
Organokines in Rheumatoid Arthritis: A Critical Review

 ,
, 

 ,
,  , , , ,
, , , ,
Abstract

1. Introduction
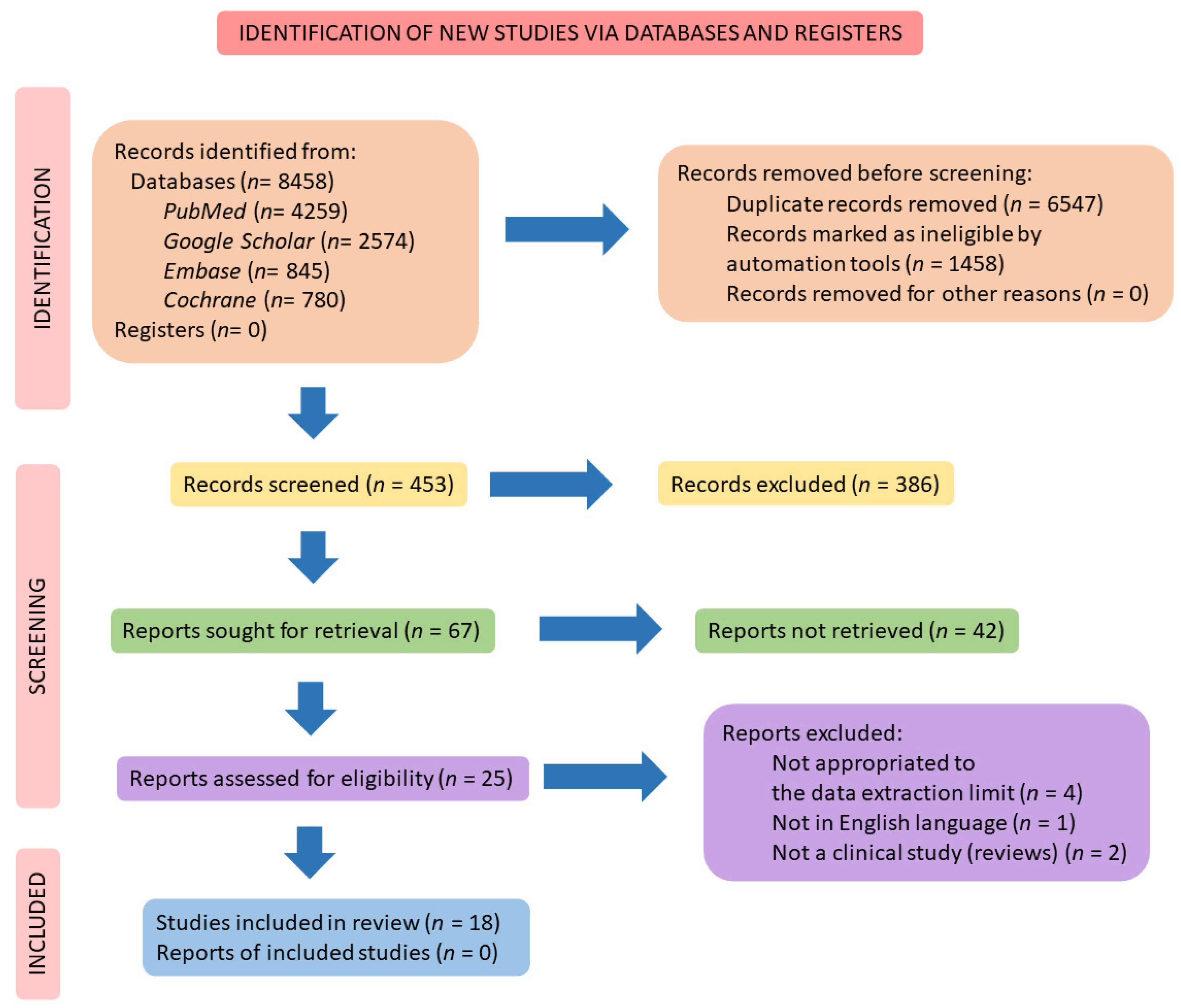
2. Materials and Methods
2.1. Focal Question
2.2. Language
2.3. Databases
2.4. Study Selection
2.5. Data Extraction
3. Results
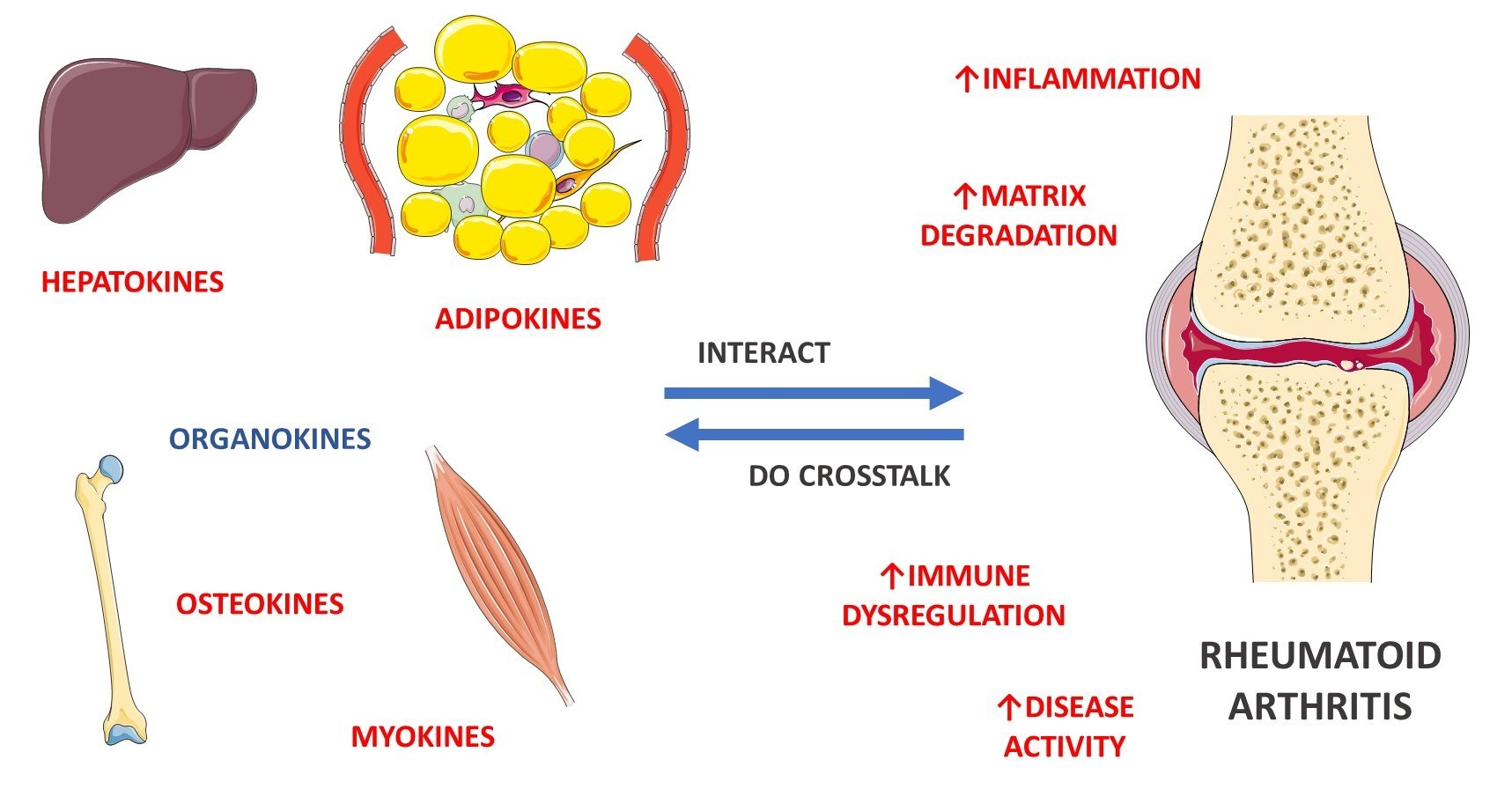
4. General Aspects of RA and the Role of Myokines, Osteokines, Adipokines, and Hepatokines
4.1. Pathophysiological Aspects of RA
4.2. Myokines in RA
4.2.1. Myostatin
4.2.2. Irisin
4.3. Osteokines in RA
4.3.1. Osteoprotegerin
4.3.2. Osteocalcin
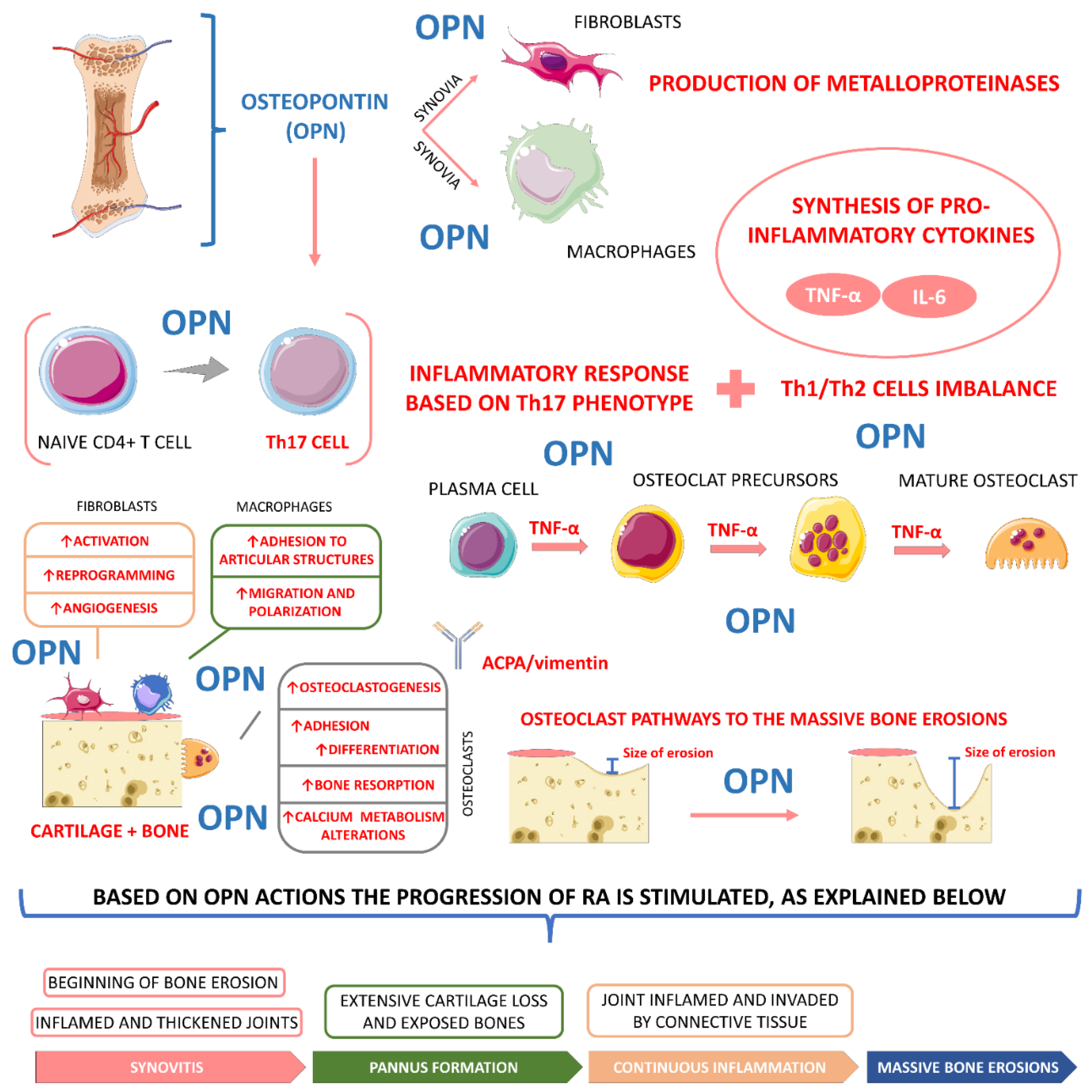
4.3.3. Osteopontin
4.3.4. Sclerostin
4.3.5. Bone Morphogenetic Proteins
4.3.6. Osteonectin (Acidic and Cysteine-Rich Secreted Protein, SPARC)
4.4. Adipokines in RA
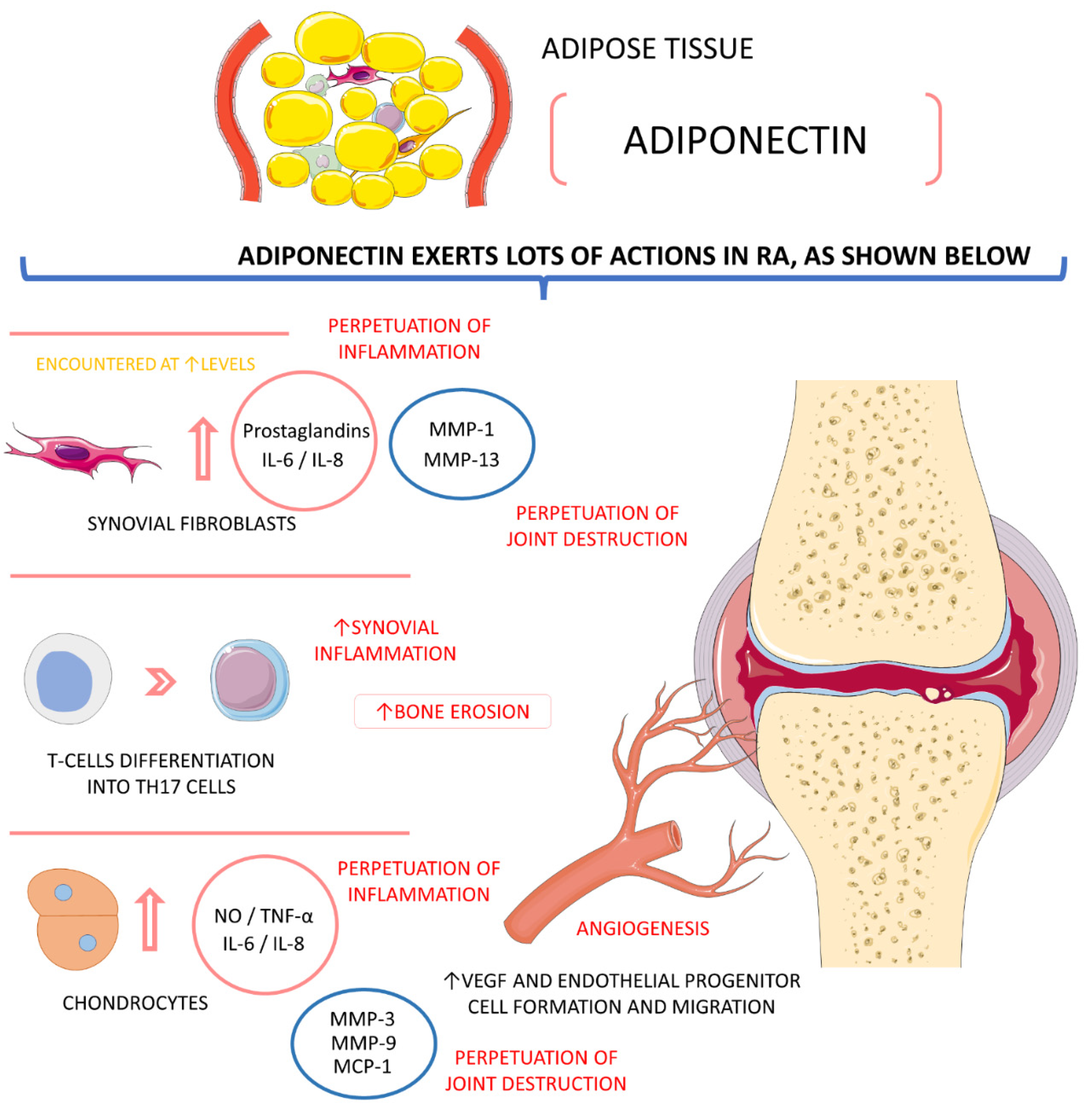
4.4.1. Adiponectin
4.4.2. Leptin
4.4.3. Visfatin (Pre-B-Cell Colony-Enhancing Factor—PBEF)
4.4.4. Omentin
4.4.5. Resistin
4.4.6. Chemerin
4.4.7. Vaspin
4.4.8. Apelin
4.5. Hepatokines in RA
4.5.1. Leukocyte Cell-Derived Chemotaxin-2 (LECT2)
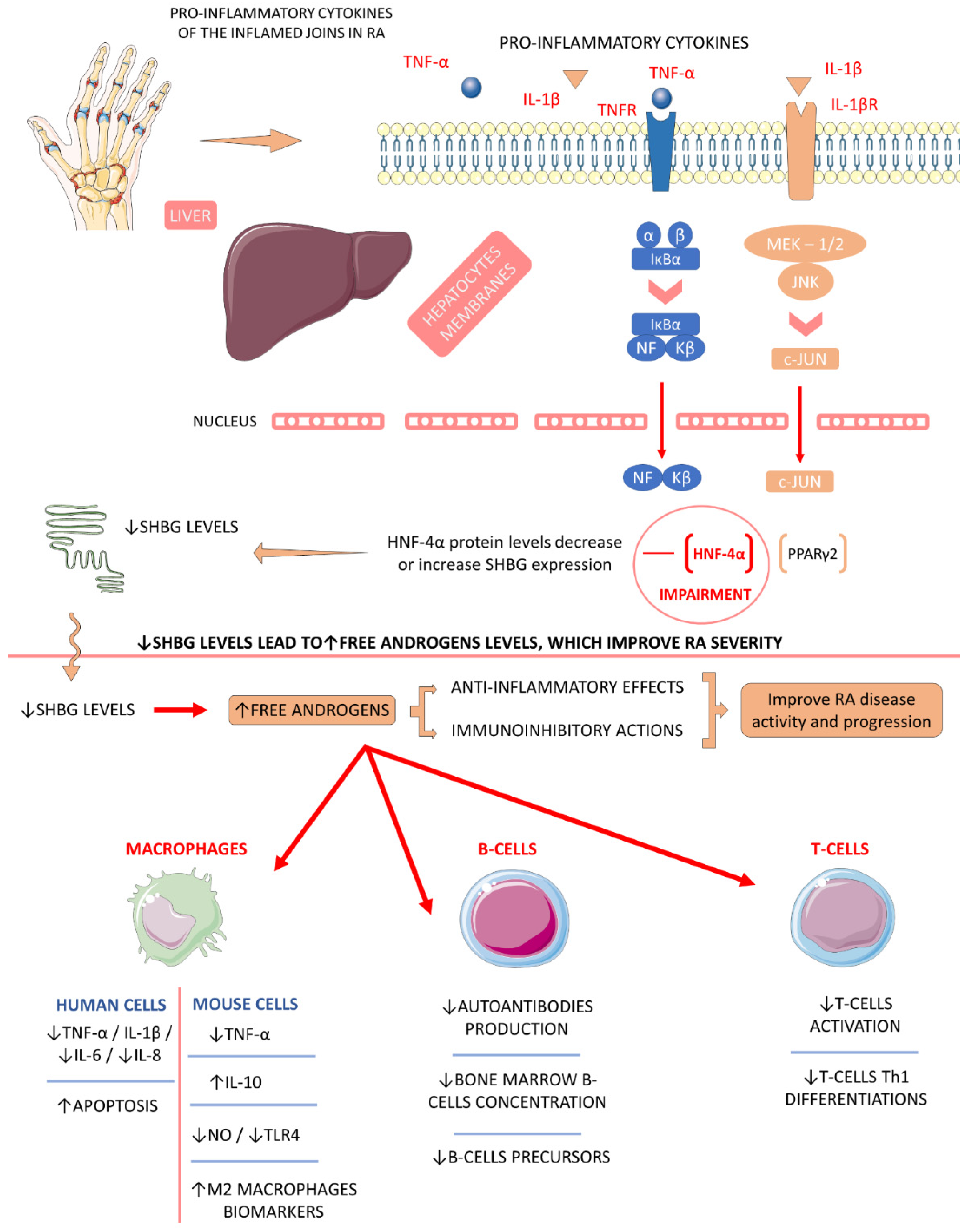
4.5.2. Sex Hormone-Binding Globulin (SHBG)
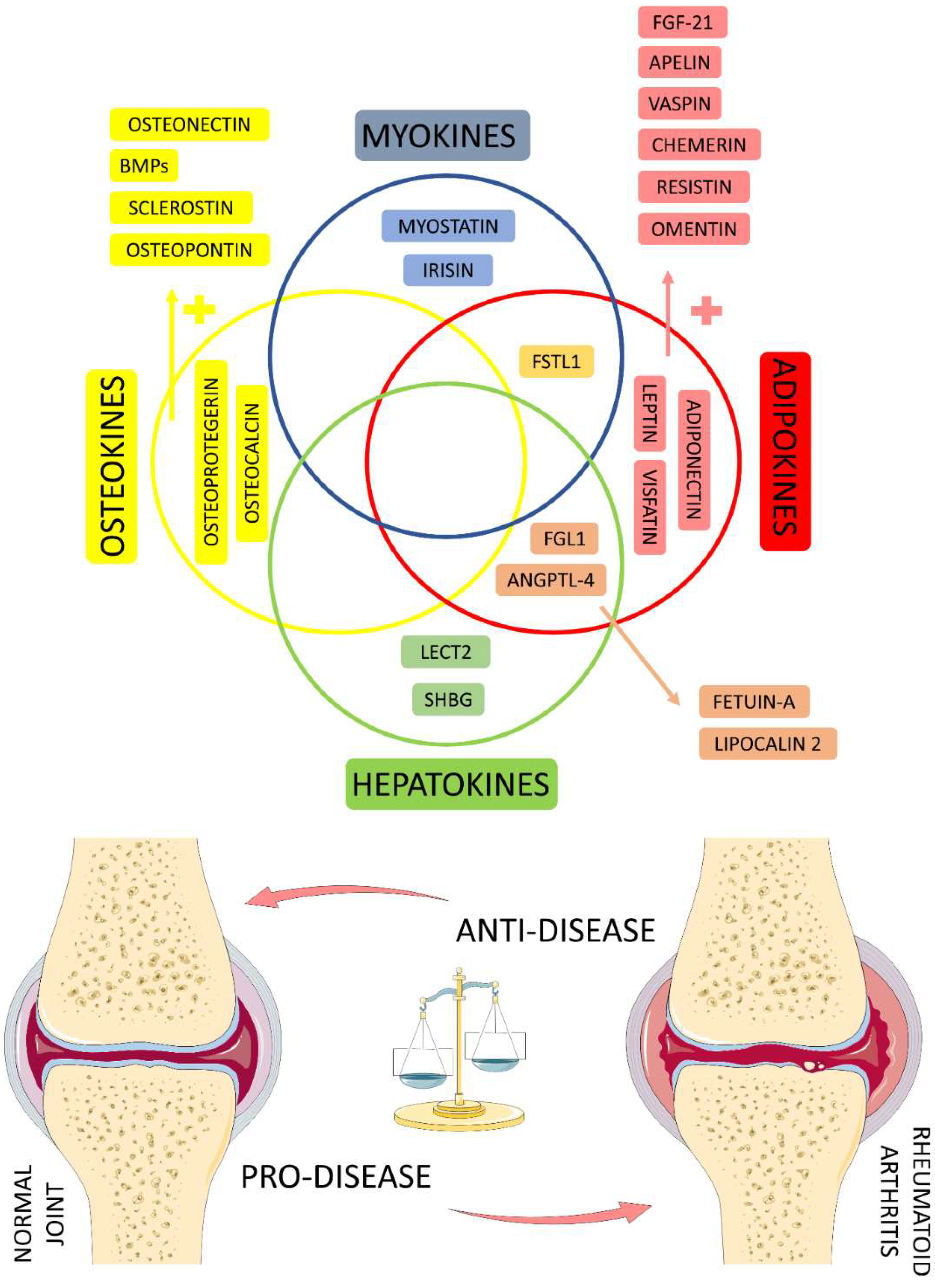
4.6. Organokines Released by Different Organs: Possible Crosstalk?
4.6.1. Fibrinogen-like Protein 1 (FGL1)
4.6.2. Angiopoietin-like 4 (ANGPTL-4)
4.6.3. Fetuin-A
4.6.4. Lipocalin 2
4.6.5. Follistatin-like 1 (FSTL1)
4.7. Studies Evaluating the Role of Organokines in RA Patients
4.8. Future Perspectives about the Role of Organokines and RA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karami, J.; Aslani, S.; Tahmasebi, M.N.; Mousavi, M.J.; Sharafat Vaziri, A.; Jamshidi, A.; Farhadi, E.; Mahmoudi, M. Epigenetics in rheumatoid arthritis; fibroblast-like synoviocytes as an emerging paradigm in the pathogenesis of the disease. Immunol. Cell Biol. 2020, 98, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Edilova, M.I.; Akram, A.; Abdul-Sater, A.A. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed. J. 2021, 44, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yu, G.; Yang, K.; Li, J.; Hao, W.; Chen, H. The Efficacy of Antioxidative Stress Therapy on Oxidative Stress Levels in Rheumatoid Arthritis: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Oxidative Med. Cell. Longev. 2021, 2021, 3302886. [Google Scholar] [CrossRef] [PubMed]
- Mititelu, R.R.; Pădureanu, R.; Băcănoiu, M.; Pădureanu, V.; Docea, A.O.; Calina, D.; Barbulescu, A.L.; Buga, A.M. Inflammatory and Oxidative Stress Markers-Mirror Tools in Rheumatoid Arthritis. Biomedicines 2020, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.-F.; Bungau, S.G. Management of Rheumatoid Arthritis: An Overview. Cells 2021, 10, 2857. [Google Scholar] [CrossRef]
- Chung, H.S.; Choi, K.M. Organokines in disease. Adv. Clin. Chem. 2020, 94, 261–321. [Google Scholar] [CrossRef]
- de Oliveira Dos Santos, A.R.; de Oliveira Zanuso, B.; Miola, V.F.B.; Barbalho, S.M.; Santos Bueno, P.C.; Flato, U.A.P.; Detregiachi, C.R.P.; Buchaim, D.V.; Buchaim, R.L.; Tofano, R.J.; et al. Adipokines, Myokines, and Hepatokines: Crosstalk and Metabolic Repercussions. Int. J. Mol. Sci. 2021, 22, 2639. [Google Scholar] [CrossRef]
- Huang, C.C.; Law, Y.Y.; Liu, S.C.; Hu, S.L.; Lin, J.A.; Chen, C.J.; Wang, S.W.; Tang, C.H. Adiponectin Promotes VEGF Expression in Rheumatoid Arthritis Synovial Fibroblasts and Induces Endothelial Progenitor Cell Angiogenesis by Inhibiting miR-106a-5p. Cells 2021, 10, 2627. [Google Scholar] [CrossRef]
- Neumann, E.; Hasseli, R.; Ohl, S.; Lange, U.; Frommer, K.W.; Müller-Ladner, U. Adipokines and Autoimmunity in Inflammatory Arthritis. Cells 2021, 10, 216. [Google Scholar] [CrossRef]
- Fatel, E.C.d.S.; Rosa, F.T.; Simão, A.N.C.; Dichi, I. Adipokines in rheumatoid arthritis. Adv. Rheumatol. 2018, 58, 25. [Google Scholar] [CrossRef]
- Santos, J.P.M.d.; Maio, M.C.d.; Lemes, M.A.; Laurindo, L.F.; Haber, J.F.d.S.; Bechara, M.D.; Prado, P.S.d.; Rauen, E.C.; Costa, F.; Pereira, B.C.d.A.; et al. Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future. Int. J. Mol. Sci. 2022, 23, 498. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Ann. Intern. Med. 2009, 151, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Cheleschi, S.; Tenti, S.; Bedogni, G.; Fioravanti, A. Circulating Mir-140 and leptin improve the accuracy of the differential diagnosis between psoriatic arthritis and rheumatoid arthritis: A case-control study. Transl. Res. 2022, 239, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Mi, S.; Zhu, J.; Jin, W.; Li, Y.; Wang, T.; Li, Y.; Fan, C. No Causal Association Between Adiponectin and the Risk of Rheumatoid Arthritis: A Mendelian Randomization Study. Front. Genet. 2021, 12, 670282. [Google Scholar] [CrossRef] [PubMed]
- Wahba, A.S.; Ibrahim, M.E.; Abo-elmatty, D.M.; Mehanna, E.T. Association of the Adipokines Chemerin, Apelin, Vaspin and Omentin and Their Functional Genetic Variants with Rheumatoid Arthritis. J. Pers. Med. 2021, 11, 976. [Google Scholar] [CrossRef]
- Wahlin, B.; Ramnemark, A.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S.; Södergren, A. Osteoprotegerin and osteocalcin are associated with atherosclerosis in patients with rheumatoid arthritis: A prospective cohort study. Clin. Exp. Rheumatol. 2021, 39, 1402–1409. [Google Scholar]
- Liu, S.; Guo, Y.; Lu, L.; Lu, J.; Ke, M.; Xu, T.; Lu, Y.; Chen, W.; Wang, J.; Kong, D.; et al. Fibrinogen-Like Protein 1 Is a Novel Biomarker for Predicting Disease Activity and Prognosis of Rheumatoid Arthritis. Front. Immunol. 2020, 11, 579228. [Google Scholar] [CrossRef]
- Qu, Z.; Huang, J.; Yang, F.; Hong, J.; Wang, W.; Yan, S. Sex hormone-binding globulin and arthritis: A Mendelian randomization study. Arthritis Res. 2020, 22, 118. [Google Scholar] [CrossRef]
- Murillo-Saich, J.D.; Vazquez-Villegas, M.L.; Ramirez-Villafaña, M.; Saldaña-Cruz, A.M.; Aceves-Aceves, J.A.; Gonzalez-Lopez, L.; Guma, M.; Gamez-Nava, J.I. Association of myostatin, a cytokine released by muscle, with inflammation in rheumatoid arthritis: A cross-sectional study. Medicine 2021, 100, e24186. [Google Scholar] [CrossRef]
- Scherer, H.U.; Häupl, T.; Burmester, G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102400. [Google Scholar] [CrossRef]
- van Delft, M.A.M.; Huizinga, T.W.J. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef] [PubMed]
- Damerau, A.; Gaber, T. Modeling Rheumatoid Arthritis In Vitro: From Experimental Feasibility to Physiological Proximity. Int. J. Mol. Sci. 2020, 21, 7916. [Google Scholar] [CrossRef] [PubMed]
- Rigby, W.; Buckner, J.H.; Louis Bridges, S., Jr.; Nys, M.; Gao, S.; Polinsky, M.; Ray, N.; Bykerk, V. HLA-DRB1 risk alleles for RA are associated with differential clinical responsiveness to abatacept and adalimumab: Data from a head-to-head, randomized, single-blind study in autoantibody-positive early RA. Arthritis Res. 2021, 23, 245. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Yang, C.; Wu, G.; Yu, X.; Tian, Q.; Chen, D.; Cao, B.; Zhao, L.; Xu, N.; Jin, S.; et al. The protein-protein interaction between connective tissue growth factor and annexin A2 is relevant to pannus formation in rheumatoid arthritis. Arthritis Res. 2021, 23, 266. [Google Scholar] [CrossRef]
- Saferding, V.; Blüml, S. Innate immunity as the trigger of systemic autoimmune diseases. J. Autoimmun. 2020, 110, 102382. [Google Scholar] [CrossRef] [PubMed]
- Celen, H.; Dens, A.C.; Ronsmans, S.; Michiels, S.; De Langhe, E. Airborne pollutants as potential triggers of systemic autoimmune rheumatic diseases: A narrative review. Acta Clin. Belg. 2021. [Google Scholar] [CrossRef]
- Adami, G.; Viapiana, O.; Rossini, M.; Orsolini, G.; Bertoldo, E.; Giollo, A.; Gatti, D.; Fassio, A. Association between environmental air pollution and rheumatoid arthritis flares. Rheumatology 2021, 60, 4591–4597. [Google Scholar] [CrossRef]
- Taylan, A.; Akinci, B.; Toprak, B.; Birlik, M.; Arslan, F.D.; Ekerbicer, H.; Gundogdu, B.; Colak, A.; Engin, B. Association of Leptin Levels and Disease Activity in Patients with Early Rheumatoid Arthritis. Arch. Med. Res. 2021, 52, 544–553. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Tuure, L.; Nieminen, R.; Laasonen, L.; Leirisalo-Repo, M.; Moilanen, E. Pretreatment resistin levels are associated with erosive disease in early rheumatoid arthritis treated with disease-modifying anti-rheumatic drugs and infliximab. Scand. J. Rheumatol. 2021, 51, 1–6. [Google Scholar] [CrossRef]
- Fang, Q.; Zhou, C.; Nandakumar, K.S. Molecular and Cellular Pathways Contributing to Joint Damage in Rheumatoid Arthritis. Mediat. Inflamm. 2020, 2020, 3830212. [Google Scholar] [CrossRef]
- Maślińska, M.; Trędzbor, B.; Krzystanek, M. Dysbiosis, gut-blood barrier rupture and autoimmune response in rheumatoid arthritis and schizophrenia. Reumatologia 2021, 59, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Rossini, M.; Viapiana, O.; Orsolini, G.; Bertoldo, E.; Pontalti, M.; Benini, C.; Fracassi, E.; Giollo, A.; Gatti, D.; et al. Environmental Air Pollution Is a Predictor of Poor Response to Biological Drugs in Chronic Inflammatory Arthritides. ACR Open Rheumatol. 2021, 3, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, N.; Apparailly, F.; Courties, G. Synovial macrophages: From ordinary eaters to extraordinary multitaskers. Trends Immunol. 2021, 42, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-M.; Zhao, Y.-P.; Zhao, Y.; Deng, S.-L.; Yu, K. Regulation of Myostatin on the Growth and Development of Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 785712. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Behl, T.; Kumar, A.; Khullar, G.; Arora, S. Role of Nrf2 in rheumatoid arthritis. Curr. Res. Transl. Med. 2020, 68, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Z.; Wang, D.; Huang, C.; Xu, J.; Liu, C.; Yang, C. Muscle-brain communication in pain: The key role of myokines. Brain Res. Bull. 2021, 179, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, A.N.; Kironenko, T.A.; Milovanova, K.G.; Orlova, A.A.; Dyakova, E.Y.; Kalinnikova Yu, G.; Kabachkova, A.V.; Chibalin, A.V.; Kapilevich, L.V. Treadmill Training Effect on the Myokines Content in Skeletal Muscles of Mice With a Metabolic Disorder Model. Front. Physiol. 2021, 12, 709039. [Google Scholar] [CrossRef]
- Elliott, B.; Renshaw, D.; Getting, S.; Mackenzie, R. The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol. 2012, 205, 324–340. [Google Scholar] [CrossRef]
- Su, C.M.; Hu, S.L.; Sun, Y.; Zhao, J.; Dai, C.; Wang, L.; Xu, G.; Tang, C.H. Myostatin induces tumor necrosis factor-α expression in rheumatoid arthritis synovial fibroblasts through the PI3K-Akt signaling pathway. J. Cell Physiol. 2019, 234, 9793–9801. [Google Scholar] [CrossRef]
- Fennen, M.; Weinhage, T.; Kracke, V.; Intemann, J.; Varga, G.; Wehmeyer, C.; Foell, D.; Korb-Pap, A.; Pap, T.; Dankbar, B. A myostatin-CCL20-CCR6 axis regulates Th17 cell recruitment to inflamed joints in experimental arthritis. Sci. Rep. 2021, 11, 14145. [Google Scholar] [CrossRef]
- Castillero, E.; Nieto-Bona, M.P.; Fernández-Galaz, C.; Martín, A.I.; López-Menduiña, M.; Granado, M.; Villanúa, M.A.; López-Calderón, A. Fenofibrate, a PPAR{alpha} agonist, decreases atrogenes and myostatin expression and improves arthritis-induced skeletal muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E790–E799. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Bone: Targeting myostatin could prevent bone destruction in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Dankbar, B.; Fennen, M.; Brunert, D.; Hayer, S.; Frank, S.; Wehmeyer, C.; Beckmann, D.; Paruzel, P.; Bertrand, J.; Redlich, K.; et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 2015, 21, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Colaianni, G.; Cinti, S.; Colucci, S.; Grano, M. Irisin and musculoskeletal health. Ann. N. Y. Acad. Sci. 2017, 1402, 5–9. [Google Scholar] [CrossRef]
- Korta, P.; Pocheć, E.; Mazur-Biały, A. Irisin as a Multifunctional Protein: Implications for Health and Certain Diseases. Medicina 2019, 55, 485. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Anastasilakis, A.D.; Efstathiadou, Z.A.; Makras, P.; Perakakis, N.; Kountouras, J.; Mantzoros, C.S. Irisin in metabolic diseases. Endocrine 2018, 59, 260–274. [Google Scholar] [CrossRef]
- Fardellone, P.; Séjourné, A.; Paccou, J.; Goëb, V. Bone remodelling markers in rheumatoid arthritis. Mediat. Inflamm. 2014, 2014, 484280. [Google Scholar] [CrossRef]
- Kirk, B.; Feehan, J.; Lombardi, G.; Duque, G. Muscle, Bone, and Fat Crosstalk: The Biological Role of Myokines, Osteokines, and Adipokines. Curr. Osteoporos. Rep. 2020, 18, 388–400. [Google Scholar] [CrossRef]
- Yu, Z.; Ling, Z.; Lu, L.; Zhao, J.; Chen, X.; Xu, P.; Zou, X. Regulatory Roles of Bone in Neurodegenerative Diseases. Front. Aging Neurosci. 2020, 12, 467. [Google Scholar] [CrossRef]
- Gonzalez-Gil, A.M.; Elizondo-Montemayor, L. The Role of Exercise in the Interplay between Myokines, Hepatokines, Osteokines, Adipokines, and Modulation of Inflammation for Energy Substrate Redistribution and Fat Mass Loss: A Review. Nutrients 2020, 12, 1899. [Google Scholar] [CrossRef]
- Jura-Półtorak, A.; Szeremeta, A.; Olczyk, K.; Zoń-Giebel, A.; Komosińska-Vassev, K. Bone Metabolism and RANKL/OPG Ratio in Rheumatoid Arthritis Women Treated with TNF-α Inhibitors. J. Clin. Med. 2021, 10, 2905. [Google Scholar] [CrossRef] [PubMed]
- Remuzgo-Martínez, S.; Genre, F.; López-Mejías, R.; Ubilla, B.; Mijares, V.; Pina, T.; Corrales, A.; Blanco, R.; Martín, J.; Llorca, J.; et al. Expression of osteoprotegerin and its ligands, RANKL and TRAIL, in rheumatoid arthritis. Sci. Rep. 2016, 6, 29713. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, Y.; Lu, J.; Xu, J. Osteoprotegerin and RANKL in the pathogenesis of rheumatoid arthritis-induced osteoporosis. Rheumatol. Int. 2012, 32, 3397–3403. [Google Scholar] [CrossRef] [PubMed]
- López-Mejias, R.; Ubilla, B.; Genre, F.; Corrales, A.; Hernández, J.L.; Ferraz-Amaro, I.; Tsang, L.; Llorca, J.; Blanco, R.; González-Juanatey, C.; et al. Osteoprotegerin Concentrations Relate Independently to Established Cardiovascular Disease in Rheumatoid Arthritis. J. Rheumatol. 2015, 42, 39. [Google Scholar] [CrossRef]
- Nava-Valdivia, C.A.; Ponce-Guarneros, J.M.; Saldaña-Cruz, A.M.; Corona-Sanchez, E.G.; Ramirez-Villafaña, M.; Perez-Guerrero, E.E.; Murillo-Saich, J.D.; Contreras-Haro, B.; Vazquez-Villegas, M.L.; Gonzalez-Ponce, F.; et al. Assessment of Serum sRANKL, sRANKL/OPG Ratio, and Other Bone Turnover Markers with the Estimated 10-Year Risk of Major and Hip Osteoporotic Fractures in Rheumatoid Arthritis: A Cross-Sectional Study. Biomed. Res. Int. 2021, 2021, 5567666. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Liu, G. Association Between Osteoprotegerin Gene Polymorphisms and Rheumatoid Arthritis Susceptibility: A Meta-analysis. Arch. Med. Res. 2016, 47, 134–141. [Google Scholar] [CrossRef]
- Kawashiri, S.-Y.; Endo, Y.; Nishino, A.; Okamoto, M.; Tsuji, S.; Takatani, A.; Shimizu, T.; Sumiyoshi, R.; Koga, T.; Iwamoto, N.; et al. Association between serum bone biomarker levels and therapeutic response to abatacept in patients with rheumatoid arthritis (RA): A multicenter, prospective, and observational RA ultrasound cohort study in Japan. BMC Musculoskelet. Disord. 2021, 22, 506. [Google Scholar] [CrossRef]
- Wang, P.; Li, S.; Liu, L.-N.; Lv, T.-T.; Li, X.-M.; Li, X.-P.; Pan, H.-F. Circulating osteoprotegerin levels are elevated in rheumatoid arthritis: A systematic review and meta-analysis. Clin. Rheumatol. 2017, 36, 2193–2200. [Google Scholar] [CrossRef]
- He, C.; He, W.; Hou, J.; Chen, K.; Huang, M.; Yang, M.; Luo, X.; Li, C. Bone and Muscle Crosstalk in Aging. Front. Cell Dev. Biol. 2020, 8, 585644. [Google Scholar] [CrossRef]
- Weitzmann, M.N. Bone and the Immune System. Toxicol. Pathol. 2017, 45, 911–924. [Google Scholar] [CrossRef]
- Panagopoulos, P.K.; Lambrou, G.I. Bone erosions in rheumatoid arthritis: Recent developments in pathogenesis and therapeutic implications. J. Musculoskelet. Neuronal Interact. 2018, 18, 304–319. [Google Scholar] [PubMed]
- Liu, L.-N.; Mao, Y.-M.; Zhao, C.-N.; Wang, H.; Yuan, F.-F.; Li, X.-M.; Pan, H.-F. Circulating Levels of Osteoprotegerin, Osteocalcin and Osteopontin in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Immunol. Investig. 2019, 48, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Colaianni, G.; Storlino, G.; Sanesi, L.; Colucci, S.; Grano, M. Myokines and Osteokines in the Pathogenesis of Muscle and Bone Diseases. Curr. Osteoporos. Rep. 2020, 18, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Corrado, A.; Maruotti, N.; Cici, D.; Cantatore, F.P. Osteoblast role in the pathogenesis of rheumatoid arthritis. Mol. Biol. Rep. 2021, 48, 2843–2852. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef]
- Ye, X.H.; Cheng, J.L.; Liu, R.P. Osteoprotegerin polymorphisms in Chinese Han patients with rheumatoid arthritis. Genet. Mol. Res. 2015, 14, 6569–6577. [Google Scholar] [CrossRef]
- Lu, W.; Xiao, W.; Xie, W.; Fu, X.; Pan, L.; Jin, H.; Yu, Y.; Zhang, Y.; Li, Y. The Role of Osteokines in Sarcopenia: Therapeutic Directions and Application Prospects. Front. Cell Dev. Biol. 2021, 9, 735374. [Google Scholar] [CrossRef]
- Vlot, M.C.; den Heijer, M.; de Jongh, R.T.; Vervloet, M.G.; Lems, W.F.; de Jonge, R.; Obermayer-Pietsch, B.; Heijboer, A.C. Clinical utility of bone markers in various diseases. Bone 2018, 114, 215–225. [Google Scholar] [CrossRef]
- Gulyás, K.; Horváth, Á.; Végh, E.; Pusztai, A.; Szentpétery, Á.; Pethö, Z.; Váncsa, A.; Bodnár, N.; Csomor, P.; Hamar, A.; et al. Effects of 1-year anti-TNF-α therapies on bone mineral density and bone biomarkers in rheumatoid arthritis and ankylosing spondylitis. Clin. Rheumatol. 2020, 39, 167–175. [Google Scholar] [CrossRef]
- Luukkonen, J.; Pascual, L.M.; Patlaka, C.; Lång, P.; Turunen, S.; Halleen, J.; Nousiainen, T.; Valkealahti, M.; Tuukkanen, J.; Andersson, G.; et al. Increased amount of phosphorylated proinflammatory osteopontin in rheumatoid arthritis synovia is associated to decreased tartrate-resistant acid phosphatase 5B/5A ratio. PLoS ONE 2017, 12, e0182904. [Google Scholar] [CrossRef]
- Yang, X.L.; Hu, Z.D.; Wu, Q.; Liu, X.; Liu, Q.J.; Zhang, Y.C.; Yang, Q.R. Association of polymorphisms in SPARC and NLRP2 genes with rheumatoid arthritis in a Chinese Han population. Mod. Rheumatol. 2015, 25, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Fontanella, R.A.; Scisciola, L.; Rizzo, M.R.; Surina, S.; Sardu, C.; Marfella, R.; Paolisso, G.; Barbieri, M. Adiponectin Related Vascular and Cardiac Benefits in Obesity: Is There a Role for an Epigenetically Regulated Mechanism? Front. Cardiovasc. Med. 2021, 8, 768026. [Google Scholar] [CrossRef]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. 2020, 26, e919159. [Google Scholar] [CrossRef] [PubMed]
- Carrión, M.; Frommer, K.W.; Pérez-García, S.; Müller-Ladner, U.; Gomariz, R.P.; Neumann, E. The Adipokine Network in Rheumatic Joint Diseases. Int. J. Mol. Sci. 2019, 20, 4091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Luo, W.; Li, Y.; Gao, S.; Lei, G. Role of osteopontin in rheumatoid arthritis. Rheumatol. Int. 2015, 35, 589–595. [Google Scholar] [CrossRef]
- Mehta, B.B.; Sharma, S.; Vasishta, R.K.; Sen, R.K.; Sharma, A.; Luthra-Guptasarma, M. Blocking osteopontin-fibronectin interactions reduce extracellular fibronectin deployment and arthritic immunopathology. Int. Immunopharmacol. 2018, 55, 297–305. [Google Scholar] [CrossRef]
- Lories, R.J.; Luyten, F.P. Bone morphogenetic protein signaling and arthritis. Cytokine Growth Factor Rev. 2009, 20, 467–473. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Sozzani, S. Adipokines as Potential Biomarkers in Rheumatoid Arthritis. Mediat. Inflamm. 2014, 2014, 425068. [Google Scholar] [CrossRef]
- Chemel, M.; Brion, R.; Segaliny, A.I.; Lamora, A.; Charrier, C.; Brulin, B.; Maugars, Y.; Le Goff, B.; Heymann, D.; Verrecchia, F. Bone Morphogenetic Protein 2 and Transforming Growth Factor β1 Inhibit the Expression of the Proinflammatory Cytokine IL-34 in Rheumatoid Arthritis Synovial Fibroblasts. Am. J. Pathol. 2017, 187, 156–162. [Google Scholar] [CrossRef]
- Francischetti, E.A.; Dezonne, R.S.; Pereira, C.M.; de Moraes Martins, C.J.; Celoria, B.M.J.; de Oliveira, P.A.C.; de Abreu, V.G. Insights Into the Controversial Aspects of Adiponectin in Cardiometabolic Disorders. Horm. Metab. Res. 2020, 52, 695–707. [Google Scholar] [CrossRef]
- Su, C.-M.; Chiang, Y.-C.; Huang, C.-Y.; Hsu, C.-J.; Fong, Y.-C.; Tang, C.-H. Osteopontin Promotes Oncostatin M Production in Human Osteoblasts: Implication of Rheumatoid Arthritis Therapy. J. Immunol. 2015, 195, 3355. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, H.; Kobayashi, H.; Kanno, T.; Asano, T.; Saito, R.; Sato, S.; Suzuki, E.; Watanabe, H.; Ohira, H. Plasma osteopontin is correlated with bone resorption markers in rheumatoid arthritis patients. Int. J. Rheum. Dis. 2014, 17, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Xu, L.; Sun, X.; Wang, Y.; Xuan, W.; Zhang, Q.; Zhao, P.; Wu, Q.; Liu, R.; Che, N.; et al. Adiponectin aggravates bone erosion by promoting osteopontin production in synovial tissue of rheumatoid arthritis. Arthritis Res. 2018, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Lukač, N.; Katavić, V.; Novak, S.; Šućur, A.; Filipović, M.; Kalajzić, I.; Grčević, D.; Kovačić, N. What do we know about bone morphogenetic proteins and osteochondroprogenitors in inflammatory conditions? Bone 2020, 137, 115403. [Google Scholar] [CrossRef]
- Ait Eldjoudi, D.; Cordero Barreal, A.; Gonzalez-Rodríguez, M.; Ruiz-Fernández, C.; Farrag, Y.; Farrag, M.; Lago, F.; Capuozzo, M.; Gonzalez-Gay, M.A.; Mera Varela, A.; et al. Leptin in Osteoarthritis and Rheumatoid Arthritis: Player or Bystander? Int. J. Mol. Sci. 2022, 23, 2859. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C. Circulating adiponectin and visfatin levels in rheumatoid arthritis and their correlation with disease activity: A meta-analysis. Int. J. Rheum. Dis. 2018, 21, 664–672. [Google Scholar] [CrossRef]
- Pietrzyk, B.; Smertka, M.; Chudek, J. Sclerostin: Intracellular mechanisms of action and its role in the pathogenesis of skeletal and vascular disorders. Adv. Clin. Exp. Med. 2017, 26, 1283–1291. [Google Scholar] [CrossRef]
- Verschueren, P.C.; Lories, R.J.; Daans, M.; Théate, I.; Durez, P.; Westhovens, R.; Luyten, F.P. Detection, identification and in vivo treatment responsiveness of bone morphogenetic protein (BMP)-activated cell populations in the synovium of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 117–123. [Google Scholar] [CrossRef]
- Mohammed Ali, D.M.; Al-Fadhel, S.Z.; Al-Ghuraibawi, N.H.A.; Al-Hakeim, H.K. Serum chemerin and visfatin levels and their ratio as possible diagnostic parameters of rheumatoid arthritis. Reumatologia 2020, 58, 67–75. [Google Scholar] [CrossRef]
- Franco-Trepat, E.; Alonso-Pérez, A.; Guillán-Fresco, M.; Jorge-Mora, A.; Gualillo, O.; Gómez-Reino, J.J.; Gómez Bahamonde, R. Visfatin as a therapeutic target for rheumatoid arthritis. Expert Opin. Targets 2019, 23, 607–618. [Google Scholar] [CrossRef]
- Sato, H.; Muraoka, S.; Kusunoki, N.; Masuoka, S.; Yamada, S.; Ogasawara, H.; Imai, T.; Akasaka, Y.; Tochigi, N.; Takahashi, H.; et al. Resistin upregulates chemokine production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. 2017, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Kamihagi, K.; Satakeda, H.; Katayama, M.; Pan, H.; Okamoto, H.; Noshiro, M.; Takahashi, K.; Yoshihara, Y.; Shimmei, M.; et al. Enhancement of SPARC (osteonectin) synthesis in arthritic cartilage. Increased levels in synovial fluids from patients with rheumatoid arthritis and regulation by growth factors and cytokines in chondrocyte cultures. Arthritis Rheum. 1996, 39, 539–551. [Google Scholar] [CrossRef]
- Kaji, H. Effects of myokines on bone. Bonekey Rep. 2016, 5, 826. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Botti, L.; Gulino, A.; Lecis, D.; Bassani, B.; Portararo, P.; Milani, M.; Cancila, V.; De Cecco, L.; Dugo, M.; et al. SPARC regulation of PMN clearance protects from pristane-induced lupus and rheumatoid arthritis. iScience 2021, 24, 102510. [Google Scholar] [CrossRef] [PubMed]
- Senolt, L.; Housa, D.; Vernerová, Z.; Jirásek, T.; Svobodová, R.; Veigl, D.; Anderlová, K.; Müller-Ladner, U.; Pavelka, K.; Haluzík, M. Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann. Rheum. Dis. 2007, 66, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Badoer, E. Cardiovascular and Metabolic Crosstalk in the Brain: Leptin and Resistin. Front. Physiol. 2021, 12, 639417. [Google Scholar] [CrossRef]
- Choi, H.M.; Doss, H.M.; Kim, K.S. Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int. J. Mol. Sci. 2020, 21, 1219. [Google Scholar] [CrossRef]
- Szumilas, K.; Szumilas, P.; Słuczanowska-Głąbowska, S.; Zgutka, K.; Pawlik, A. Role of Adiponectin in the Pathogenesis of Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 8265. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Fujio, K. Emerging role of leptin in joint inflammation and destruction. Immunol. Med. 2022, 45, 27–34. [Google Scholar] [CrossRef]
- Tian, G.; Liang, J.-N.; Wang, Z.-Y.; Zhou, D. Emerging role of leptin in rheumatoid arthritis. Clin. Exp. Immunol. 2014, 177, 557–570. [Google Scholar] [CrossRef]
- Picó, C.; Palou, M. Leptin and Metabolic Programming. Nutrients 2022, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Pulito-Cueto, V.; Remuzgo-Martínez, S.; Genre, F.; Calvo-Alén, J.; Aurrecoechea, E.; Llorente, I.; Triguero-Martinez, A.; Blanco, R.; Llorca, J.; Ruiz-Lucea, E.; et al. Anti-IL-6 therapy reduces leptin serum levels in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2020, 38, 1201–1205. [Google Scholar] [PubMed]
- Wang, Z.; Huang, X.; Ye, X.; Li, X.; Wei, J. Roles of leptin on the key effector cells of rheumatoid arthritis. Immunol. Lett. 2021, 233, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Graessler, J.; Verlohren, M.; Graessler, A.; Zeissig, A.; Kuhlisch, E.; Kopprasch, S.; Schroeder, H.-E. Association of chondromodulin-II Val58Ile polymorphism with radiographic joint destruction in rheumatoid arthritis. J. Rheumatol. 2005, 32, 1654. [Google Scholar] [PubMed]
- Okumura, A.; Saito, T.; Otani, I.; Kojima, K.; Yamada, Y.; Ishida-Okawara, A.; Nakazato, K.; Asano, M.; Kanayama, K.; Iwakura, Y.; et al. Suppressive role of leukocyte cell-derived chemotaxin 2 in mouse anti-type II collagen antibody-induced arthritis. Arthritis Rheum. 2008, 58, 413–421. [Google Scholar] [CrossRef]
- Meier, F.M.; Frommer, K.W.; Peters, M.A.; Brentano, F.; Lefèvre, S.; Schröder, D.; Kyburz, D.; Steinmeyer, J.; Rehart, S.; Gay, S.; et al. Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J. Biol. Chem. 2012, 287, 28378–28385. [Google Scholar] [CrossRef]
- Castan-Laurell, I.; Dray, C.; Valet, P. The therapeutic potentials of apelin in obesity-associated diseases. Mol. Cell Endocrinol. 2021, 529, 111278. [Google Scholar] [CrossRef]
- Cutolo, M.; Seriolo, B.; Villaggio, B.; Pizzorni, C.; Craviotto, C.; Sulli, A. Androgens and estrogens modulate the immune and inflammatory responses in rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2002, 966, 131–142. [Google Scholar] [CrossRef]
- Cutolo, M.; Straub, R.H. Sex steroids and autoimmune rheumatic diseases: State of the art. Nat. Rev. Rheumatol. 2020, 16, 628–644. [Google Scholar] [CrossRef]
- Su, C.-M.; Hsu, C.-J.; Tsai, C.-H.; Huang, C.-Y.; Wang, S.-W.; Tang, C.-H. Resistin Promotes Angiogenesis in Endothelial Progenitor Cells Through Inhibition of MicroRNA206: Potential Implications for Rheumatoid Arthritis. Stem Cells 2015, 33, 2243–2255. [Google Scholar] [CrossRef]
- Treeck, O.; Buechler, C.; Ortmann, O. Chemerin and Cancer. Int. J. Mol. Sci. 2019, 20, 3750. [Google Scholar] [CrossRef] [PubMed]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Masuko, K. Angiopoietin-like 4: A molecular link between insulin resistance and rheumatoid arthritis. J. Orthop. Res. 2017, 35, 939–943. [Google Scholar] [CrossRef]
- Swales, C.; Athanasou, N.A.; Knowles, H.J. Angiopoietin-Like 4 Is Over-Expressed in Rheumatoid Arthritis Patients: Association with Pathological Bone Resorption. PLoS ONE 2014, 9, e109524. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.N.; Jung, C.H. The Role of Anti-Inflammatory Adipokines in Cardiometabolic Disorders: Moving beyond Adiponectin. Int. J. Mol. Sci. 2021, 22, 13529. [Google Scholar] [CrossRef]
- Kaya Sezginer, E.; Kırlangıç, Ö.F.; Eşkin Tanrıverdi, M.D.; Topçu, H.O.; Gür, S. Analysis of Changes in Serum Levels and Gene Expression Profiles of Novel Adipocytokines (Omentin, Vaspin, Irisin and Visfatin) and Their Correlation with Serum C-reactive Protein Levels in Women Diagnosed with Endometriosis. Turk. J. Pharm. Sci. 2022, 19, 48–53. [Google Scholar] [CrossRef]
- Kurowska, P.; Mlyczyńska, E.; Dawid, M.; Jurek, M.; Klimczyk, D.; Dupont, J.; Rak, A. Review: Vaspin (SERPINA12) Expression and Function in Endocrine Cells. Cells 2021, 10, 1710. [Google Scholar] [CrossRef]
- Icer, M.A.; Yıldıran, H. Effects of fetuin-A with diverse functions and multiple mechanisms on human health. Clin. Biochem. 2021, 88, 1–10. [Google Scholar] [CrossRef]
- Nguyen, M.V.C.; Courtier, A.; Adrait, A.; Defendi, F.; Couté, Y.; Baillet, A.; Guigue, L.; Gottenberg, J.-E.; Dumestre-Pérard, C.; Brun, V.; et al. Fetuin-A and thyroxin binding globulin predict rituximab response in rheumatoid arthritis patients with insufficient response to anti-TNFα. Clin. Rheumatol. 2020, 39, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Matuszewska, A.; Szechiński, J. Evaluation of selected bone metabolism markers in rheumatoid arthritis patients. Adv. Clin. Exp. Med. 2013, 22, 193–202. [Google Scholar]
- Kucukoglu, O.; Sowa, J.P.; Mazzolini, G.D.; Syn, W.K.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.W.; Zemel, B.S.; Taratuta, E.G.; Baker, J.F. Circulating Fibroblast Growth Factor-21 Levels in Rheumatoid Arthritis: Associations With Disease Characteristics, Body Composition, and Physical Functioning. J. Rheumatol. 2021, 48, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, S.; Liu, Y.; Tian, G.; Yuan, Q.; Bai, F.; Wang, W.; Zhang, Z.; Ren, G.; Zhang, Y.; et al. Fibroblast growth factor 21 (FGF21) ameliorates collagen-induced arthritis through modulating oxidative stress and suppressing nuclear factor-kappa B pathway. Int. Immunopharmacol. 2015, 25, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; He, J.; Li, S.; Song, L.; Guo, X.; Yao, W.; Zou, D.; Gao, X.; Liu, Y.; Bai, F.; et al. Fibroblast growth factor 21 (FGF21) inhibits macrophage-mediated inflammation by activating Nrf2 and suppressing the NF-κB signaling pathway. Int. Immunopharmacol. 2016, 38, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Opoku, Y.; Liu, Z.; Liu, H.; Afrifa, J.; Koranteng, H.; Ren, G.; Li, D. Fibroblast Growth Factor–21 Ameliorates Rheumatoid Arthritis by Maintaining Articular Integrity. Int. J. Pept. Res. Ther. 2020, 26, 651–659. [Google Scholar] [CrossRef]
- Opoku, Y.K.; Liu, Z.; Afrifa, J.; Khoso, M.H.; Ren, G.; Li, D. Therapeutic Role of Fibroblast Growth Factor 21 (FGF21) in the Amelioration of Chronic Diseases. Int. J. Pept. Res. Ther. 2020, 26, 107–119. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Xu, N.; Wei, Q.; Wu, M.; Li, X.; Zheng, P.; Sun, S.; Jin, Y.; Zhang, G.; et al. Follistatin-like protein 1 is elevated in systemic autoimmune diseases and correlated with disease activity in patients with rheumatoid arthritis. Arthritis Res. 2011, 13, R17. [Google Scholar] [CrossRef]
- Ni, S.; Li, C.; Xu, N.; Liu, X.; Wang, W.; Chen, W.; Wang, Y.; van Wijnen, A.J. Follistatin-like protein 1 induction of matrix metalloproteinase 1, 3 and 13 gene expression in rheumatoid arthritis synoviocytes requires MAPK, JAK/STAT3 and NF-κB pathways. J. Cell Physiol. 2018, 234, 454–463. [Google Scholar] [CrossRef]
- Osiecka-Iwan, A.; Hyc, A.; Radomska-Lesniewska, D.M.; Rymarczyk, A.; Skopinski, P. Antigenic and immunogenic properties of chondrocytes. Implications for chondrocyte therapeutic transplantation and pathogenesis of inflammatory and degenerative joint diseases. Cent. Eur. J. Immunol. 2018, 43, 209–219. [Google Scholar] [CrossRef]
- Zhu, S.; Bennett, S.; Li, Y.; Liu, M.; Xu, J. The molecular structure and role of LECT2 or CHM-II in arthritis, cancer, and other diseases. J. Cell Physiol. 2022, 237, 480–488. [Google Scholar] [CrossRef]
- Zheng, H.; Miyakawa, T.; Sawano, Y.; Asano, A.; Okumura, A.; Yamagoe, S.; Tanokura, M. Crystal Structure of Human Leukocyte Cell-derived Chemotaxin 2 (LECT2) Reveals a Mechanistic Basis of Functional Evolution in a Mammalian Protein with an M23 Metalloendopeptidase Fold. J. Biol. Chem. 2016, 291, 17133–17142. [Google Scholar] [CrossRef] [PubMed]
- Slowik, V.; Apte, U. Leukocyte Cell-Derived Chemotaxin-2: It’s Role in Pathophysiology and Future in Clinical Medicine. Clin. Transl. Sci. 2017, 10, 249–259. [Google Scholar] [CrossRef]
- Lashkari, M.; Noori, A.; Oveisi, S.; Kheirkhah, M. Association of serum testosterone and dehydroepiandrosterone sulfate with rheumatoid arthritis: A case control study. Electron. Physician 2018, 10, 6500–6505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kornicka-Garbowska, K.; Bourebaba, L.; Röcken, M.; Marycz, K. Sex Hormone Binding Globulin (SHBG) Mitigates ER Stress in Hepatocytes In Vitro and Ex Vivo. Cells 2021, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V. Muscle-bone crosstalk by organokines-the new “hope molecules”. J. World Fed. Orthod. 2022, 11, 47–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-H.; Qi, L.-W.; Alolga, R.; Liu, Q. Implication of the hepatokine, fibrinogen-like protein 1 in liver diseases, metabolic disorders and cancer: The need to harness its full potential. Int. J. Biol. Sci. 2022, 18, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Li, H.Y.; Ou, H.Y.; Wu, P.; Wang, S.H.; Chang, C.J.; Lin, S.Y.; Wu, C.L.; Wu, H.T. Role of placental fibrinogen-like protein 1 in gestational diabetes. Transl. Res. 2020, 218, 73–80. [Google Scholar] [CrossRef]
- Wu, H.T.; Chen, S.C.; Fan, K.C.; Kuo, C.H.; Lin, S.Y.; Wang, S.H.; Chang, C.J.; Li, H.Y. Targeting fibrinogen-like protein 1 is a novel therapeutic strategy to combat obesity. FASEB J. 2020, 34, 2958–2967. [Google Scholar] [CrossRef]
- Gamez-Nava, J.I.; Ramirez-Villafaña, M.; Cons-Molina, F.; Gomez-Ramirez, E.E.; Esparza-Guerrero, Y.; Saldaña-Cruz, A.M.; Sanchez-Rodriguez, E.N.; Jacobo-Cuevas, H.; Totsuka-Sutto, S.E.; Perez-Guerrero, E.E.; et al. Serum irisin concentrations and osteoporotic vertebral fractures in women with rheumatoid arthritis: A cross-sectional study. Medicine 2022, 101, e28799. [Google Scholar] [CrossRef]
- Tao, S.S.; Dan, Y.L.; Wu, G.C.; Zhang, Q.; Zhang, T.P.; Fan, Y.G.; Pan, H.F. Association of Leptin Gene Polymorphisms with Rheumatoid Arthritis in a Chinese Population. Biomed. Res. Int. 2020, 2020, 3789319. [Google Scholar] [CrossRef]
- Chen, Y.M.; Chen, P.K.; Chang, C.K.; Lin, C.C.; Chen, H.H.; Lan, J.L.; Chang, S.H.; Chen, D.Y. Association of Apolipoprotein E Polymorphism with Adipokines and Cardiovascular Disease Risk in Rheumatoid Arthritis Patients. Life 2020, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Yan, G.; Chin, Y.; Hwee, F.A.; Kersten, S.; Tan, N.S. Angiopoietin-like 4: A decade of research. Biosci. Rep. 2011, 32, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, C.; Van de Wiele, T.; Verstraete, W.; Bracke, M.; Vanhoecke, B. Angiopoietin-like protein 4: Health effects, modulating agents and structure–function relationships. Expert Rev. Proteom. 2012, 9, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H. Multiple Roles of Angiopoietin-Like 4 in Osteolytic Disease. Front. Endocrinol. 2017, 8, 80. [Google Scholar] [CrossRef]
- Makoveichuk, E.; Ruge, T.; Nilsson, S.; Södergren, A.; Olivecrona, G. High Concentrations of Angiopoietin-Like Protein 4 Detected in Serum from Patients with Rheumatoid Arthritis Can Be Explained by Non-Specific Antibody Reactivity. PLoS ONE 2017, 12, e0168922. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Suárez, Y. ANGPTL4: A multifunctional protein involved in metabolism and vascular homeostasis. Curr. Opin. Hematol. 2020, 27, 206–231. [Google Scholar] [CrossRef]
- Harman, H.; Tekeoğlu, İ.; Gürol, G.; Sağ, M.S.; Karakeçe, E.; Çİftçİ, İ.H.; Kamanlı, A.; Nas, K. Comparison of fetuin-A and transforming growth factor beta 1 levels in patients with spondyloarthropathies and rheumatoid arthritis. Int. J. Rheum. Dis. 2017, 20, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Gulkesen, A.; Akgol, G.; Poyraz, A.K.; Aydin, S.; Denk, A.; Yildirim, T.; Kaya, A. Lipocalin 2 as a clinical significance in rheumatoid arthritis. Cent. Eur. J. Immunol. 2017, 42, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef]
- Mattiotti, A.; Prakash, S.; Barnett, P.; van den Hoff, M.J.B. Follistatin-like 1 in development and human diseases. Cell Mol. Life Sci. 2018, 75, 2339–2354. [Google Scholar] [CrossRef]
- Senesi, P.; Luzi, L.; Terruzzi, I. Adipokines, Myokines, and Cardiokines: The Role of Nutritional Interventions. Int. J. Mol. Sci. 2020, 21, 8372. [Google Scholar] [CrossRef] [PubMed]
- Clutter, S.D.; Wilson, D.C.; Marinov, A.D.; Hirsch, R. Follistatin-like protein 1 promotes arthritis by up-regulating IFN-gamma. J. Immunol. 2009, 182, 234–239. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, W.Y.; Seong, S.J.; Kim, S.Y.; Lim, M.S.; Yoon, Y.R. Follistatin-like 1 promotes osteoclast formation via RANKL-mediated NF-κB activation and M-CSF-induced precursor proliferation. Cell Signal. 2016, 28, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.L.; Shi, G.R.; Xie, J.; Du, X.Z.; Yang, H. MicroRNA-27a Inhibits Cell Migration and Invasion of Fibroblast-Like Synoviocytes by Targeting Follistatin-Like Protein 1 in Rheumatoid Arthritis. Mol. Cells 2016, 39, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chu, P.; Yu, X.; Li, J.; Zhang, W.; Gong, M. ZFAS1 knockdown inhibits fibroblast-like synoviocyte proliferation, migration, invasion and inflammation, and promotes apoptosis via miR-3926/FSTL1 in rheumatoid arthritis. Exp. Med. 2021, 22, 914. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.A.; Gad, R.; Senosy, T.; Higazi, A.M.; Elshereef, R. Serum irisin level in rheumatoid arthritis patients: Relationship to disease activity, subclinical atherosclerosis, and cardiovascular risk factors. Egypt. Rheumatol. 2022, 44, 109–114. [Google Scholar] [CrossRef]
- Tekeoğlu, İ.; Harman, H.; Sağ, S.; Altındiş, M.; Kamanlı, A.; Nas, K. Levels of serum pentraxin 3, IL-6, fetuin A and insulin in patients with rheumatoid arthritis. Cytokine 2016, 83, 171–175. [Google Scholar] [CrossRef]
- Papichev, E.V.; Zavodovsky, B.V.; Polyakova, Y.V.; Seewordova, L.E.; Akhverdyan, Y.R. Novel hepatokine in rheumatoid arthritis laboratory diagnostics. Klin. Lab. Diagn. 2018, 63, 756–760. [Google Scholar] [CrossRef]
- Papichev, E.V.; Sivordova, L.E.; Polyakova, J.V.; Akhverdyan, Y.R.; Zavodovsky, B.V.; Rogatkina, T. AB005Y Fetuin-a: Clinical and laboratory associations in women with rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1228. [Google Scholar]
- Diller, M.; Frommer, K.; Dankbar, B.; Tarner, I.; Hülser, M.L.; Tsiklauri, L.; Hasseli, R.; Sauerbier, M.; Pap, T.; Rehart, S.; et al. The activin-follistatin anti-inflammatory cycle is deregulated in synovial fibroblasts. Arthritis Res. 2019, 21, 144. [Google Scholar] [CrossRef]
- Jin, Q.Y.; Zhu, Q.H.; Deng, W.; Hou, C.X.; Sun, N.N.; Han, W.; Tang, Y.T.; Wang, C.X.; Ye, J.H. Follistatin-like 1 suppresses osteoblast differentiation of bone marrow mesenchymal cells during inflammation. Arch. Oral Biol. 2022, 135, 105345. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.F.; Katz, P.; Weber, D.R.; Gould, P.; George, M.D.; Long, J.; Zemel, B.S.; Giles, J.T. Adipocytokines and Associations with Abnormal Body Composition in Rheumatoid Arthritis. Arthritis Care Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Villegas, M.L.; Gamez-Nava, J.I.; Saldaña-Cruz, A.M.; Celis, A.; Sanchez-Rodriguez, E.N.; Perez-Guerrero, E.E.; Ramirez-Villafaña, M.; Nava-Valdivia, C.A.; Contreras-Haro, B.; Vasquez-Jimenez, J.C.; et al. Functional disability is related to serum chemerin levels in rheumatoid arthritis. Sci. Rep. 2021, 11, 8360. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ponce, F.; Gamez-Nava, J.I.; Perez-Guerrero, E.E.; Saldaña-Cruz, A.M.; Vazquez-Villegas, M.L.; Ponce-Guarneros, J.M.; Huerta, M.; Trujillo, X.; Contreras-Haro, B.; Rocha-Muñoz, A.D.; et al. Serum chemerin levels: A potential biomarker of joint inflammation in women with rheumatoid arthritis. PLoS ONE 2021, 16, e0255854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Johansson, L.; Andersson-Assarsson, J.; Taube, M.; Peltonen, M.; Svensson, P.A.; Herder, C.; Rudin, A.; Carlsson, L.; Rantapää-Dahlqvist, S.; et al. Adiponectin Associates with Rheumatoid Arthritis Risk in Overweight and Obesity Independently of Other Adipokines. J. Clin. Med. 2021, 10, 2791. [Google Scholar] [CrossRef]
- Chen, J.; Xie, Z.; Bin, Z. The Association Between Serum Leptin Levels and Cardiovascular Events in Patients with Rheumatoid Arthritis. Lab. Med. 2021, 52, 86–92. [Google Scholar] [CrossRef]
- Gamal, R.M.; Mohamed, M.E.; Hammam, N.; El Fetoh, N.A.; Rashed, A.M.; Furst, D.E. Preliminary study of the association of serum irisin levels with poor sleep quality in rheumatoid arthritis patients. Sleep Med. 2020, 67, 71–76. [Google Scholar] [CrossRef]
- Gibas-Dorna, M.; Piątek, J.; Kupsz, J.; Bernatek, M.; Krauss, H.; Sowińska, A.; Kołodziejski, P.; Owoc, A.; Bojar, I. Relationship between adipokines and lipid profile in postmenopausal women with different apolipoprotein E genotypes. Women Health 2017, 57, 891–904. [Google Scholar] [CrossRef]
- Baker, J.F.; England, B.R.; Mikuls, T.R.; Sayles, H.; Cannon, G.W.; Sauer, B.C.; George, M.D.; Caplan, L.; Michaud, K. Obesity, Weight Loss, and Progression of Disability in Rheumatoid Arthritis. Arthritis Care Res. 2018, 70, 1740–1747. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Release | Organokine | Expression in RA | General Functions | Role in RA | References |
|---|---|---|---|---|---|
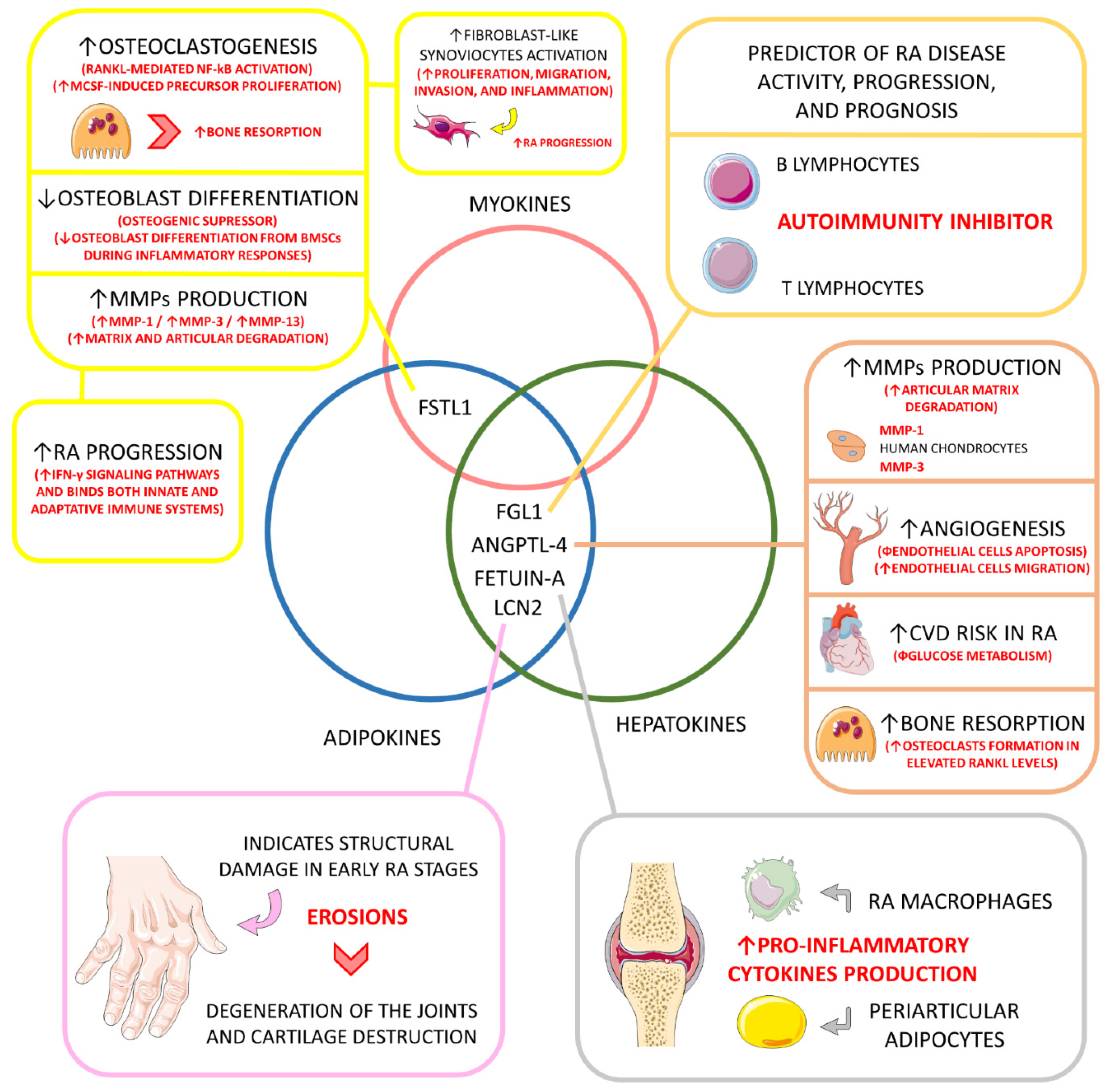
| Hepatokine and adipokine. | Fibrinogen-like protein 1 (FGL1) (HEPASSOCIN) | ↑, 10-times higher compared to healthy individuals. | Mitogenic to human hepatocytes (↑ liver regeneration); Rescue role in hyperglycemic crisis; Liver protector; Lipogenic (↑ lipogenesis); ↑ Preadipocyte proliferation; Immune activation; Probable role in cancer development; ↑ IR, probable role in NAFLD. | Predictor of disease activity, progression, and prognosis; Probable role in inhibiting autoimmunity under up-regulated circulation (major inhibition of LAG-3); | [26,139,140,156] |
| Adipokine and hepatokine. | Angiopoietin-like 4 (ANGPTL-4) | ↑, principally in the synovial fluid, bones, and cartilages. | Regulator of plasma triglycerides metabolism by inhibiting the lipoprotein lipase, principally under fasting conditions; Prevents excessive lipid uptake by the heart and skeletal muscles. | ↑ Production of MMP-1 and MMP-3 by chondrocytes (↑ cartilage matrix degradation and catabolism); In inflammation-derived hypoxia, and ANGPTL-4 stimulates osteoclasts to bone resorption (principally in elevated RANKL levels); Probably promotes angiogenesis (inhibiting endothelial apoptosis and stimulating endothelial cell migration); ↑ Cardiovascular risk among RA patients, principally due to impaired glucose metabolism. | [113,114,144,145] |
| Adipokine and hepatokine. | Fetuin-A | ↑ or ↓ compared to controls depending on the population. | ↑ Insulin resistance in the liver and skeletal muscles; Pro-adipogenic and suppressor of adiponectin secretion and effects. | It may induce pro-inflammatory cytokines production in adipocytes and macrophages involved in RA structures. | [11,147,157,158,159] |
| Adipokine and hepatokine. | Lipocalin 2 (LCN2) | ↑, compared to controls. | Inflammatory biomarker; Role in apoptosis, cell differentiation, fatty acid oxidation, iron transport, inflammation regulation, immune response, and metabolism in general. | Serum levels of LCN2 can be used to indicate structural damage, such as erosions in the early stage of the disease, but cannot be used to monitor disease activity and are not directly related to inflammatory activation; LCN2 can also play a role in the degeneration of joints in RA and could be a biomarker of cartilage degradation in arthritic diseases. | [148,149] |
| Adipokine and myokine. | Follistatin-like 1 (FSTL1) | ↑, compared to controls. | Involved in fatty acid and glucose oxidation; Involved in cardiomyocyte hypertrophy, cardiomyocyte apoptosis, and vascular smooth muscle cell proliferation and migration; It can influence endothelial cells’ survival, migration, and differentiation; It can be involved in tumor growth and metastasis; In inflammatory diseases, it can augment pro-inflammatory cytokines secretion. | Higher levels reflect articular degradation and inflammation in RA; Osteoclastogenic via RANKL-mediated NF-κB activation and M-CSF-induced precursor proliferation; Osteogenic suppressor by ↓ osteoblast differentiation of BMSCs during inflammation; Probably involved in RA fibroblast-like synoviocytes proliferation, migration, invasion, and inflammation; ↑ RA progression by up-regulating IFN-γ signaling pathways and bridging innate and adaptive immune responses; ↑ MMP-1, MMP-3, and MMP-13 gene expression via MAPK, JAK/STAT3, and NF-κB signaling pathways. | [127,128,150,152,153,154,155,160,161] |
| References | Organokine (S) Evaluated | Study Design | Sample | Evaluations and Interventions in RA | Primary Efficacy Outcome/ Correlation |
|---|---|---|---|---|---|
| Cheleschi et al., 2022 [13] | Adiponectin, chemerin, leptin, resistin, and visfatin. | Case-control study. Italy. | 50 RA patients (50–67, 15♂ and 35♀), 50 affected by PsA (55–63 y, 22♂ and 28♀), and 50 controls (40–59, 19♂ and 31♀). | Assessment of the relationships between adipokines and microRNAs in the discrimination between RA and PsA. | Leptin and microRNA-140 were increased in serum of PsA compared to RA or controls and therefore can be biomarkers used to discriminate PsA from RA. |
| Wahba et al., 2021 [15] | Chemerin, apelin, vaspin, and omentin. | Observational cross-sectional study. Egypt. | 150 RA patients (60♂ and 90♀, 44.29 ± 9.4 y) + 150 (75♂ and 75♀, 42.07 ± 11.3 y) healthy individuals. | Assessment of the roles of chemerin, apelin, vaspin, and omentin in RA pathophysiology among patients and their genetic variants, named rs17173608, rs2235306 rs2236242, and rs2274907. | Chemerin and vaspin levels were higher in RA patients and associated with clinical and laboratory parameters of the disease. Further, apelin and omentin levels were lower. |
| Gould et al., 2021 [122] | FGF-21. | Observational and longitudinal study. USA. | 113 RA participants aged between 18–70 y were primarily enrolled, and 84 attended the follow-ups. | Assessment of associations between FGF-21 and adverse changes in body composition longitudinally and physical functioning in individuals affected by RA. | FGF-21 levels were positively associated with obesity and secretion of pro-inflammatory cytokines in RA and with worsening physical function in RA. |
| Baker et al., 2021 [162] | Adiponectin leptin and FGF-21. | Observational cross-sectional study. USA. | 419 older-aged patients diagnosed with RA. | Determine associations between adipokines levels and abnormal body composition among patients with RA. | Grater fat mass associated with lower adiponectin and higher FGF-21 serum levels. Adipokines associates with both excessive adiposity and low lean mass in individuals with RA. |
| Vazquez-Villegas et al., 2021 [163] | Chemerin. | Observational cross-sectional study. Mexico. | 82 women diagnosed with RA (43 with functional disabilities and 39 without functional disabilities, 35–77 y and 30–79 y, respectively). | Elucidate if chemerin serum levels are associated with functional disabilities among women affected by RA. | Higher chemerin serum levels are significantly associated with functional disabilities among RA women, whereas no other blood biomarkers correlated with loss of function are present. |
| Gonzalez-Ponce et al., 2021 [164] | Chemerin. | Observational cross-sectional study. Mexico. | 210 RA women patients (56.59 ± 11.25 y). | Evaluate whether serum chemerin is a biomarker of disease activity among RA patients. | Higher chemerin serum levels increase the risk of moderate and severe RA disease, supporting chemerin as a joint inflammatory biomarker in RA. |
| Zhang et al., 2021 [165] | Adiponectin, leptin, resistin, and visfatin. | Case-control cohort study. Sweden; | 82 obese individuals that developed RA during follow-up matched with 410 controls + 88 additional matched pairs. | Identity if adiponectin, leptin, resistin, and visfatin serum levels associate with RA risk and if adiponectin performs an association with RA risk that is independent of the other adipokines. | In the included obese individuals, higher levels of adiponectin were associated with increased risk for developing RA but not higher levels of leptin, resistin, or visfatin. |
| Wahlin et al., 2021 [16] | Osteoprotegerin and osteocalcin. | Prospective cohort study. Sweden. | 79 patients newly diagnosed with RA (≤60 y at diagnosis). | Evaluate possible interactions between subclinical atherosclerosis and markers of bone turnover, regulators of bone formation, and BMD in patients with RA. | Subclinical atherosclerosis in individuals on new-onset with RA is associated with osteoprotegerin and osteocalcin but not with markers reflecting ongoing bone turnover or BMD. |
| Hanzhu Chen et al., 2021 [14] | Adiponectin. | Mendelian randomization study. Multicenter. | 14,361 cases and 43,923 controls. Results were analyzed with data from another 11,437 cases and 604,953 controls to decrease bias risk. | Assessment of a possible causal relationship between adiponectin and RA risk. | There was no evidence to support a causal association between adiponectin on RA risk and of RA on circulating levels of adiponectin. |
| Jiliang Chen et al., 2021 [166] | Leptin. | Cohort analytical study. China. | 223 RA patients (68.1 (57.2–75.6 y, 115♂ and 108♀)). | Explore whether leptin is associated with increased cardiovascular risk. | Elevated serum leptin levels were significantly associated with the prediction of cardiovascular events among RA individuals. |
| Taylan et al., 2021 [28] | Leptin. | Cohort analytical study. Turkey. | 47 RA patients with early disease (54 ± 15 y, 13♂ and 34♀) and 25 controls (51 ± 14, 3♂ and 22♀). | Explore the relationship between serum leptin levels and disease activity among RA patients. | Leptin serum levels were significant and directly associated with RA disease activity, and treatment with DMARDs decreased the levels of this adipokine. |
| Vuolteenaho et al., 2021 [29] | Resistin. | Post hoc analyses of an investigator-initiated, multicenter, controlled study. Finland. | 99 early, DMARDs-naïve RA patients. | Investigation of whether resistin could be associated with not only inflammation but also disease activity among DMARDs-naïve RA patients as well as disease progression. | Serum pretreatment resistin levels were associated with an increased risk of erosive disease in the early stages of RA disease. |
| Liu et al., 2020 [17] | Fibrinogen-like protein 1 (FGL1) (HEPASSOCIN). | Large-scale cohort two-centered study. China. | 1244 participants divided into 5 cohorts: #1 had 35 RAP with MTHDA + 60 healthy; #2 had 38 RAP with MTHDA + 15 RAP in remission/with LDA + 28 healthy; #3 had 221 RAP with MTHDA + 84 RAP with LDA + 102 RAP in remission, + 47 RAP with MTHDA, and 233 healthy; #4 had 82 RAP with MTHDA before DMARD treatment + 23 RAP with MTHDA + 26 RAP with LDA + 33 RA in remission after DMARD intervention; and #5 had 35 healthy individuals + 23 RAP in remission + 24 RAP with low to high disease activity + patients without RAP. | Evaluation of biomarkers that can precisely indicate and monitor RA disease activity and provide adequate therapeutics. | FGL1 could positively predict RA disease activity and its prognosis. Clinically, FGL1 was considered useful for predicting RA progression. |
| Qu et al., 2020 [18] | Sex hormone-binding globulin (SHBG) | Mendelian randomization study. China. | In all, data from 28,837 European ancestry individuals were assessed. This study also analyzed Mendelian association of SHBG and osteoarthritis and ankylosing spondylitis. | Determine whether the concentrations of SHBG are correlated with the elevated risk for developing RA. | Positive causal effects of circulating SHBG levels were found to determine RA development risk with no sex specifications. |
| Murillo-Saich et al., 2021 [19] | Myostatin. | Cross-sectional study. Mexico. | 127 women were enrolled without any rheumatological diagnosis (24–85 y) and 84 RA women (24–89 y). | Evaluate the association between serum myostatin levels and inflammatory parameters among RA women. | Myostatin associated significantly with RA disease activity by augmented inflammatory biomarkers, suggesting myostatin roles in muscle wasting and inflammation among RA women. |
| Gamez-Nava et al., 2022 [139] | Irisin. | Cross-sectional study. Mexico. | 148 women with RA (≥45 y) and 97 control women. | Evaluate the association between serum irisin levels and osteoporotic vertebral fracture among RA diseased women. | Decreased levels of irisin associated significantly with the occurrence of osteoporotic vertebral fracture. |
| Gamal et al., 2020 [167] | Irisin. | Cross-sectional study. Egypt. | 58 RA patients (44.12 ± 11.78 y) and 30 matched controls. | Evaluate if irisin levels were correlated with good sleep quality in RA individuals compared to controls. | Irisin levels were decreased in RA patients with poor sleep quality compared to controls. |
| Soliman et al., 2022 [156] | Irisin. | Cross-sectional study. Egypt. | 60 RA patients (47.03 ± 9.5 y) and 30 healthy individuals. | Asses if serum irisin levels in RA patients correlate significantly with cardiovascular risk factors and subclinical atherosclerosis occurrence. | Decreases in irisin serum levels correlated significantly with increased cardiovascular risk factors occurrence and subclinical atherosclerosis prediction. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laurindo, L.F.; de Maio, M.C.; Barbalho, S.M.; Guiguer, E.L.; Araújo, A.C.; de Alvares Goulart, R.; Flato, U.A.P.; Júnior, E.B.; Detregiachi, C.R.P.; dos Santos Haber, J.F.; et al. Organokines in Rheumatoid Arthritis: A Critical Review. Int. J. Mol. Sci. 2022, 23, 6193. https://doi.org/10.3390/ijms23116193
Laurindo LF, de Maio MC, Barbalho SM, Guiguer EL, Araújo AC, de Alvares Goulart R, Flato UAP, Júnior EB, Detregiachi CRP, dos Santos Haber JF, et al. Organokines in Rheumatoid Arthritis: A Critical Review. International Journal of Molecular Sciences. 2022; 23(11):6193. https://doi.org/10.3390/ijms23116193
Chicago/Turabian StyleLaurindo, Lucas Fornari, Mariana Canevari de Maio, Sandra Maria Barbalho, Elen Landgraf Guiguer, Adriano Cressoni Araújo, Ricardo de Alvares Goulart, Uri Adrian Prync Flato, Edgar Baldi Júnior, Cláudia Rucco Penteado Detregiachi, Jesselina Francisco dos Santos Haber, and et al. 2022. "Organokines in Rheumatoid Arthritis: A Critical Review" International Journal of Molecular Sciences 23, no. 11: 6193. https://doi.org/10.3390/ijms23116193
APA StyleLaurindo, L. F., de Maio, M. C., Barbalho, S. M., Guiguer, E. L., Araújo, A. C., de Alvares Goulart, R., Flato, U. A. P., Júnior, E. B., Detregiachi, C. R. P., dos Santos Haber, J. F., Bueno, P. C. S., Girio, R. S. J., Eleutério, R. G., & Bechara, M. D. (2022). Organokines in Rheumatoid Arthritis: A Critical Review. International Journal of Molecular Sciences, 23(11), 6193. https://doi.org/10.3390/ijms23116193