiology using respiratory-calibrated MRI. Neuroimage 2012, 60, 582–591. [Google Scholar] [CrossRef]
- Jain, V.; Langham, M.C.; Wehrli, F.W. MRI estimation of global brain oxygen consumption rate. J. Cereb. Blood Flow Metab. 2010, 30, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ge, Y. Quantitative evaluation of oxygenation in venous vessels using T2-Relaxation-Under-Spin-Tagging MRI. Magn. Reson. Med. 2008, 60, 357–363. [Google Scholar] [CrossRef]
- An, H.; Lin, W.; Celik, A.; Lee, Y.Z. Quantitative measurements of cerebral metabolic rate of oxygen utilization using MRI: A volunteer study. NMR Biomed. 2001, 14, 441–447. [Google Scholar] [CrossRef]
- Christen, T.; Bolar, D.S.; Zaharchuk, G. Imaging brain oxygenation with MRI using blood oxygenation approaches: Methods, validation, and clinical applications. AJNR Am. J. Neuroradiol. 2013, 34, 1113–1123. [Google Scholar] [CrossRef]
- Hoge, R.D. Calibrated FMRI. Neuroimage 2012, 62, 930–937. [Google Scholar] [CrossRef]
- Germuska, M.; Chandler, H.L.; Stickland, R.C.; Foster, C.; Fasano, F.; Okell, T.W.; Steventon, J.; Tomassini, V.; Murphy, K.; Wise, R.G. Dual-calibrated fMRI measurement of absolute cerebral metabolic rate of oxygen consumption and effective oxygen diffusivity. Neuroimage 2019, 184, 717–728. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Panahi, Y.; Javadi, B.; Sahebkar, A. The Underlying Role of Oxidative Stress in Neurodegeneration: A Mechanistic Review. CNS Neurol. Disord. Drug Targets 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox. Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Lassegue, B.; Ushio-Fukai, M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox. Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Chance, B.; Schoener, B.; Oshino, R.; Itshak, F.; Nakase, Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J. Biol. Chem. 1979, 254, 4764–4771. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Hulbert, A.J.; Pamplona, R.; Buffenstein, R.; Buttemer, W.A. Life and death: Metabolic rate, membrane composition, and life span of animals. Physiol. Rev. 2007, 87, 1175–1213. [Google Scholar] [CrossRef]
- Chomyn, A.; Attardi, G. MtDNA mutations in aging and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 519–529. [Google Scholar] [CrossRef]
- Park, C.B.; Larsson, N.G. Mitochondrial DNA mutations in disease and aging. J. Cell. Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Nekhaeva, E.; Bodyak, N.B.; Khrapko, K. Mutation and intracellular clonal expansion of mitochondrial genomes: Two synergistic components of the aging process? Mech. Ageing Dev. 2003, 124, 49–53. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Zhang, Z.; Li, H.; Yu, Q.; Douglas, J.T.; Bratasz, A.; Kuppusamy, P.; Yan, S.S. Increased Electron Paramagnetic Resonance Signal Correlates with Mitochondrial Dysfunction and Oxidative Stress in an Alzheimer’s disease Mouse Brain. J. Alzheimers Dis. 2016, 51, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.J.; Kim, H.J.; Shin, C.Y.; Han, S.H. Melatonin Potentiates the Neuroprotective Properties of Resveratrol Against Beta-Amyloid-Induced Neurodegeneration by Modulating AMP-Activated Protein Kinase Pathways. J. Clin. Neurol. 2010, 6, 127–137. [Google Scholar] [CrossRef]
- Cui, K.; Luo, X.; Xu, K.; Ven Murthy, M.R. Role of oxidative stress in neurodegeneration: Recent developments in assay methods for oxidative stress and nutraceutical antioxidants. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 771–799. [Google Scholar] [CrossRef]
- Berkowitz, B.A. Oxidative stress measured in vivo without an exogenous contrast agent using QUEST MRI. J. Magn. Reson. 2018, 291, 94–100. [Google Scholar] [CrossRef]
- Terpstra, M.; Tkac, I.; Rao, R.; Gruetter, R. Quantification of vitamin C in the rat brain in vivo using short echo-time 1H MRS. Magn. Reson. Med. 2006, 55, 979–983. [Google Scholar] [CrossRef]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels—A novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Shukla, D.; Mandal, P.K.; Ersland, L.; Gruner, E.R.; Tripathi, M.; Raghunathan, P.; Sharma, A.; Chaithya, G.R.; Punjabi, K.; Splaine, C. A Multi-Center Study on Human Brain Glutathione Conformation using Magnetic Resonance Spectroscopy. J. Alzheimers Dis. 2018, 66, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Cho, T.; Jantaratnotai, N.; Wang, Y.T.; McGeer, E.; McGeer, P.L. Depletion of GSH in glial cells induces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J. 2010, 24, 2533–2545. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Dwivedi, D.; Megha, K.; Mishra, R.; Mandal, P.K. Glutathione in Brain: Overview of Its Conformations, Functions, Biochemical Characteristics, Quantitation and Potential Therapeutic Role in Brain Disorders. Neurochem. Res. 2020, 45, 1461–1480. [Google Scholar] [CrossRef]
- Mailloux, R.J.; McBride, S.L.; Harper, M.E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602. [Google Scholar] [CrossRef]
- Shen, D.; Dalton, T.P.; Nebert, D.W.; Shertzer, H.G. Glutathione redox state regulates mitochondrial reactive oxygen production. J. Biol. Chem. 2005, 280, 25305–25312. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.J.; Pappa, A.; Panayiotidis, M.I. The central role of glutathione in the pathophysiology of human diseases. Arch. Physiol. Biochem. 2007, 113, 234–258. [Google Scholar] [CrossRef]
- Sinha, R.; Sinha, I.; Calcagnotto, A.; Trushin, N.; Haley, J.S.; Schell, T.D.; Richie, J.P., Jr. Oral supplementation with liposomal glutathione elevates body stores of glutathione and markers of immune function. Eur. J. Clin. Nutr. 2018, 72, 105–111. [Google Scholar] [CrossRef]
- Mandal, P.K.; Shukla, D.; Tripathi, M.; Ersland, L. Cognitive Improvement with Glutathione Supplement in Alzheimer’s Disease: A Way Forward. J. Alzheimers Dis. 2019, 68, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Moumen, R.; Nouvelot, A.; Duval, D.; Lechevalier, B.; Viader, F. Plasma superoxide dismutase and glutathione peroxidase activity in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1997, 151, 35–39. [Google Scholar] [CrossRef]
- Paschalis, V.; Theodorou, A.A.; Margaritelis, N.V.; Kyparos, A.; Nikolaidis, M.G. N-acetylcysteine supplementation increases exercise performance and reduces oxidative stress only in individuals with low levels of glutathione. Free Radic. Biol. Med. 2018, 115, 288–297. [Google Scholar] [CrossRef]
- Choi, I.Y.; Andronesi, O.C.; Barker, P.; Bogner, W.; Edden, R.A.E.; Kaiser, L.G.; Lee, P.; Marjanska, M.; Terpstra, M.; de Graaf, R.A. Spectral editing in (1) H magnetic resonance spectroscopy: Experts’ consensus recommendations. NMR Biomed. 2021, 34, e4411. [Google Scholar] [CrossRef]
- Shukla, D.; Mandal, P.K.; Tripathi, M.; Vishwakarma, G.; Mishra, R.; Sandal, K. Quantitation of in vivo brain glutathione conformers in cingulate cortex among age-matched control, MCI, and AD patients using MEGA-PRESS. Hum. Brain Mapp. 2020, 41, 194–217. [Google Scholar] [CrossRef]
- Mischley, L.K.; Conley, K.E.; Shankland, E.G.; Kavanagh, T.J.; Rosenfeld, M.E.; Duda, J.E.; White, C.C.; Wilbur, T.K.; De La Torre, P.U.; Padowski, J.M. Central nervous system uptake of intranasal glutathione in Parkinson’s disease. NPJ. Parkinsons Dis. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Nakamoto, Y.; Yoneda, M. PET Imaging for Oxidative Stress in Neurodegenerative Disorders Associated with Mitochondrial Dysfunction. Antioxidants 2020, 9, 861. [Google Scholar] [CrossRef]
- Yoshii, Y.; Yoneda, M.; Ikawa, M.; Furukawa, T.; Kiyono, Y.; Mori, T.; Yoshii, H.; Oyama, N.; Okazawa, H.; Saga, T.; et al. Radiolabeled Cu-ATSM as a novel indicator of overreduced intracellular state due to mitochondrial dysfunction: Studies with mitochondrial DNA-less rho0 cells and cybrids carrying MELAS mitochondrial DNA mutation. Nucl. Med. Biol. 2012, 39, 177–185. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Liddell, J.R.; Lim, S.; Paterson, B.M.; Cater, M.A.; Savva, M.S.; Mot, A.I.; James, J.L.; Trounce, I.A.; White, A.R.; et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copperII. Proc. Natl. Acad. Sci. USA 2012, 109, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Okazawa, H.; Kudo, T.; Kuriyama, M.; Fujibayashi, Y.; Yoneda, M. Evaluation of striatal oxidative stress in patients with Parkinson’s disease using [62Cu]ATSM PET. Nucl. Med. Biol. 2011, 38, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Schenck, J.F.; Zimmerman, E.A. High-field magnetic resonance imaging of brain iron: Birth of a biomarker? NMR Biomed. 2004, 17, 433–445. [Google Scholar] [CrossRef]
- Haacke, E.M.; Ayaz, M.; Khan, A.; Manova, E.S.; Krishnamurthy, B.; Gollapalli, L.; Ciulla, C.; Kim, I.; Petersen, F.; Kirsch, W. Establishing a baseline phase behavior in magnetic resonance imaging to determine normal vs. abnormal iron content in the brain. J. Magn. Reson. Imaging 2007, 26, 256–264. [Google Scholar] [CrossRef]
- Biasiotto, G.; Filosto, M.; Zanella, I. Editorial: Iron and Neurodegeneration. Front. Neurosci. 2019, 13, 1382. [Google Scholar] [CrossRef]
- Dietrich, O.; Levin, J.; Ahmadi, S.A.; Plate, A.; Reiser, M.F.; Botzel, K.; Giese, A.; Ertl-Wagner, B. MR imaging differentiation of Fe2+ and Fe3+ based on relaxation and magnetic susceptibility properties. Neuroradiology 2017, 59, 403–409. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, 3, CD007176. [Google Scholar] [CrossRef]
- Berkowitz, B.A.; Podolsky, R.H.; Childers, K.L.; Gow, A.; Schneider, B.L.; Lloyd, S.C.; Bosse, K.E.; Conti, A.C.; Roberts, R.; Berri, A.M.; et al. Age-related murine hippocampal CA1 laminae oxidative stress measured in vivo by QUEnch-assiSTed (QUEST) MRI: Impact of isoflurane anesthesia. Geroscience 2020, 42, 563–574. [Google Scholar] [CrossRef]
- Kuhl, A.; Dixon, A.; Hali, M.; Apawu, A.K.; Muca, A.; Sinan, M.; Warila, J.; Braun, R.D.; Berkowitz, B.A.; Holt, A.G. Novel QUEST MRI In Vivo Measurement of Noise-induced Oxidative Stress in the Cochlea. Sci. Rep. 2019, 9, 16265. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasuhn, J.; Kunert, L.; Brüggemann, N. Neuroimaging Methods to Map In Vivo Changes of OXPHOS and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 7263. https://doi.org/10.3390/ijms23137263
Prasuhn J, Kunert L, Brüggemann N. Neuroimaging Methods to Map In Vivo Changes of OXPHOS and Oxidative Stress in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2022; 23(13):7263. https://doi.org/10.3390/ijms23137263
Chicago/Turabian StylePrasuhn, Jannik, Liesa Kunert, and Norbert Brüggemann. 2022. "Neuroimaging Methods to Map In Vivo Changes of OXPHOS and Oxidative Stress in Neurodegenerative Disorders" International Journal of Molecular Sciences 23, no. 13: 7263. https://doi.org/10.3390/ijms23137263
APA StylePrasuhn, J., Kunert, L., & Brüggemann, N. (2022). Neuroimaging Methods to Map In Vivo Changes of OXPHOS and Oxidative Stress in Neurodegenerative Disorders. International Journal of Molecular Sciences, 23(13), 7263. https://doi.org/10.3390/ijms23137263