
Maternal Prenatal Inflammation Increases Brain Damage Susceptibility of Lipopolysaccharide in Adult Rat Offspring via COX-2/PGD-2/DPs Pathway Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
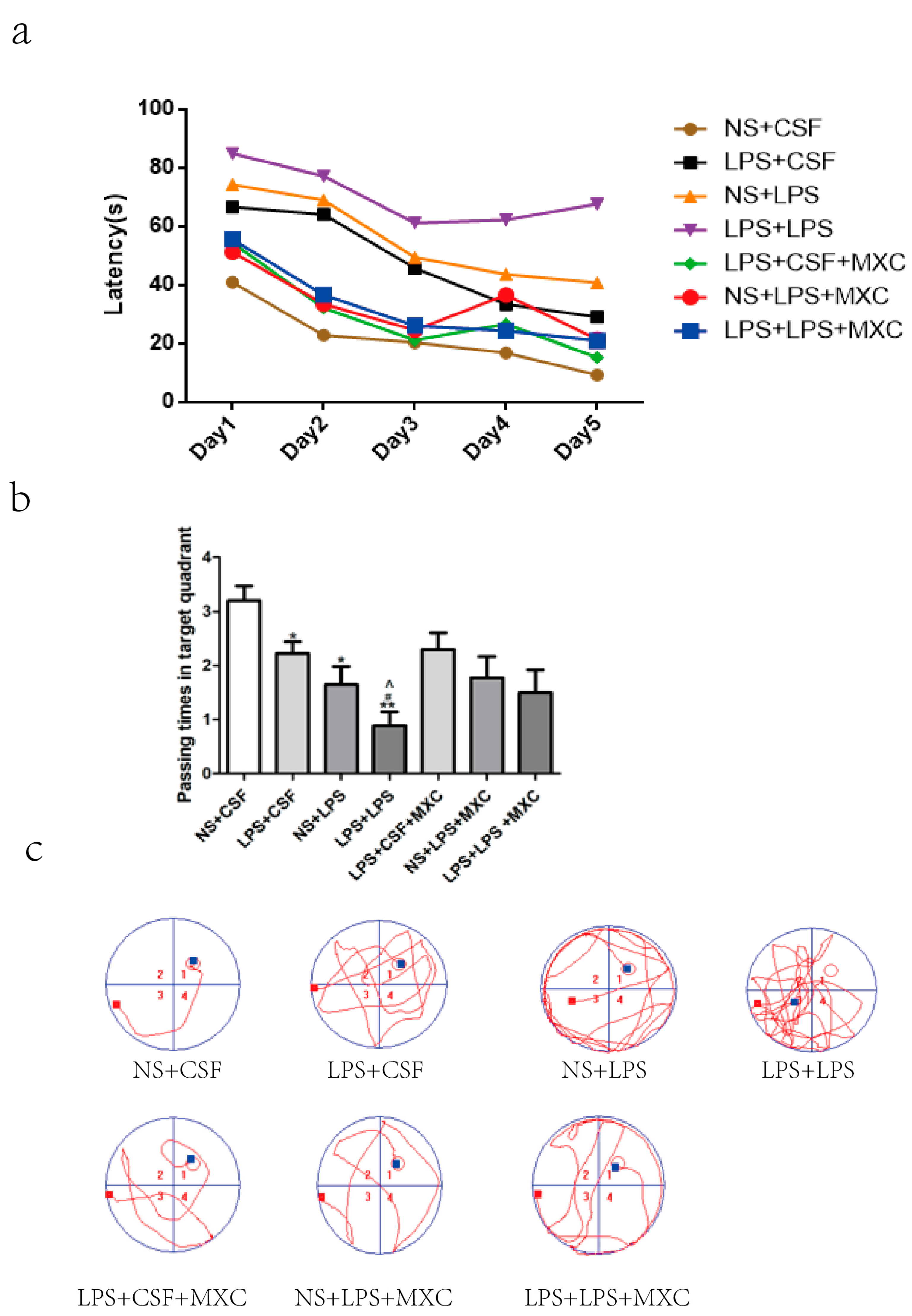
2.1. Effect of MXC on Learning Cognitive Function in LPS-Treated Offspring from Prenatal Inflammation Exposure
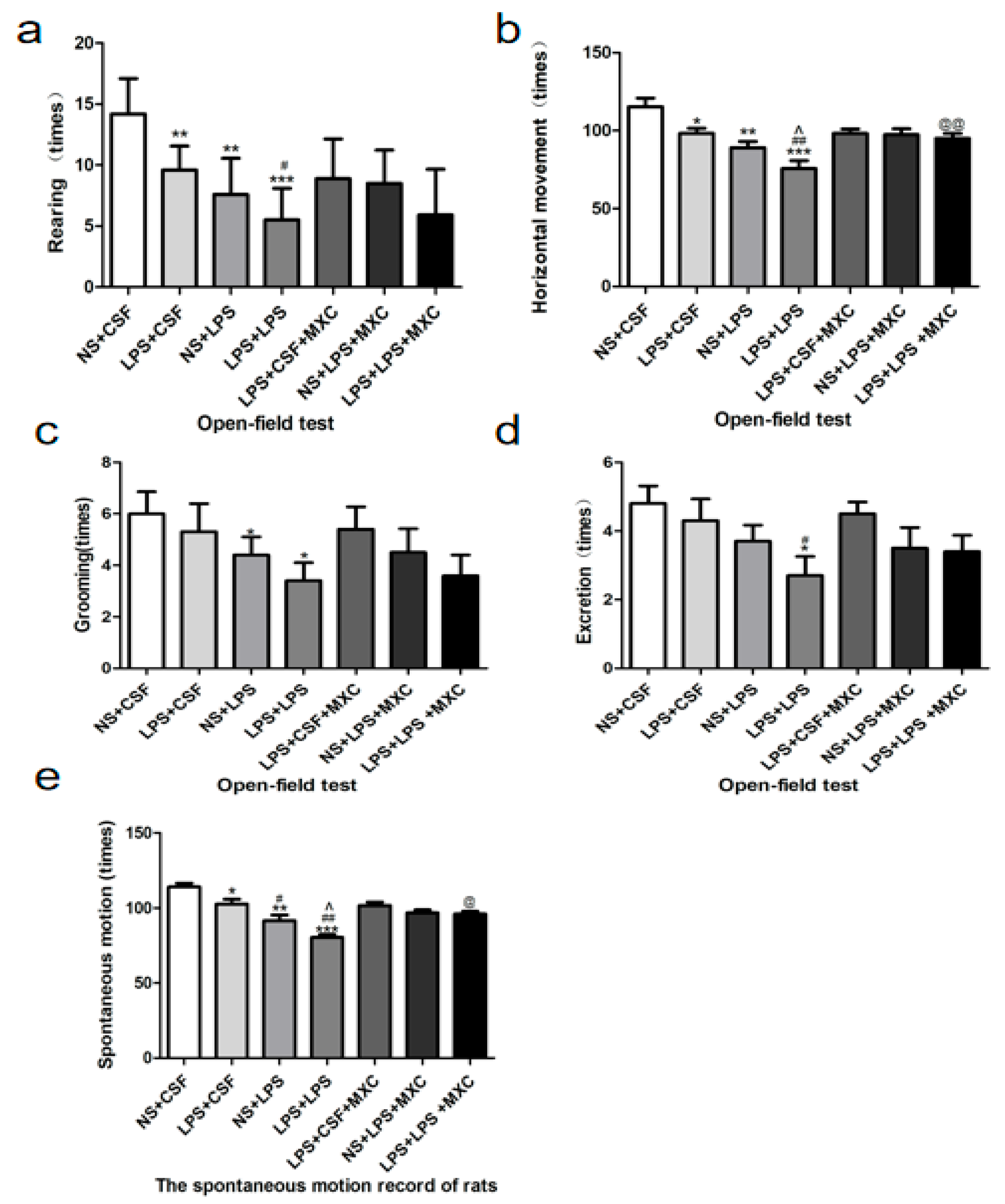
2.2. Effect of MXC on the Exploratory Behavior and Spontaneous Locomotor Activity in LPS-Treated Offspring from Prenatal Inflammation Exposure
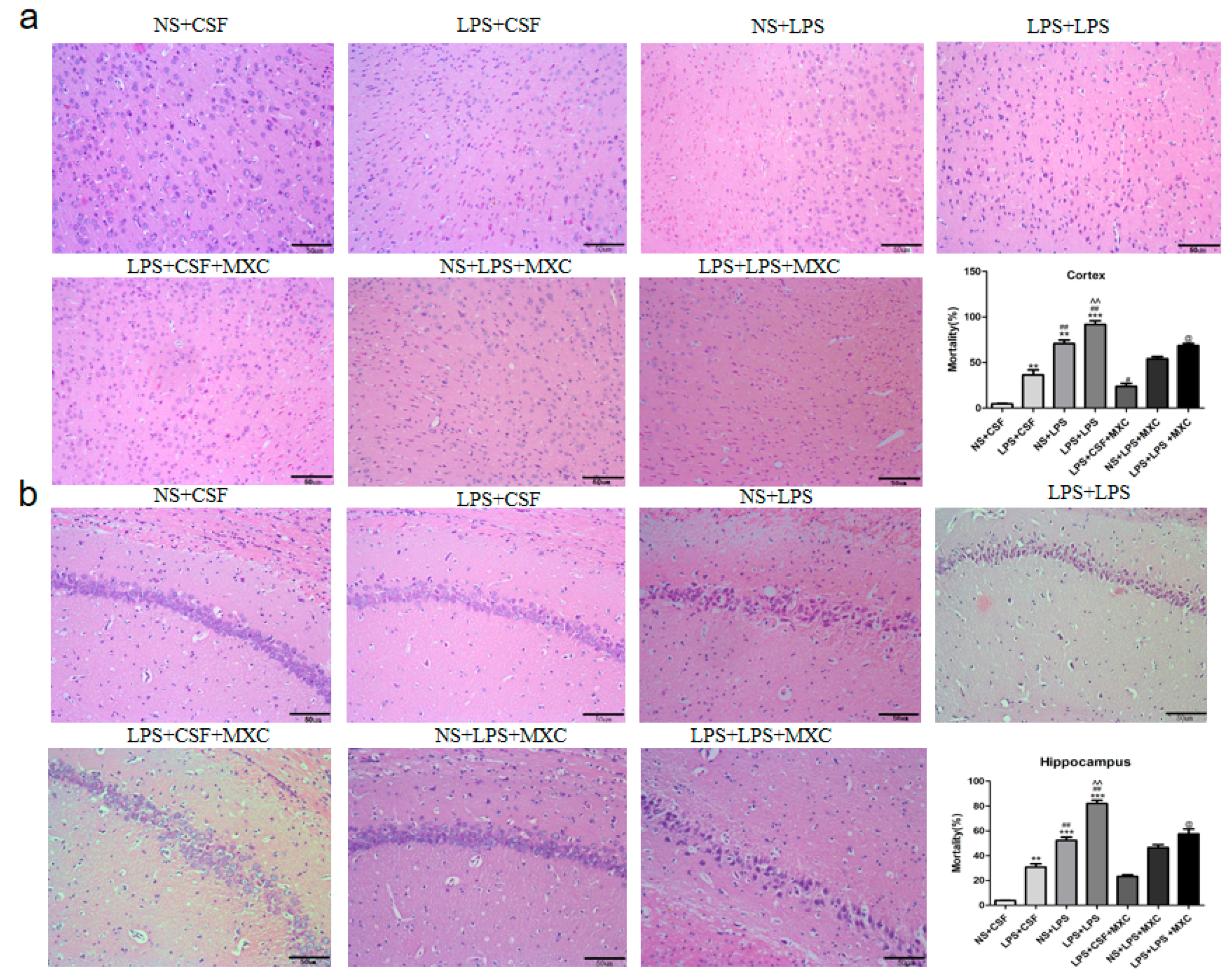
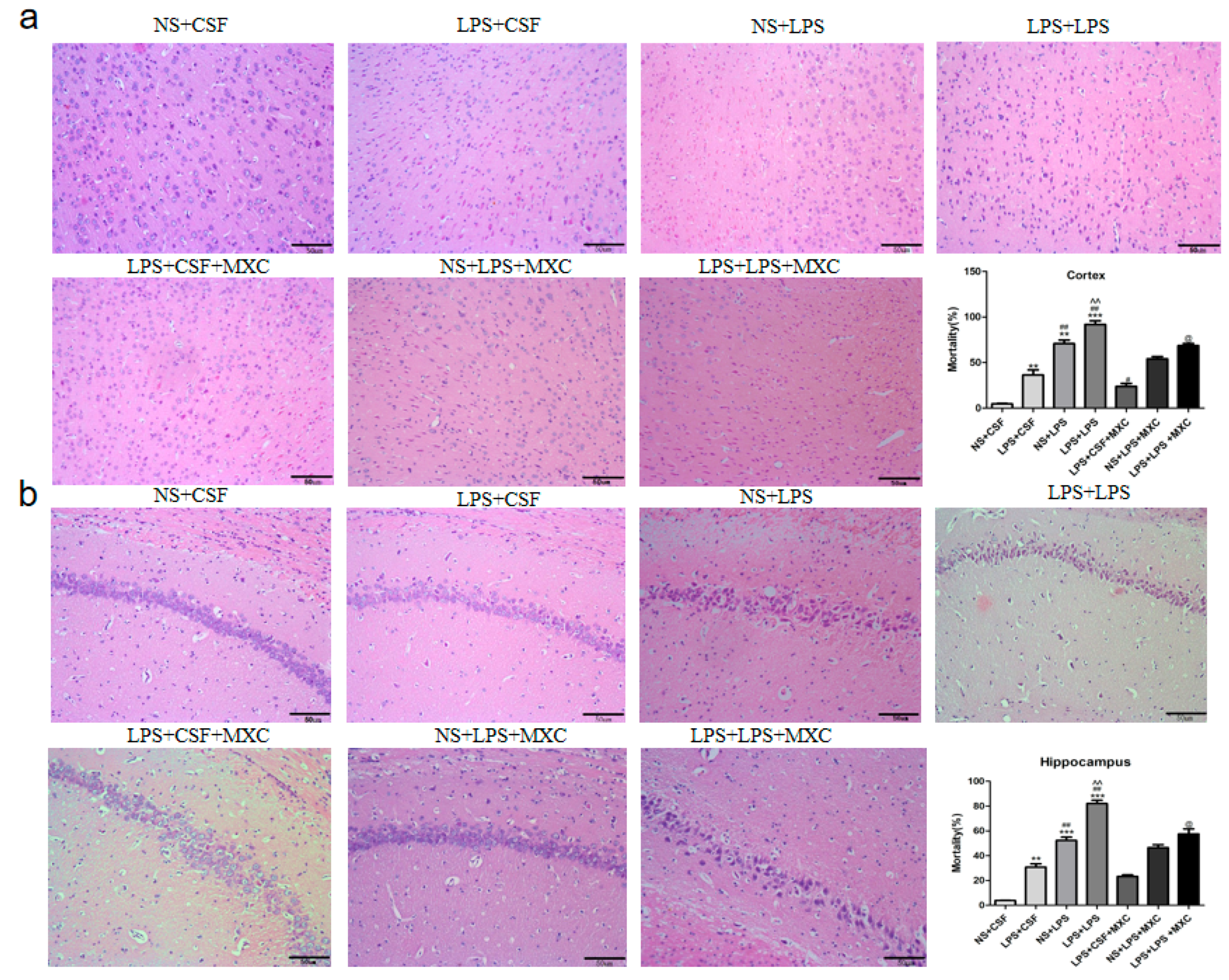
2.3. Effect of MXC on Hippocampal and Cortical Histopathology in LPS-Treated Offspring from Prenatal Inflammation Exposure
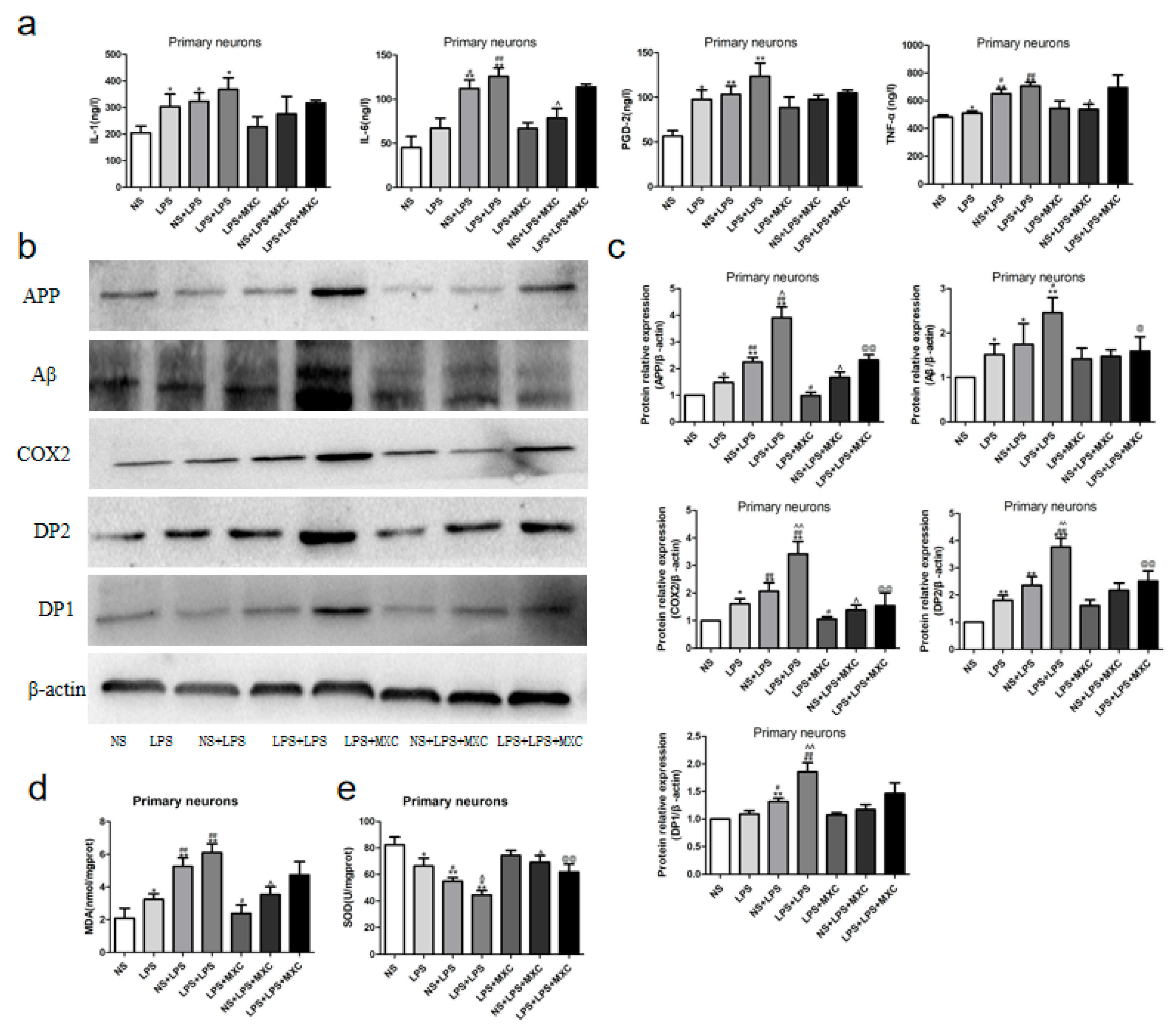
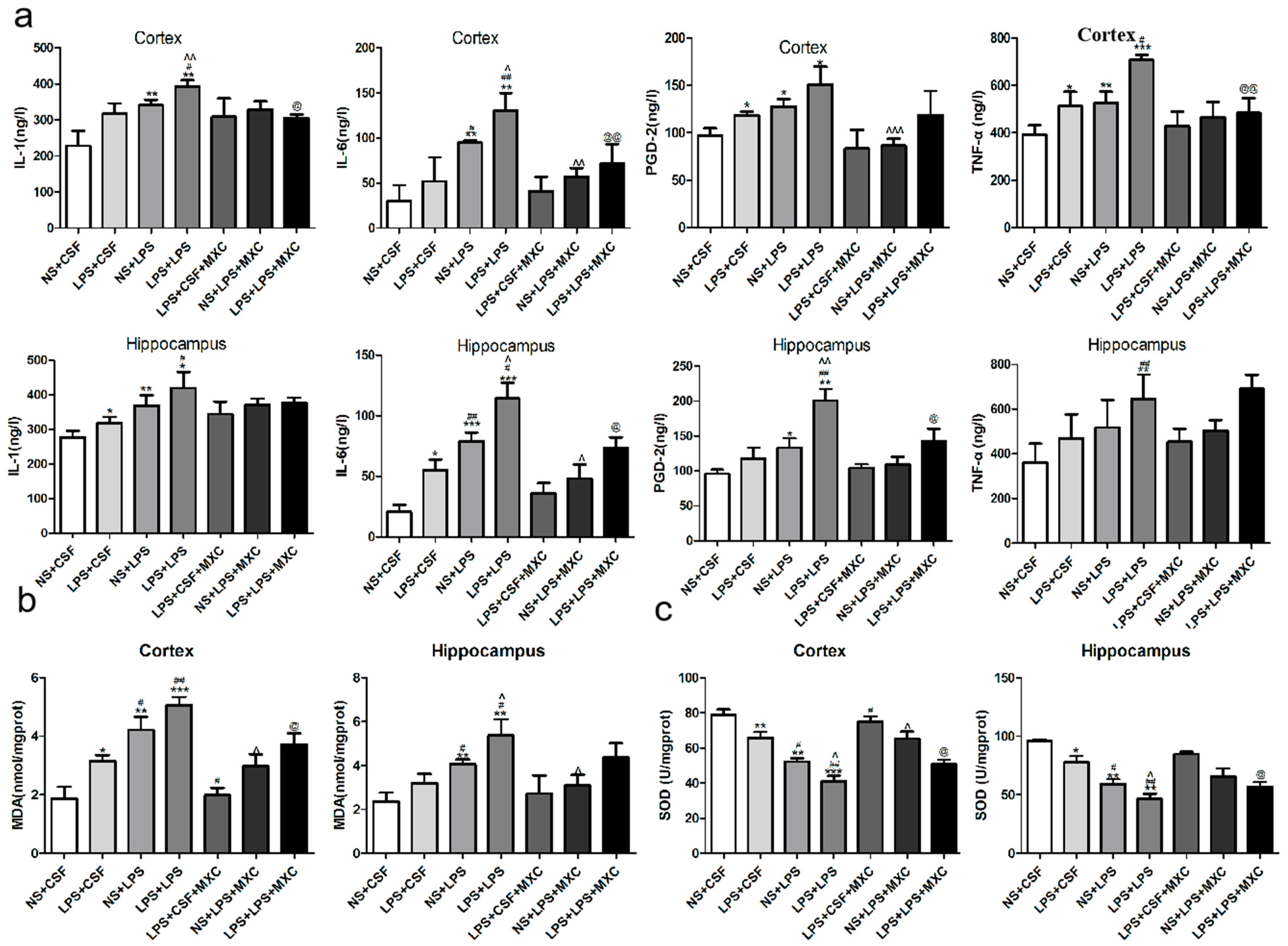
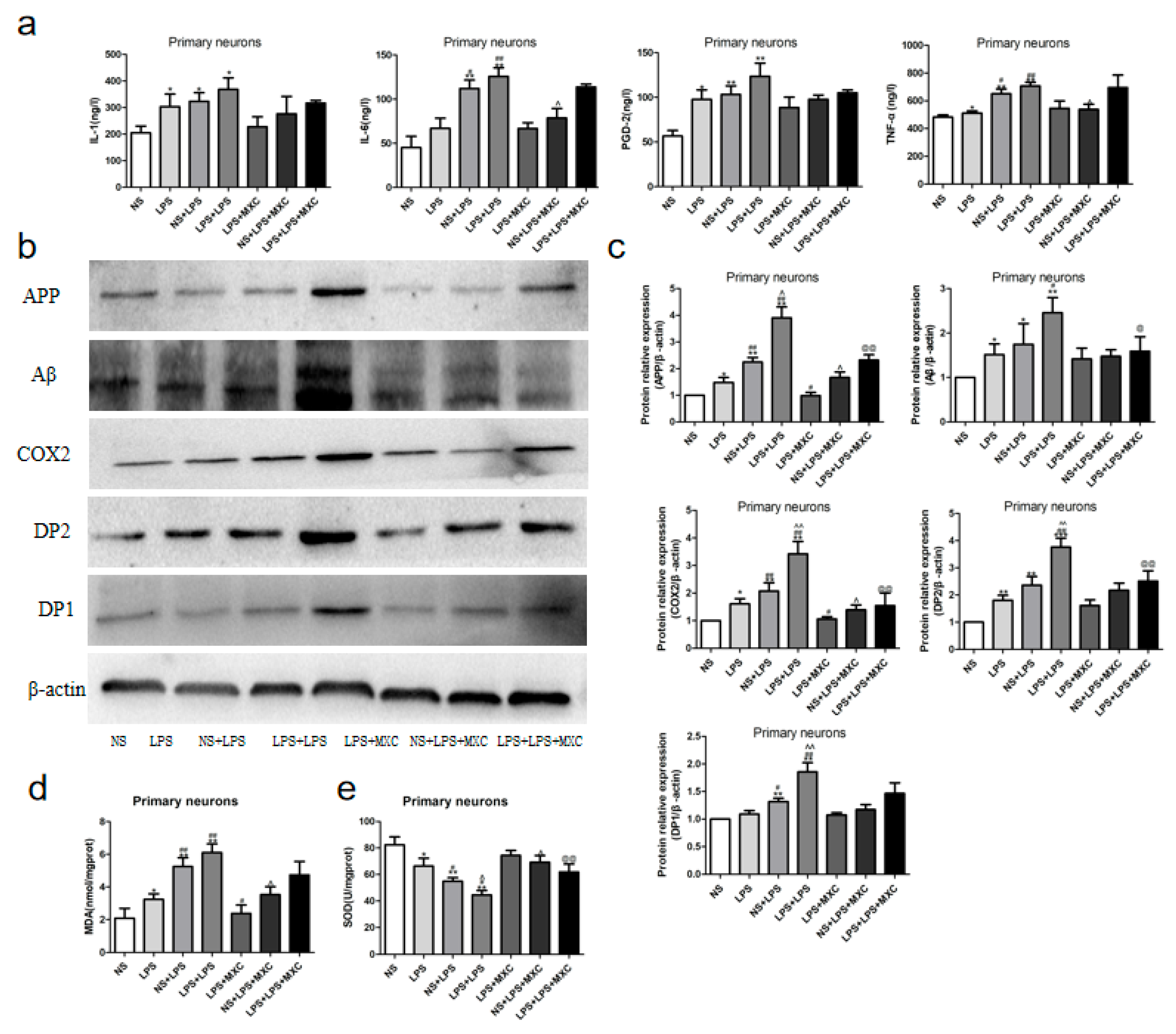
2.4. Effect of MXC on the Levels of PGD-2, TNF-α, IL-1β and IL-6 in LPS-Treated Offspring from Prenatal Inflammation Exposure
2.5. Effect of MXC on the MDA Levels and the SOD Activity in LPS-Treated Offspring from Prenatal Inflammation Exposure
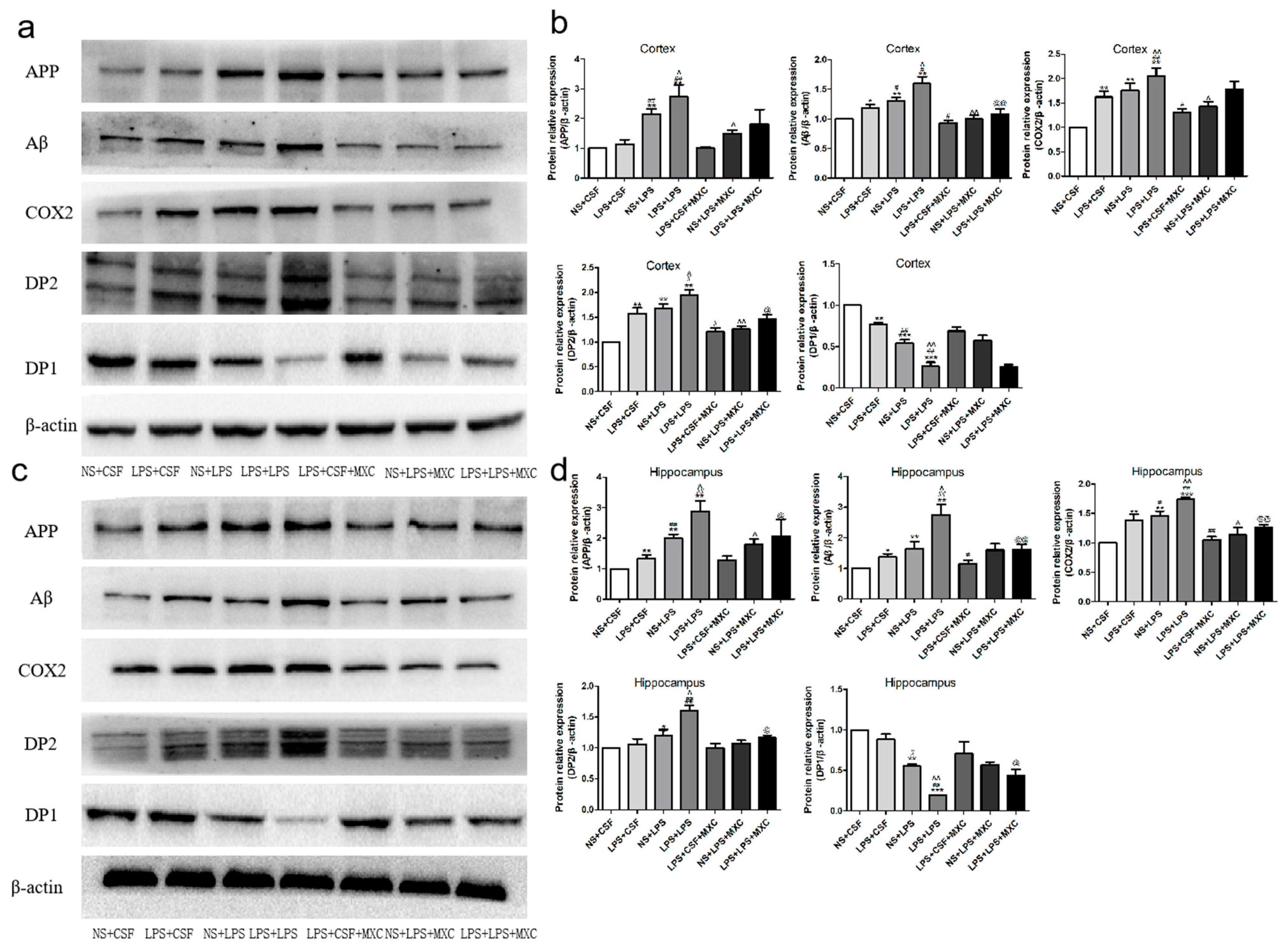
2.6. Effect of MXC on the Protein Expressions of COX-2, DP2, DP1, Aβ, and APP in LPS-Treated Offspring from Prenatal Inflammation Exposure
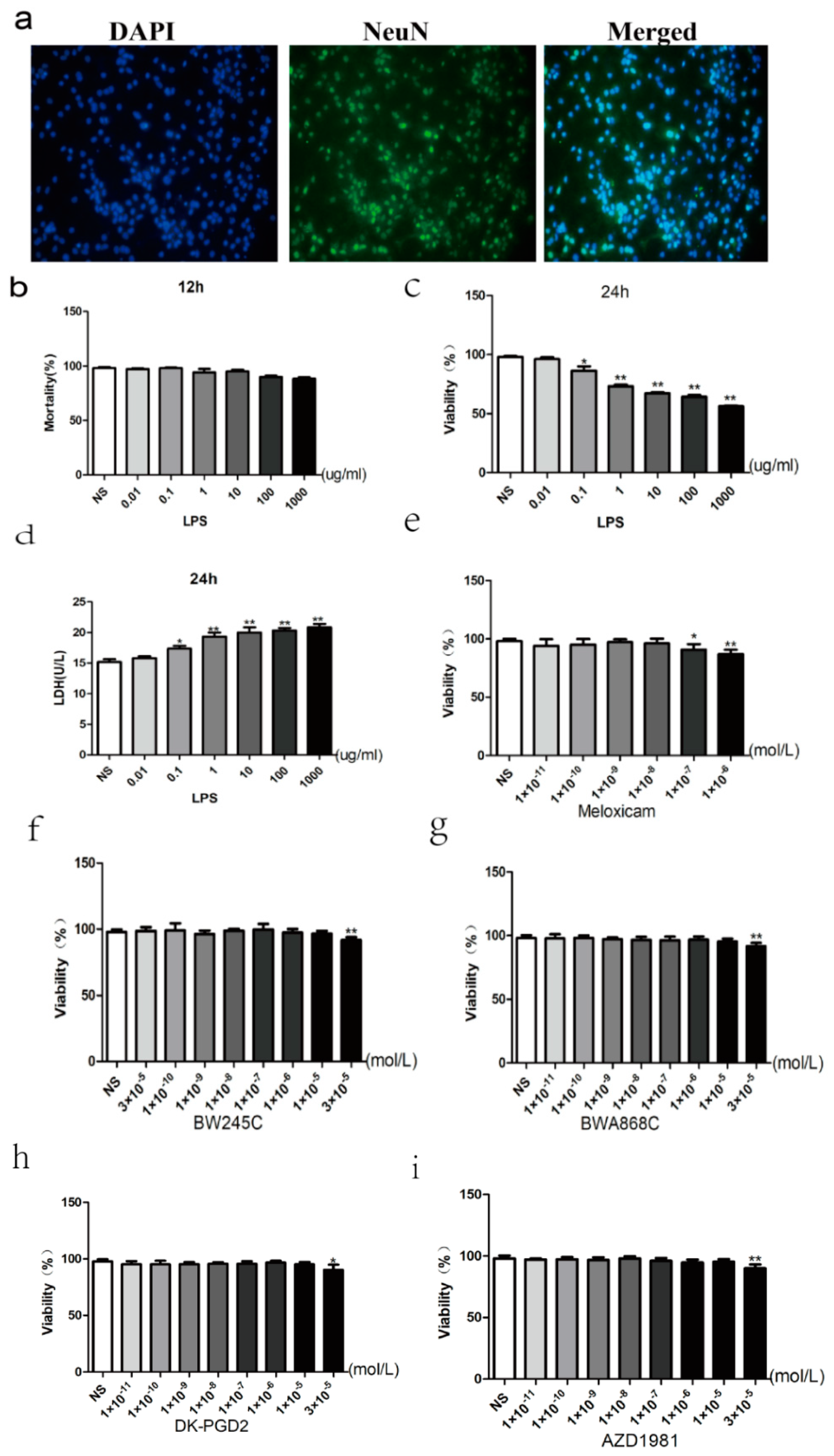
2.7. Primary Cultured and Identification of Neurons
2.8. Dose-Dependent Effect of LPS
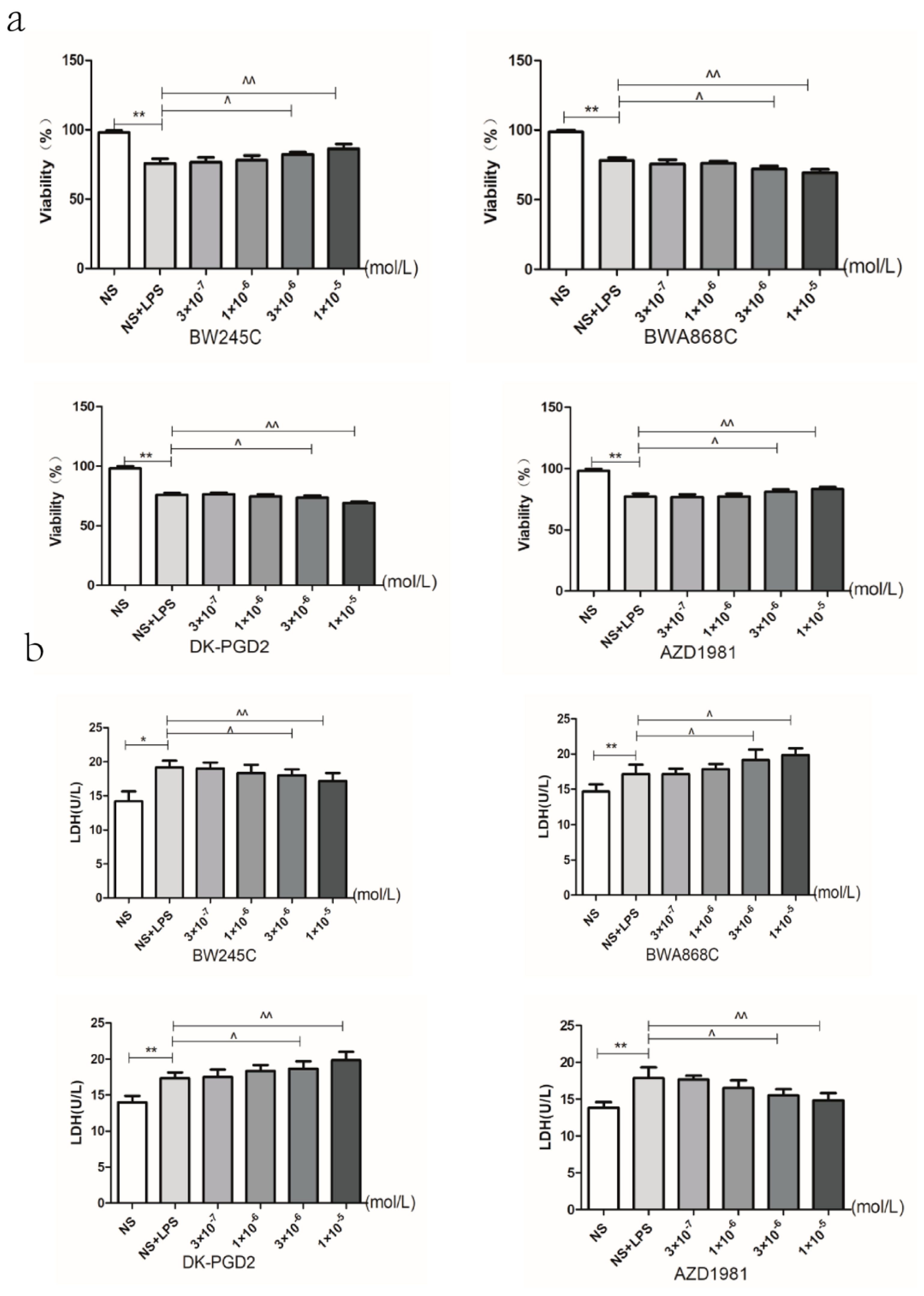
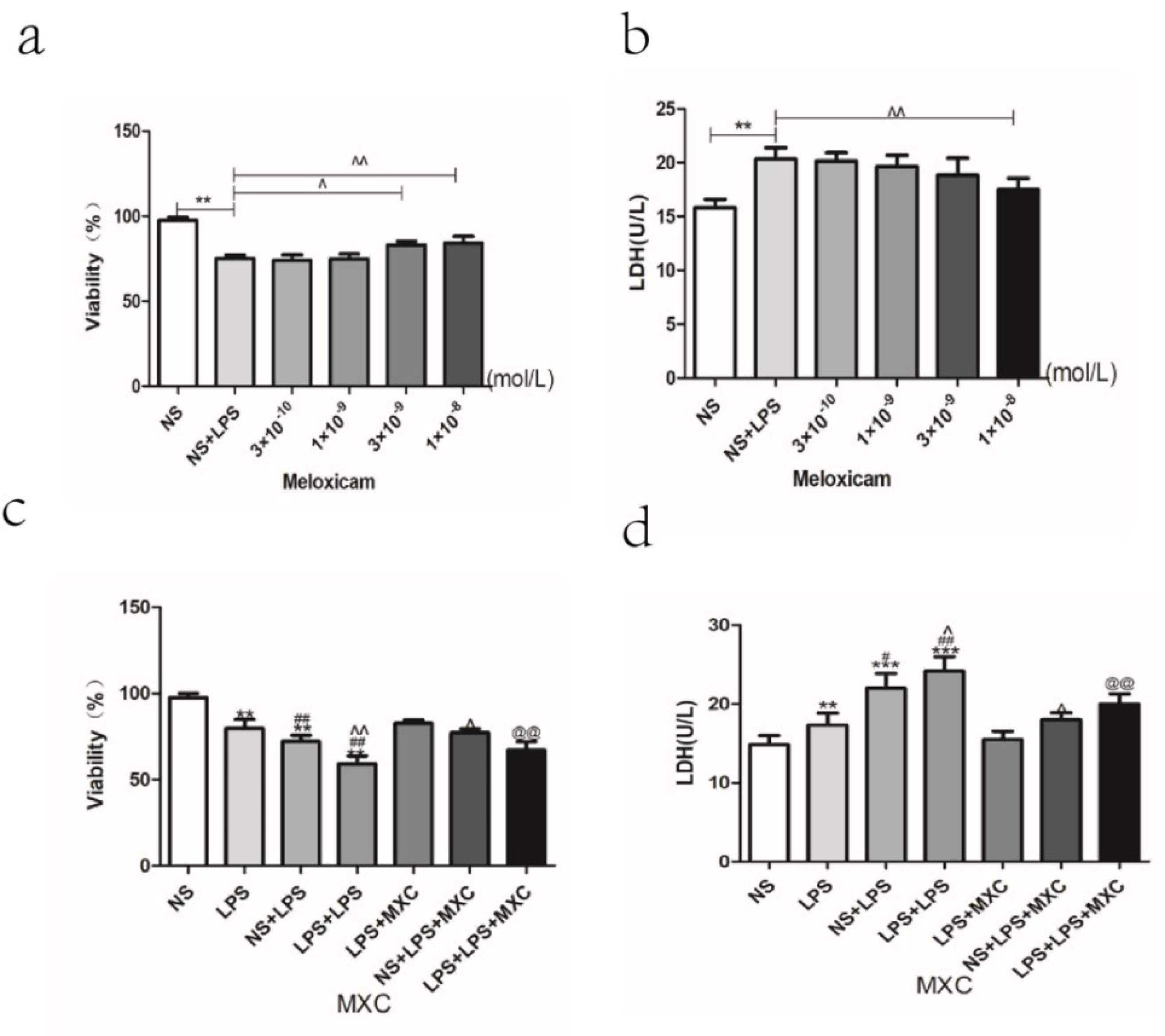
2.9. Effect of MXC on the Neuron Viability and the LDH Leakage Rate in LPS-Treated Primary Cultured Neurons of Offspring from Prenatal Inflammation Exposure
2.10. Effect of MXC on the Levels of TNF-α, IL-6, IL-1β, and PGD2 in LPS-Treated Primary Cultured Neurons of Offspring from Prenatal Inflammation Exposure
2.11. Effect of MXC on the Protein Expressions of COX-2, DP2, DP1, Aβ, and APP in LPS-Treated Primary Cultured Neurons of Offspring from Prenatal Inflammation Exposure
2.12. Effect of MXC on the Levels of MDA and the Activity of SOD in LPS-Treated Primary Cultured Neurons of Offspring from Prenatal Inflammation Exposure
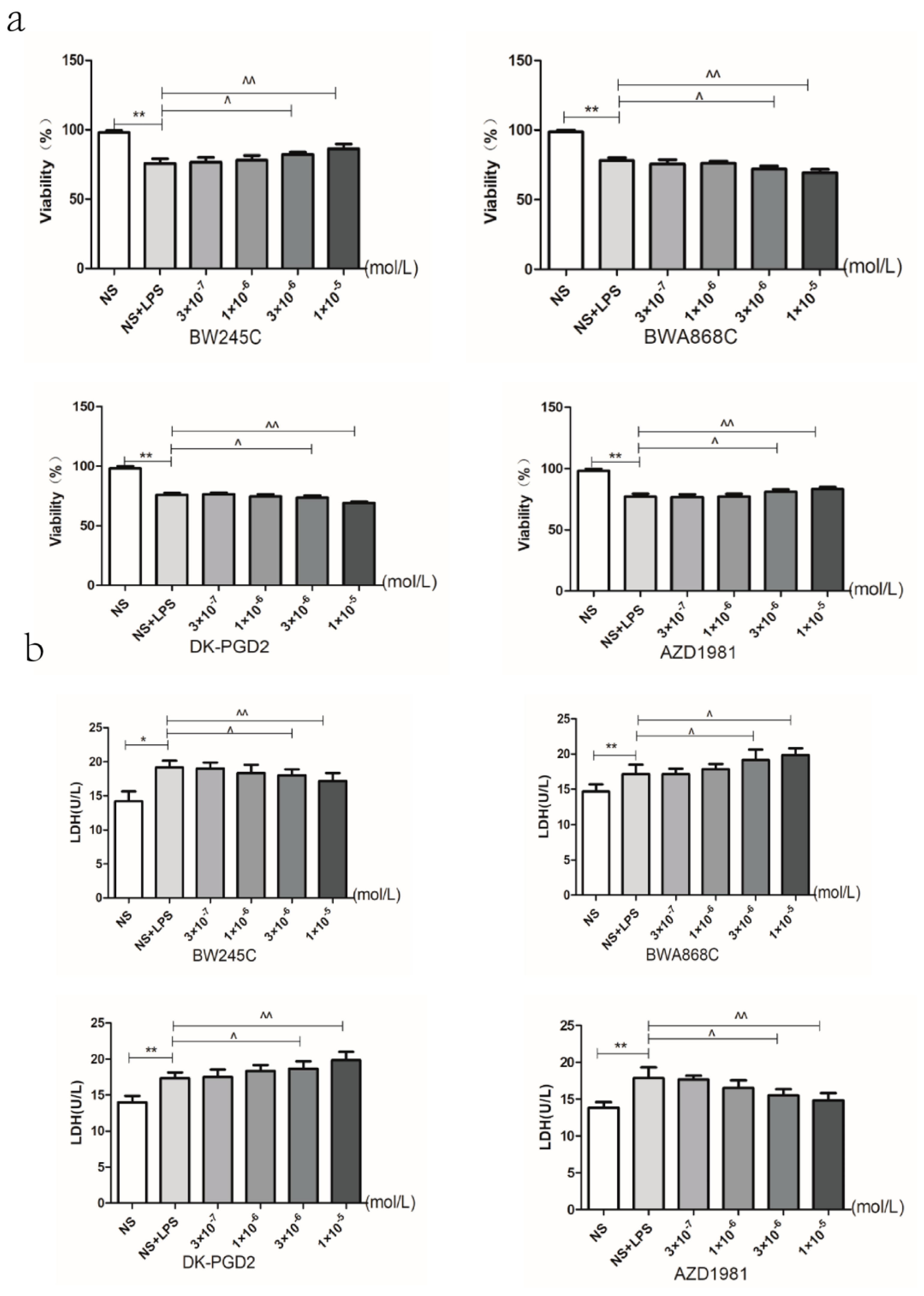
2.13. Effect of DP Agonist or Antagonist on the Neuron Viability and the LDH Leakage in LPS-Treated Primary Cultured Neurons of Offspring from Prenatal Inflammation Exposure
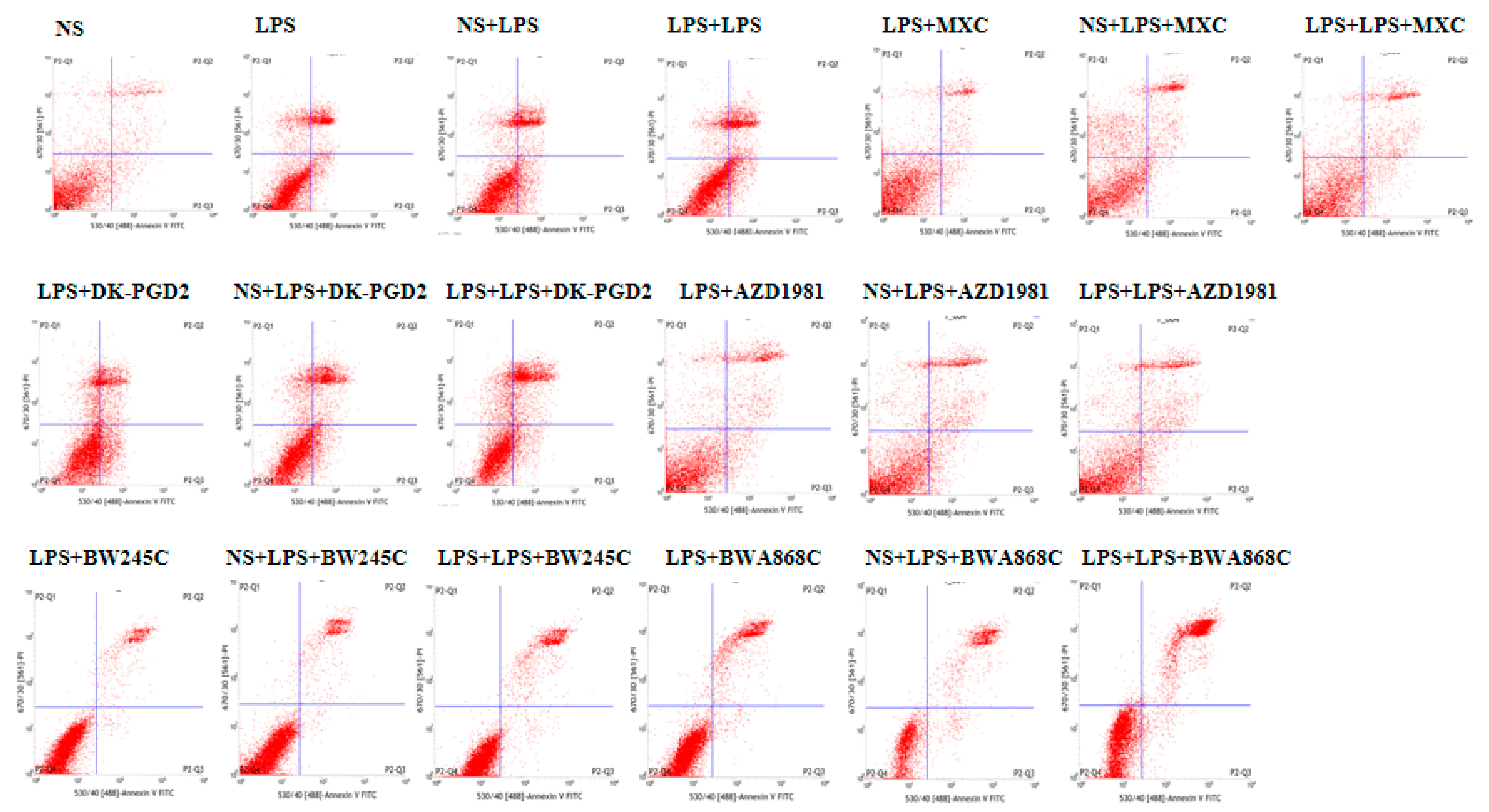
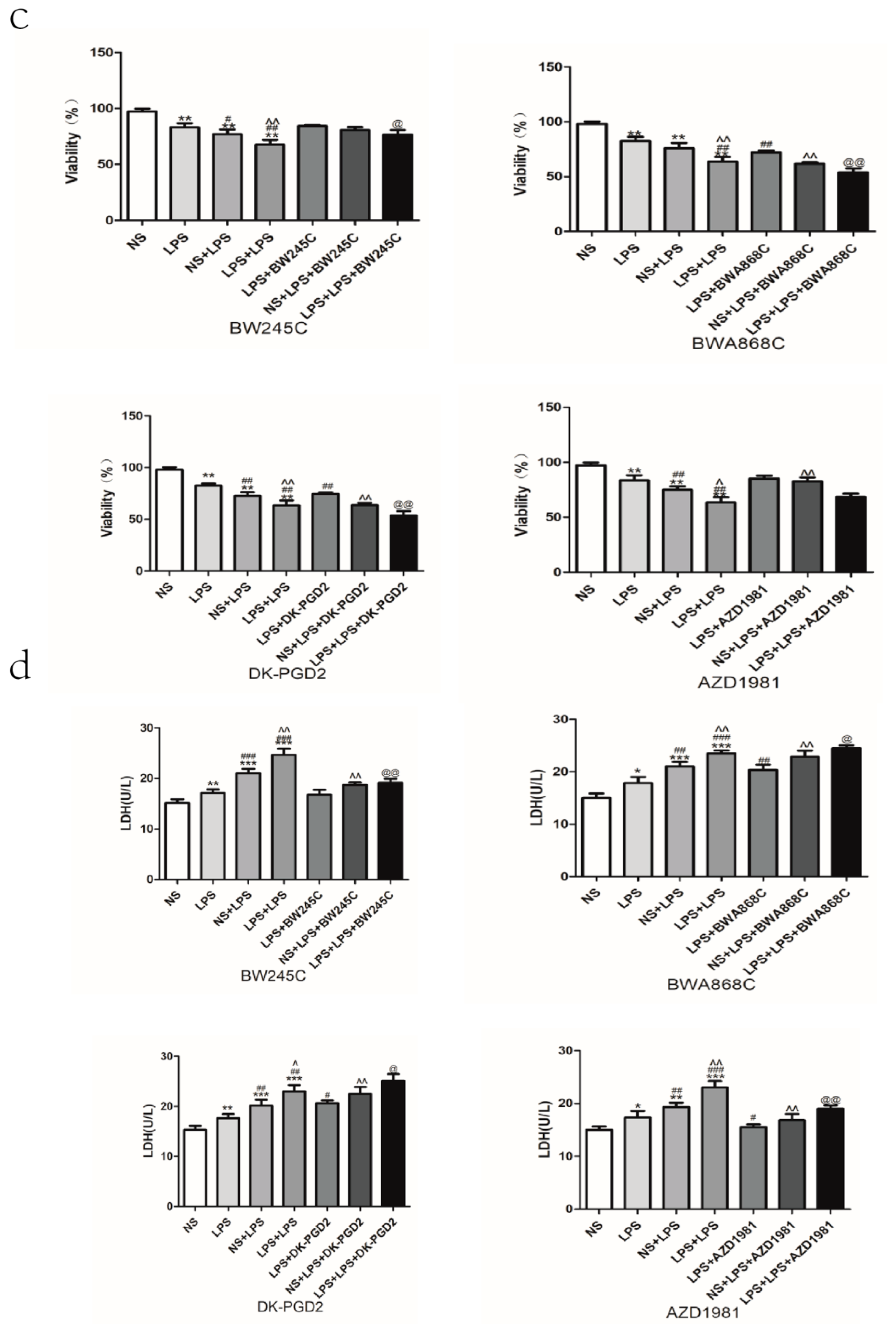
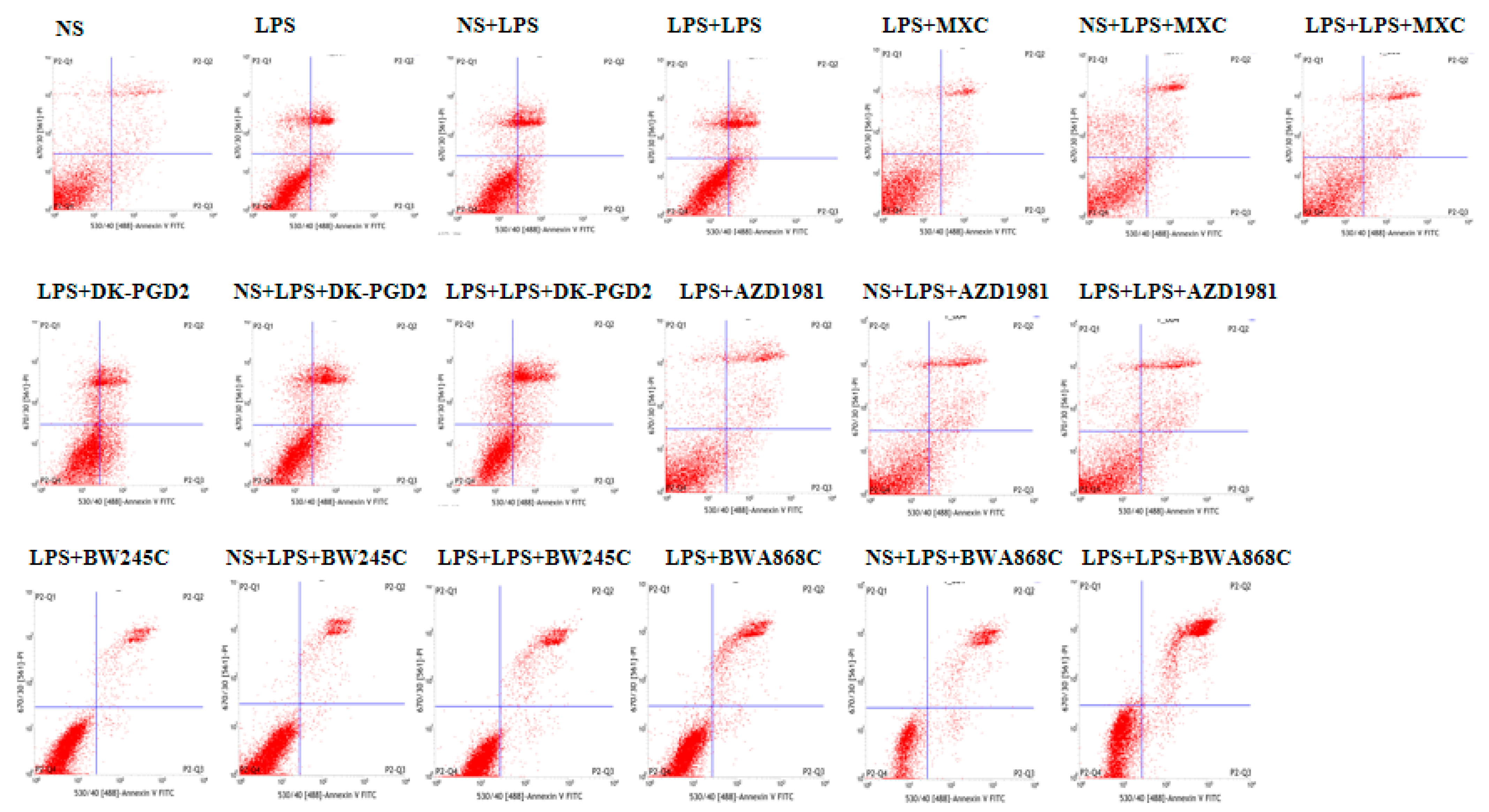
2.14. Effect of COX2 or DP Intervention on the Apoptosis in LPS-Treated Primary Cultured Neurons of Offspring from Prenatal Inflammation Exposure
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Establishment of Animal Models
4.3. Design and Treatment Groups
4.4. Morris Water Maze Test
4.5. Open-Field Test (OFT)
4.6. Rat Autonomic Activity Test
4.7. Histopathological Observation
4.8. Primary Neuron Culture
4.9. Characterization of Neuron by Immunofluorescence
4.10. Establishment of the Model of Primary Cultured Neuron Damage Induced by LPS
4.11. Detection of Neuron Viability
4.12. LDH Leakage Rate Assay
4.13. Analysis of Neuronal Apoptosis by Flow Cytometry
4.14. Oxidative Stress Level Measurement
4.15. Biochemical Assay
4.16. Western Blotting Test
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprica, M.; Hjorth, E.; Spulber, S.; Popescu, B.O.; Ankarcrona, M.; Winblad, B.; Schultzberg, M. Studies on brain volume, Alzheimer-related proteins and cytokines in mice with chronic overexpression of IL-1 receptor antagonist. J. Cell. Mol. Med. 2007, 11, 810–825. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Meijer, J.; Amanda Kiliaan, A.R.; van Swieten John, C.; Stijnen, T.; Hofman, A.; Witteman, J.C.M.; Breteler, M.M.B. Inflammatory proteins in plasma and the risk of dementia: The rotterdam study. Arch. Neurol. 2004, 61, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Miguel, R.P.J.; Maria, M.R.J. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef]
- Guo, J.; Yu, J.; Grass, D.J.; de Beer, F.C.; Kindy, M.S. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.W.; De Carli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- Hauss-Wegrzyniak, B.; Lukovic, L.; Bigaud, M.; Stoeckel, M. Brain inflammatory response induced by intracerebroventricular infusion of lipopolysaccharide: An immunohistochemical study. Brain Res. 1998, 794, 211–224. [Google Scholar] [CrossRef]
- Brown, A.S. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev. Neurobiol. 2012, 72, 1272–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sule, S.S.; Onayade, A.A. Community-based antenatal and perinatal interventions and newborn survival. Niger. J. Med. 2006, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Martinet, M.; Bullinger, A.; Perneger, T.; Pfister, R.E. Development and assessment of a sensory-motor scale for the neonate: A clinical tool at the bedside. Arch. Pediatr. 2012, 20, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.J.; Whitaker, A.M.; Smith, S.E.; Patterson, P.H.; Bauman, M.D. Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol. Psychiatry 2015, 77, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017, 549, 528. [Google Scholar] [CrossRef] [Green Version]
- Bakos, J.; Duncko, R.; Makatsori, A.; Pirnik, Z.; Kiss, A.; Jezova, D. Prenatal immune challenge affects growth, behavior, and brain dopamine in offspring. Ann. N. Y. Acad. Sci. 2004, 1018, 281–287. [Google Scholar] [CrossRef]
- Wang, X.; Rousset, C.I.; Hagberg, H.; Mallard, C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Semin. Fetal Neonatal Med. 2006, 11, 343–353. [Google Scholar] [CrossRef]
- Silver, R.M.; Edwin, S.S.; Trautman, M.S.; Simmons, D.L.; Branch, D.W.; Dudley, D.J.; Mitchell, M. Bacterial lipopolysaccharide-mediated fetal death. Production of a newly recognized form of inducible cyclooxygenase (COX-2) in murine decidua in response to lipopolysaccharide. J. Clin. Investig. 1995, 95, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Alhouayek, M.; Muccioli, G.G. COX-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef]
- Han, S.; Lee, J.H.; Kim, C.; Nam, D.; Chung, W.-S.; Lee, S.-G.; Ahn, K.S.; Cho, S.K.; Cho, M.; Ahn, K.S. Capillarisin inhibits iNOS, COX-2 expression, and proinflammatory cytokines in LPS-induced RAW 264.7 macrophages via the suppression of ERK, JNK, and NF-κB activation. Immunopharmacol. Immunotoxicol. 2013, 35, 34–42. [Google Scholar] [CrossRef]
- Minghetti, L. Role of COX-2 in inflammatory and degenerative brain diseases. In Subcellular Biochemistry; Springer: Berlin, Germany, 2007; pp. 127–141. [Google Scholar]
- Yu, L.; Jiang, R.; Su, Q.; Yu, H.; Yang, J. Hippocampal neuronal metal ion imbalance related oxidative stress in a rat model of chronic aluminum exposure and neuroprotection of meloxicam. Behav. Brain Funct. 2014, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, Y.; Stanić, D.; Tomas, D.; Jarrott, B.; Horne, M.K. Meloxicam reduces lipopolysaccharide-induced degeneration of dopaminergic neurons in the rat substantia nigra pars compacta. Neurosci. Lett. 2009, 460, 121–125. [Google Scholar] [CrossRef]
- Hammad, H.; Kool, M.; Soullié, T.; Narumiya, S.; Trottein, F.; Hoogsteden, H.C.; Lambrecht, B.N. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J. Exp. Med. 2007, 204, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Hamid, S.; Whittle, B.J.R. Interaction of prostaglandin D2 with prostacyclin, carbacyclin and the hydantoin prostaglandin, BW245C, in guinea-pig platelets. Br. J. Pharmacol. 2012, 85, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelir, E.; Arslan, S.O.; Sayan, H.; Pinar, L. Effect of rapid-eye-movement sleep deprivation on rat hypothalamic prostaglandins. Prostagland. Leukot. Essent. Fat. Acids 2005, 73, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.S.; Ahmad, M.; Maruyama, T.; Narumiya, S.; Doré, S. Prostaglandin D2 DP1 receptor is beneficial in ischemic stroke and in acute exicitotoxicity in young and old mice. Age 2010, 32, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, D.; Rash, K.; Smalstig, B.; Roberts, E.; Johnstone, E.; Sharp, J.; Panetta, J.; Little, S.; Kramer, R.; Clemens, J. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia 2015, 27, 110–128. [Google Scholar] [CrossRef]
- Sekeroglu, A.; Jacobsen, J.M.; Jansen-Olesen, I.; Gupta, S.; Sheykhzade, M.; Olesen, J.; Bhatt, D.K. Effect of PGD2 on middle meningeal artery and mRNA expression profile of L-PGD2 synthase and DP receptors in trigeminovascular system and other pain processing structures in rat brain. Pharmacol. Rep. 2017, 69, 50–56. [Google Scholar] [CrossRef]
- Jie, M.; Yang, Q.; Wei, Y.; Yang, Y.; Ji, C.; Hu, X.; Mai, S.; Kuang, S.; Tian, X.; Luo, Y.; et al. Effect of the PGD2-DP signaling pathway on primary cultured rat hippocampal neuron injury caused by aluminum overload. Sci. Rep. 2016, 6, 24646. [Google Scholar]
- Hakan, T.; Toklu, H.Z.; Biber, N.; Ozevren, H.; Solakoglu, S.; Demirturk, P.; Aker, F.V. Effect of COX-2 inhibitor meloxicam against traumatic brain injury-induced biochemical, histopathological changes and blood-brain barrier permeability. Neurol. Res. 2010, 32, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.D.; Iosif, A.M.; Smith, S.E.; Bregere, C.; Amaral, D.G.; Patterson, P.H. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol. Psychiatry 2014, 75, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneusel, I. Double-immune challenge with the viral mimic Polyi:C induces Alzheimer’s disease-like neuropathology in aged wild-type mice. Alzheimers Dement. 2011, 7, S286. [Google Scholar] [CrossRef]
- Arrode-Brusés, G.; Brusés, J.L. Maternal immune activation by poly(I:C) induces expression of cytokines IL-1β and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J. Neuroinflamm. 2012, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Deverman, B.E.; Patterson, N.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [Green Version]
- Philippens, I.H.; Ormel, P.R.; Baarends, G.; Johansson, M.; Remarque, E.J.; Doverskog, M. Acceleration of amyloidosis by inflammation in the amyloid-beta marmoset monkey model of Alzheimer’s disease. Eur. J. Neurosci. 2017, 55, 101. [Google Scholar] [CrossRef] [Green Version]
- Portavella, M.; Vargas, J.P. Emotional and spatial learning in goldfish is dependent on different telencephalic pallial systems. Eur. J. Neurosci. 2005, 21, 2800–2806. [Google Scholar] [CrossRef]
- Oehlrich, D.; Prokopcova, H.; Gijsen, H.J. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 45, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tanila, H.; Puoliväli, J.; Kadish, I.; van Groen, T. Gender differences in the amount and deposition of amyloidβ in APPswe and PS1 double transgenic mice. Neurobiol. Dis. 2003, 14, 318–327. [Google Scholar] [CrossRef]
- Castillo, E.; Leon, J.; Mazzei, G.; Abolhassani, N.; Haruyama, N.; Saito, T.; Saido, T.; Hokama, M.; Iwaki, T.; Ohara, T.; et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 2017, 7, 17762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakała, M.; Malik, A.R.; Strosznajder, J.B. Inhibitor of cyclooxygenase-2 protects against amyloid beta peptide-evoked memory impairment in mice. J. Pharmacol. Exp. Ther. 2007, 59, 164. [Google Scholar]
- Côté, S.; Carmichael, P.; Verreault, R.; Lindsay, J.; Lefebvre, J.; Laurin, D. Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer’s disease. Alzheimers Dement. 2012, 8, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Ferger, B. Inhibition of the cyclooxygenase isoenzymes COX-1 and COX-2 provide neuroprotection in the MPTP-mouse model of Parkinson’s disease. Synapses 2001, 39, 167. [Google Scholar] [CrossRef]
- Nakayama, M.; Uchimura, K.; Zhu, R.L.; Nagayama, T.; Rose, M.E.; Stetler, R.A.; Isakson, P.C.; Chen, J.; Graham, S.H. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc. Natl. Acad. Sci. USA 1998, 95, 10954–10959. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, K.; Yamagata, K.; Takemiya, T.; Kobayashi, S. COX-2 and mPGES in brain endothelial cells: Potential targets of anti-inflammatory drugs. Curr. Med. Chem. Anti-Inflamm. Anti-Allergy Agents 2002, 1, 161–166. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Hand, T.; Andreasson, K. Prostaglandin D2 mediates neuronal protection via the DP1 receptor. J. Neurochem. 2005, 92, 477–486. [Google Scholar] [CrossRef]
- Monneret, G.; Cossette, C.; Gravel, S.; Rokach, J.; Powell, W.S. 15R-methyl-prostaglandin D2 is a potent and selective CRTH2/DP2 receptor agonist in human eosinophils. J. Pharmacol. Exp. Ther. 2003, 304, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T. Prostaglandin D2 as a mediator of allergic asthma. Science 2000, 287, 2013–2017. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.H.; Dong, Z.; Yang, T. Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am. J. Physiol. 2006, 291, F1332. [Google Scholar]
- Hata, A.N.; Breyer, R.M. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Clin. Pharmacol. Ther. 2004, 103, 147–166. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Durand, M.; Lebeau Jacob, M.C.; Hogan, P.; McManus, S.; Roux, S.; De Brum-Fernandes, A.J. Prostaglandin D2 induces apoptosis of human osteoclasts by activating the CRTH2 receptor and the intrinsic apoptosis pathway. Bone 2012, 51, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Bateman, E.D.; O’Brien, C.; Rugman, P.; Luke, S.; Ivanov, S.; Uddin, M. Efficacy and safety of the CRTh2 antagonist AZD1981 as add-on therapy to inhaled corticosteroids and long-acting β2-agonists in patients with atopic asthma. Drug Des. Dev. Ther. 2018, 12, 1093–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, C.L.; Guzmán, A.; Vand, V.S.; Luheshi, G.N. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav. Immun. 2016, 63, 88–98. [Google Scholar] [CrossRef]
- Carloni, S.; Favrais, G.; Saliba, E.; Albertini, M.C.; Chalon, S.; Longini, M.; Gressens, P.; Buonocore, G.; Balduini, W. Melatonin modulates neonatal brain inflammation through endoplasmic reticulum stress, autophagy, and miR-34a/silent information regulator 1 pathway. J. Pineal Res. 2016, 61, 370–380. [Google Scholar] [CrossRef]
- Deng, X.; Li, M.; Ai, W.; He, L.; Lu, D.; Patrylo, P.R.; Cai, H.; Luo, X.; Li, Z.; Yan, X. Lipolysaccharide-induced neuroinflammation is associated with Alzheimer-like amyloidogenic axonal pathology and dendritic degeneration in rats. Adv. Alzheimers Dis. 2014, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, P.; Ren, L.; Hu, C.; Bi, J. Protective effect of melatonin on soluble Aβ1–42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimers Res. Ther. 2016, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Hiroi, R.; Neumaier, J.F. Differential effects of ovarian steroids on anxiety versus fear as measured by open field test and fear-potentiated startle. Behav. Brain Res. 2006, 166, 93. [Google Scholar] [CrossRef]
- Hampton, R.R.; Hampstead, B.M.; Murray, E.A. Selective hippocampal damage in rhesus monkeys impairs spatial memory in an open-field test. Hippocampus 2004, 14, 808. [Google Scholar] [CrossRef]
- Zhao, J.X.; Tian, Y.X.; Guo, M.L.; Wang, L.; Zhen, Y. Histopathological observation of cerebral cortex and hippocampus in the mouse with synthetic vascular dementia. Chin. J. Pathophysiol. 2000, 16, 1214–1216. [Google Scholar]
- Al-Amin, M.M.; Alam, T.; Hasan, S.M.; Hasan, A.T.; Quddus, A.H.M.R. Prenatal maternal lipopolysaccharide administration leads to age- and region-specific oxidative stress in the early developmental stage in offspring. Neuroscience 2016, 318, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Pang, A.B.; Shan, N.N.; Wang, P.; Wang, Y.K.; Li, Y.G. Primary cultivation and identification of hippocampal neurons from SD rats. J. Logist. Univ. Pap. Med. Sci. 2015, 24, 526–528. [Google Scholar] [CrossRef]
- Kim, W.-G.; Mohney, R.P.; Wilson, B.; Jeohn, G.-H.; Liu, B.; Hong, J.-S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Hong, F.; Wu, G.; Wu, J.; Feng, J. Upregulation of long noncoding RNA PEG10 associates with poor prognosis in diffuse large B cell lymphoma with facilitating tumorigenicity. Clin. Exper. Med. 2016, 16, 177–182. [Google Scholar]
- Feng, M.; Li, J.; Wang, J.; Ma, C.; Jiao, Y.; Wang, Y.; Zhang, J.; Sun, Q.; Ju, Y.; Gao, L.; et al. High glucose increases LPS-induced DC apoptosis through modulation of ERK1/2, AKT and Bax/Bcl-2. BMC Gastroenterol. 2014, 14, 98. [Google Scholar] [CrossRef] [Green Version]
- Yim, Y.S.; Park, A.; Berrios, J.; Lafourcade, M.; Pascual, L.M.; Soares, N.; Kim, J.Y.; Kim, S.; Kim, H.; Waisman, A.; et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 2017, 549, 482–487. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yao, P.; Han, W.; Luo, Y.; Li, Y.; Yang, Y.; Xia, H.; Chen, Z.; Chen, Q.; Wang, H.; et al. Maternal Prenatal Inflammation Increases Brain Damage Susceptibility of Lipopolysaccharide in Adult Rat Offspring via COX-2/PGD-2/DPs Pathway Activation. Int. J. Mol. Sci. 2022, 23, 6142. https://doi.org/10.3390/ijms23116142
Zhang J, Yao P, Han W, Luo Y, Li Y, Yang Y, Xia H, Chen Z, Chen Q, Wang H, et al. Maternal Prenatal Inflammation Increases Brain Damage Susceptibility of Lipopolysaccharide in Adult Rat Offspring via COX-2/PGD-2/DPs Pathway Activation. International Journal of Molecular Sciences. 2022; 23(11):6142. https://doi.org/10.3390/ijms23116142
Chicago/Turabian StyleZhang, Jiahua, Peishuang Yao, Wenli Han, Ying Luo, Yuke Li, Yang Yang, Hui Xia, Zhihao Chen, Qi Chen, Hong Wang, and et al. 2022. "Maternal Prenatal Inflammation Increases Brain Damage Susceptibility of Lipopolysaccharide in Adult Rat Offspring via COX-2/PGD-2/DPs Pathway Activation" International Journal of Molecular Sciences 23, no. 11: 6142. https://doi.org/10.3390/ijms23116142
APA StyleZhang, J., Yao, P., Han, W., Luo, Y., Li, Y., Yang, Y., Xia, H., Chen, Z., Chen, Q., Wang, H., Yang, L., Li, H., Hu, C., Huang, H., Peng, Z., Tan, X., Li, M., & Yang, J. (2022). Maternal Prenatal Inflammation Increases Brain Damage Susceptibility of Lipopolysaccharide in Adult Rat Offspring via COX-2/PGD-2/DPs Pathway Activation. International Journal of Molecular Sciences, 23(11), 6142. https://doi.org/10.3390/ijms23116142