
Mitochondrial rRNA Methylation by Mettl15 Contributes to the Exercise and Learning Capability in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
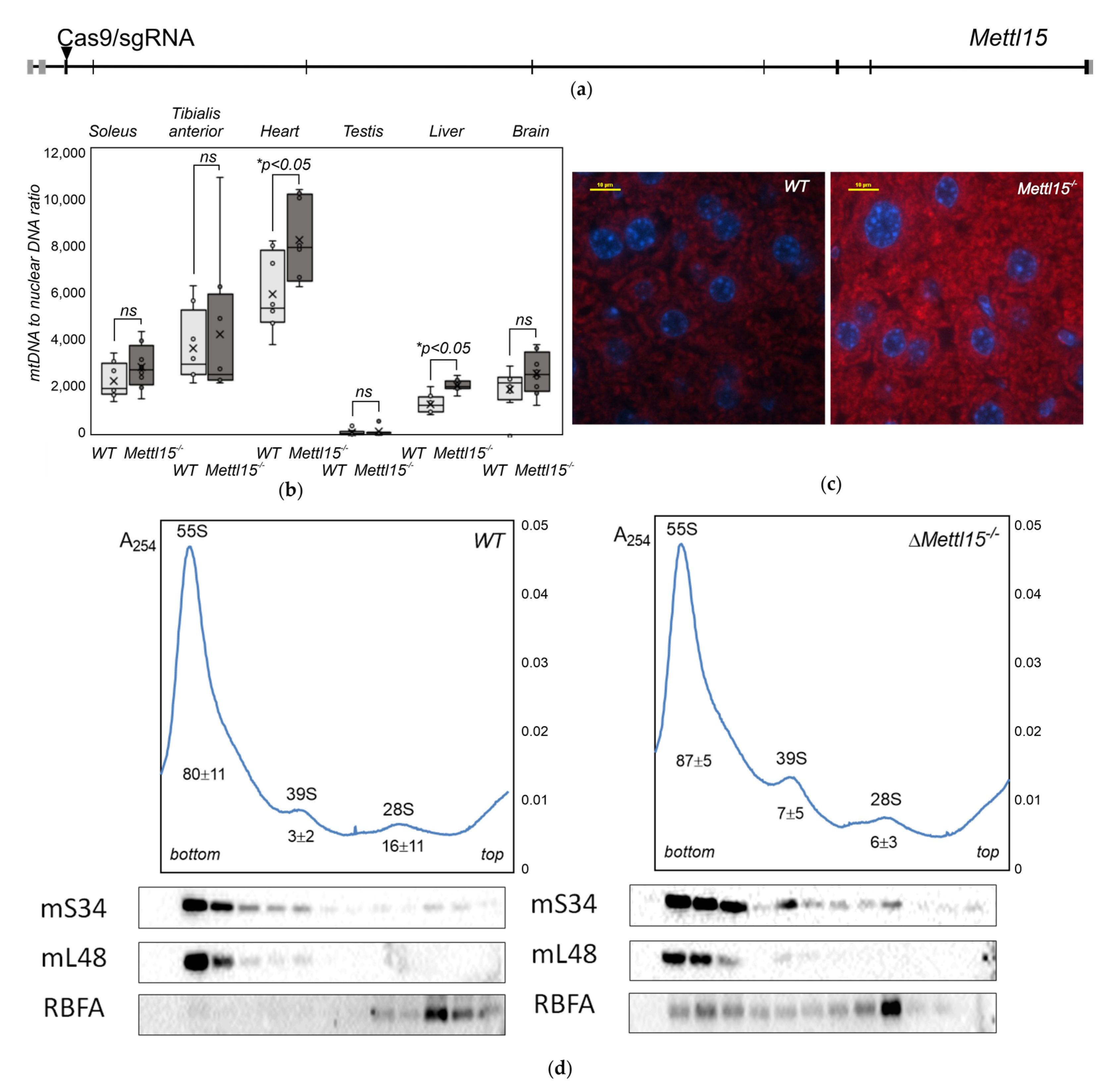
2.1. Generation of Mettl15 Knockout Mice
2.2. Abundance of Mitochondria in Mettl15−/− Knockout Mice
2.3. Alteration in the Composition of the Mitochondrial Ribosomes in Mettl15−/− Knockout Mice
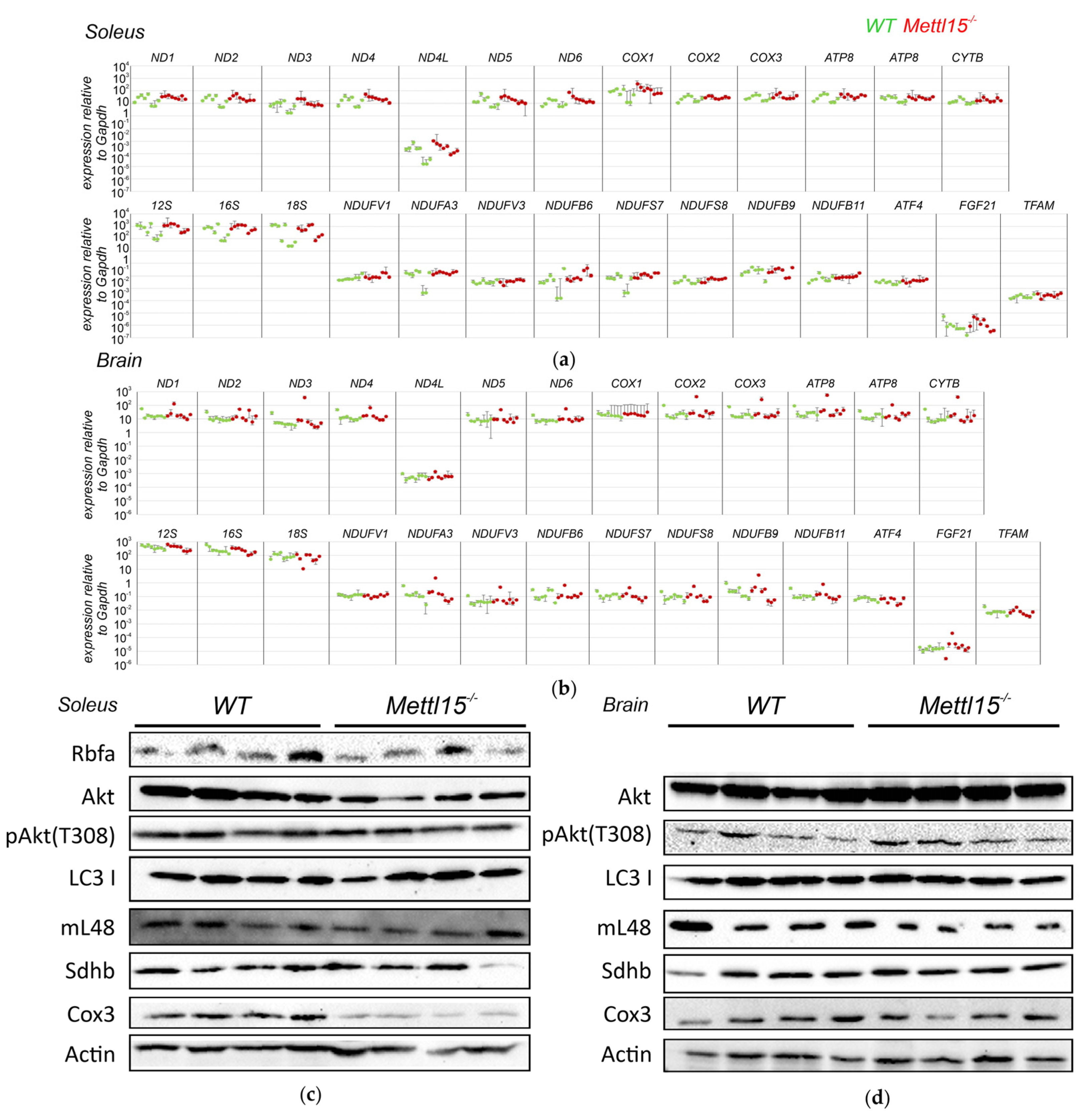
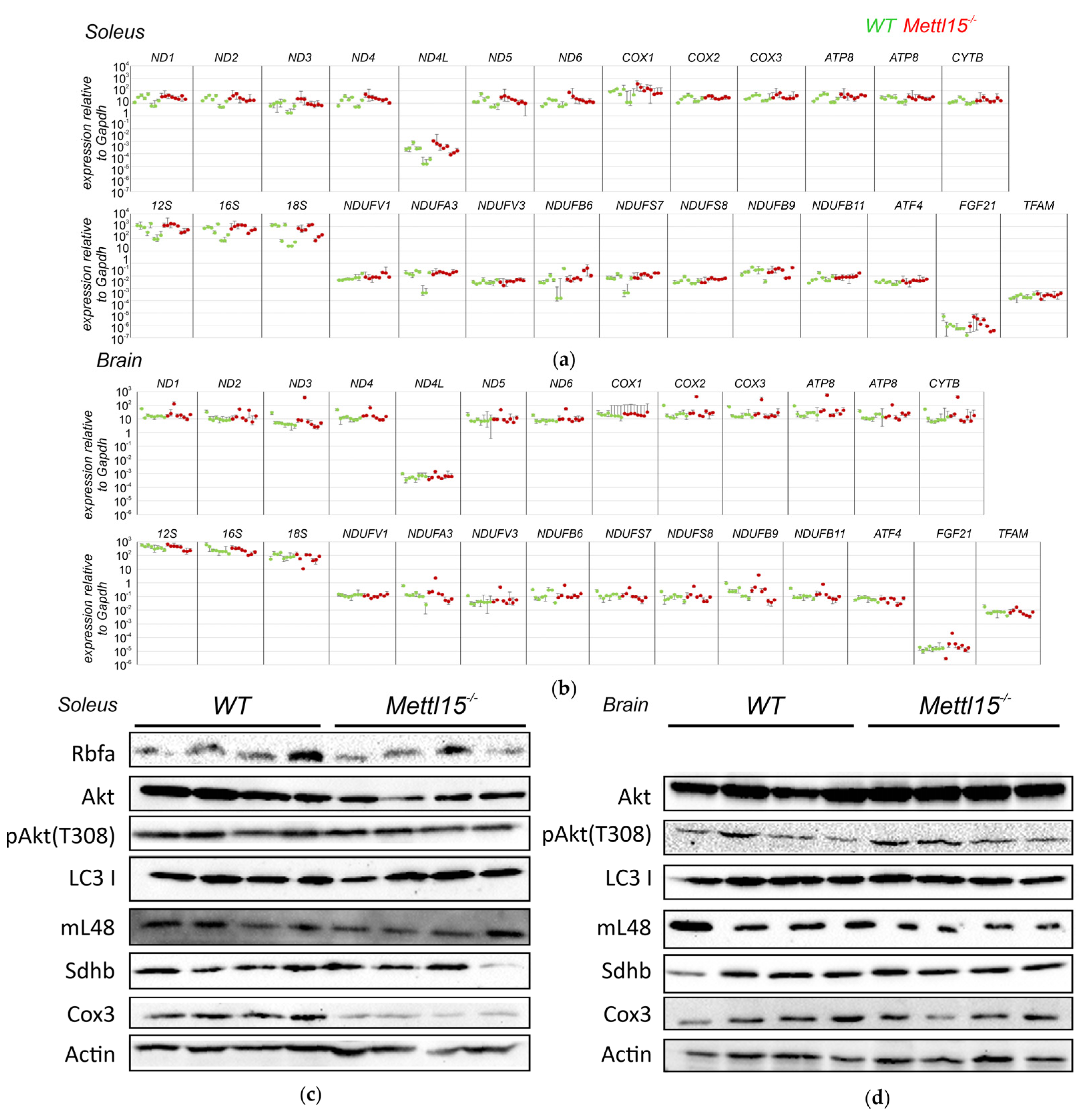
2.4. An Influence of Mettl15 Knockout on Gene Expression in Mice
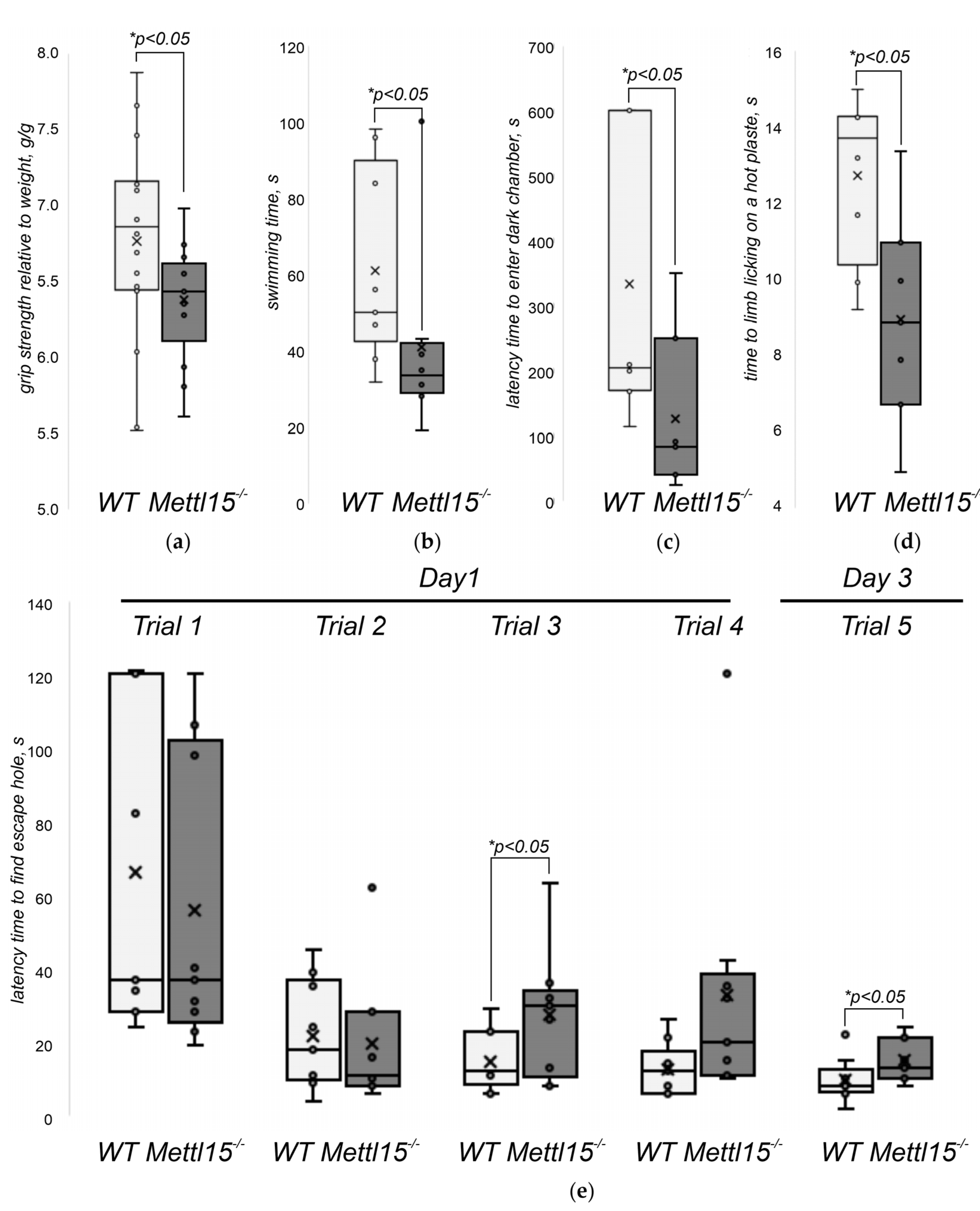
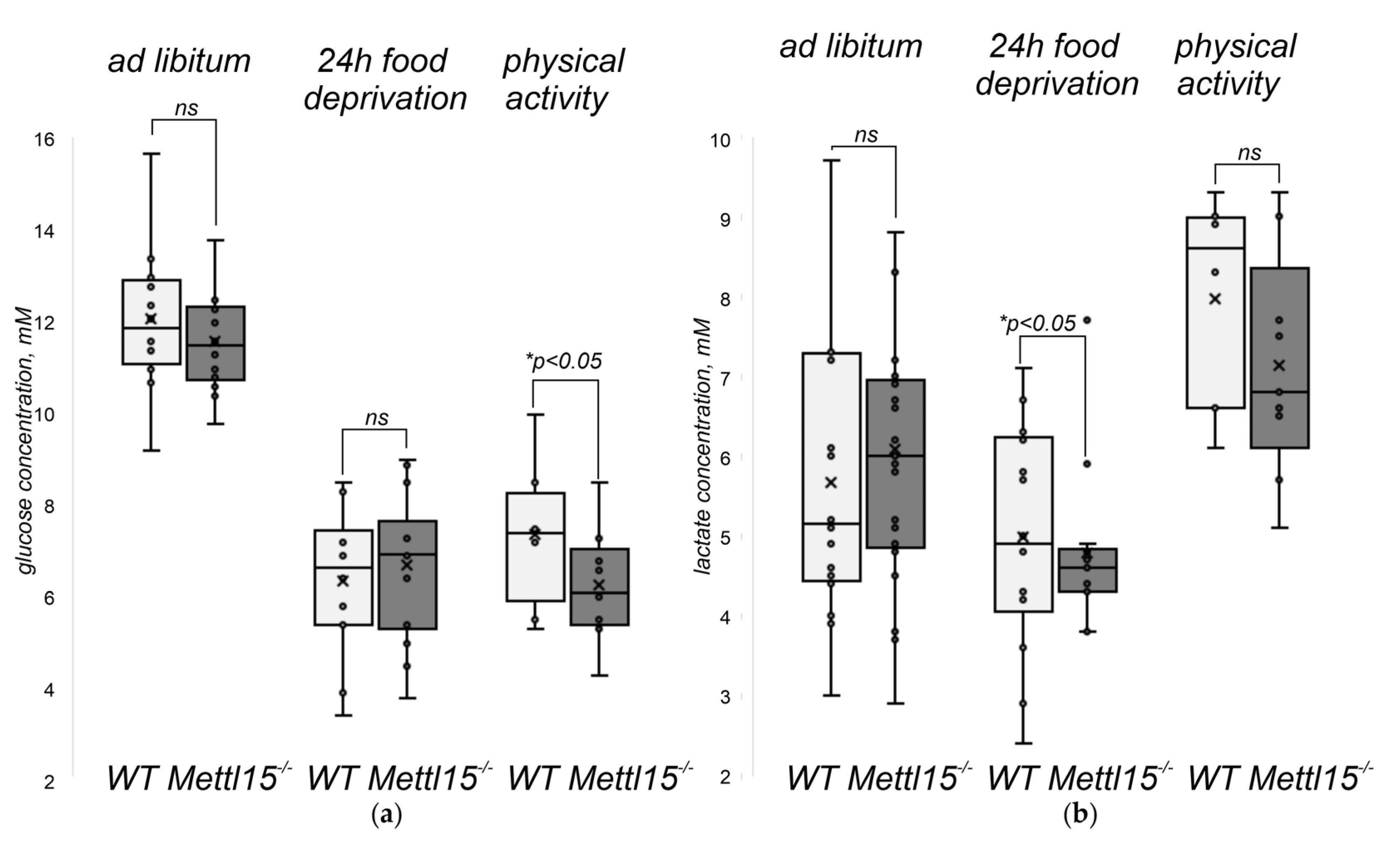
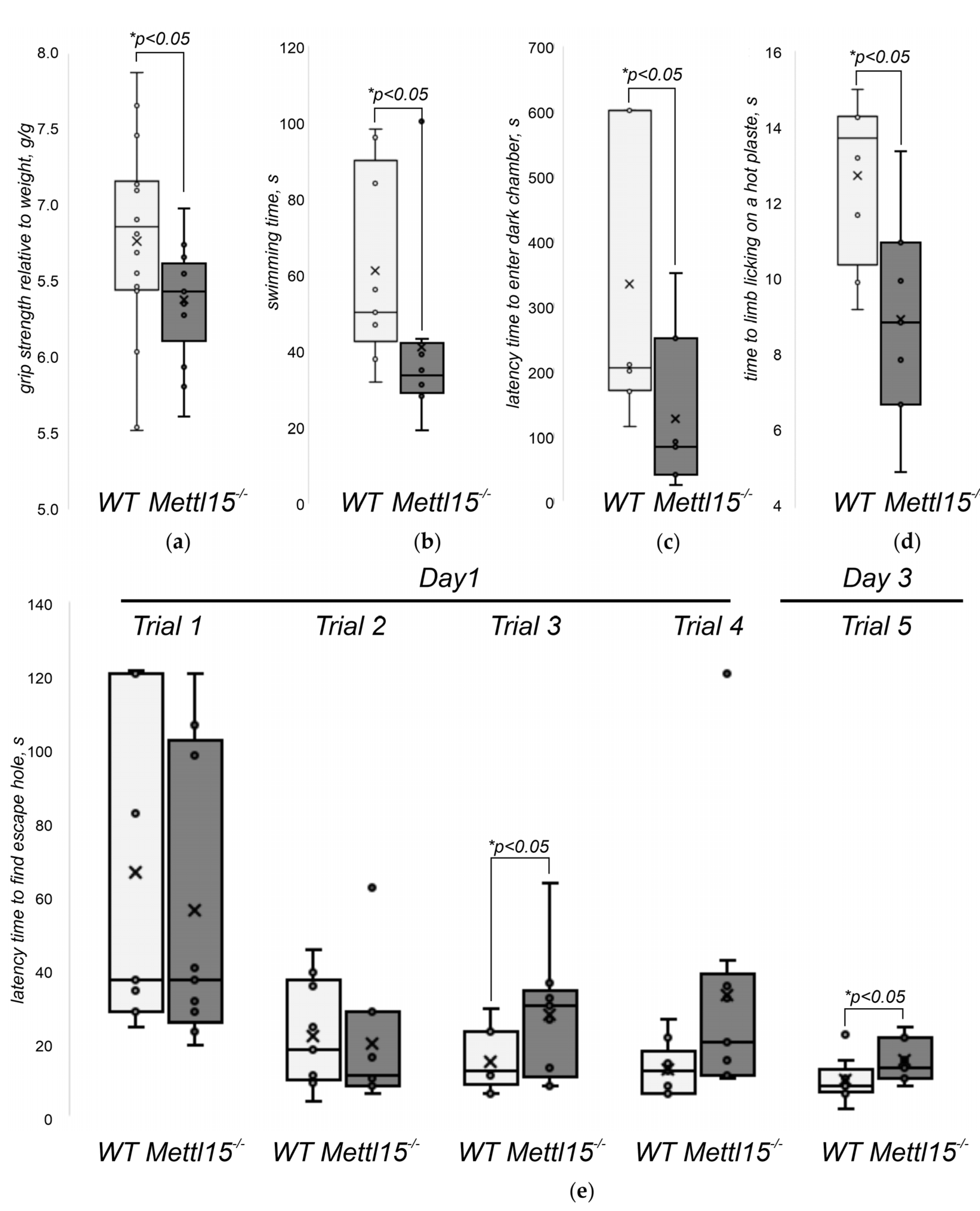
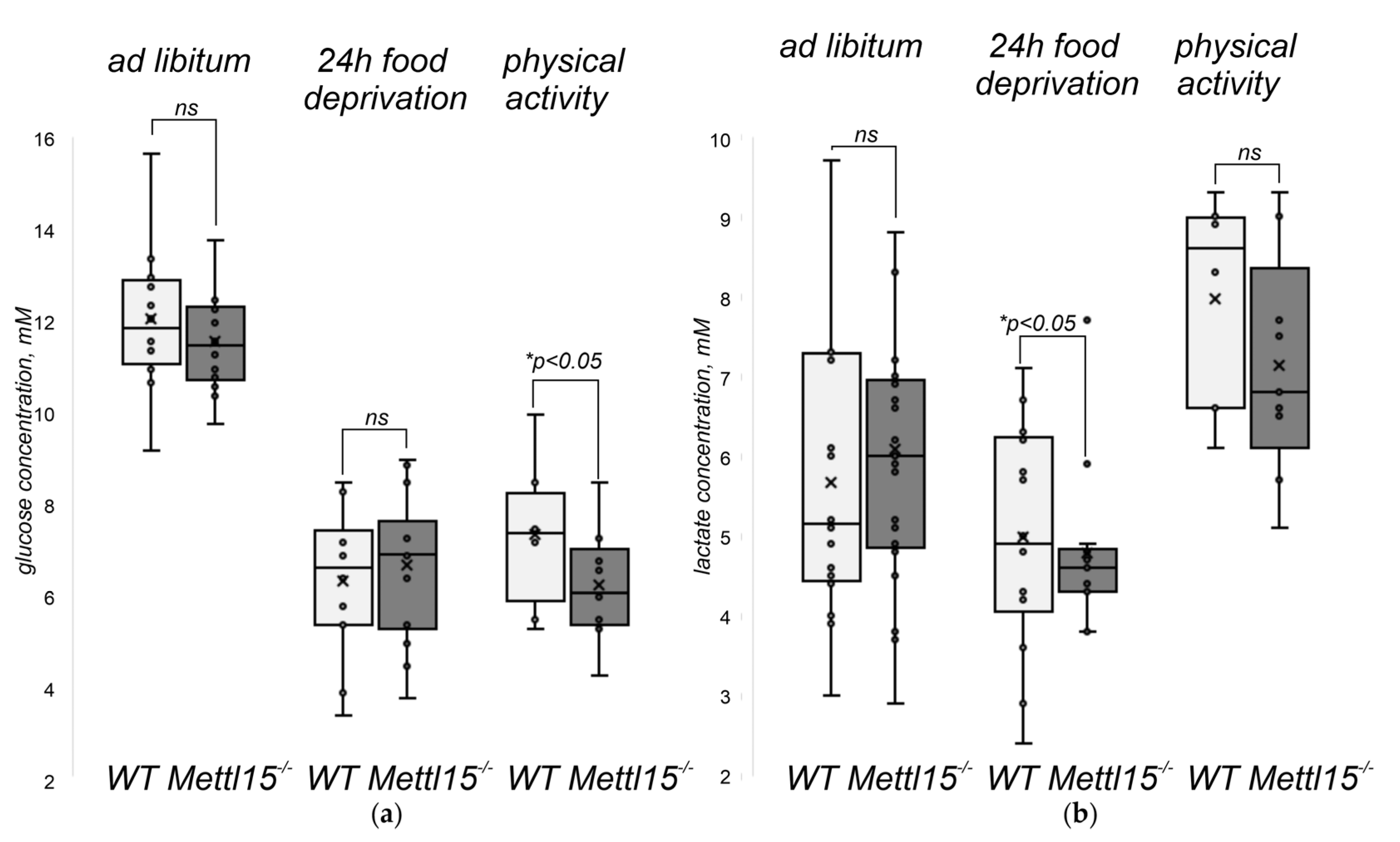
2.5. An Influence of Mettl15 Knockout on the Physiology of Mice
3. Discussion
4. Materials and Methods
4.1. Mettl15 Gene Inactivation and RNA Modification Analysis
4.2. Tests with Live Mice
4.3. Histopathological Examination
4.4. Sucrose-Gradient Profiling of Mitochondrial Ribosomes
4.5. Western Blot Analysis
4.6. Quantitative RT–PCR
4.7. Metabolome Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sagan, L. On the Origin of Mitosing Cells. 1967. J. NIH Res. 1993, 5, 65–72. [Google Scholar]
- Christian, B.E.; Spremulli, L.L. Mechanism of Protein Biosynthesis in Mammalian Mitochondria. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 1035–1054. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Amunts, A.; Brown, A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef]
- Mai, N.; Chrzanowska-Lightowlers, Z.M.A.; Lightowlers, R.N. The Process of Mammalian Mitochondrial Protein Synthesis. Cell Tissue Res. 2017, 367, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Ayyub, S.A.; Varshney, U. Translation Initiation in Mammalian Mitochondria- a Prokaryotic Perspective. RNA Biol. 2020, 17, 165–175. [Google Scholar] [CrossRef]
- Lopez Sanchez, M.I.G.; Krüger, A.; Shiriaev, D.I.; Liu, Y.; Rorbach, J. Human Mitoribosome Biogenesis and Its Emerging Links to Disease. Int. J. Mol. Sci. 2021, 22, 3827. [Google Scholar] [CrossRef]
- Goto, Y.; Nonaka, I.; Horai, S. A Mutation in the TRNA(Leu)(UUR) Gene Associated with the MELAS Subgroup of Mitochondrial Encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic Epilepsy and Ragged-Red Fiber Disease (MERRF) Is Associated with a Mitochondrial DNA TRNA(Lys) Mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An Enhanced MITOMAP with a Global MtDNA Mutational Phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef] [Green Version]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Oztas, S.; Qiu, W.Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I. Mitochondrial Ribosomal RNA Mutation Associated with Both Antibiotic-Induced and Non-Syndromic Deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef]
- Rötig, A. Human Diseases with Impaired Mitochondrial Protein Synthesis. Biochim. Biophys. Acta (BBA) Bioenerget. 2011, 1807, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial Disorders as Windows into an Ancient Organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Boczonadi, V.; Horvath, R. Mitochondria: Impaired Mitochondrial Translation in Human Disease. Int. J. Biochem. Cell Biol. 2014, 48, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Rudler, D.L.; Hughes, L.A.; Viola, H.M.; Hool, L.C.; Rackham, O.; Filipovska, A. Fidelity and Coordination of Mitochondrial Protein Synthesis in Health and Disease. J. Physiol. 2020, 599, 3449–3462. [Google Scholar] [CrossRef]
- Ferrari, A.; Del’Olio, S.; Barrientos, A. The Diseased Mitoribosome. FEBS Lett. 2021, 595, 1025–1061. [Google Scholar] [CrossRef]
- Chatzispyrou, I.A.; Alders, M.; Guerrero-Castillo, S.; Zapata Perez, R.; Haagmans, M.A.; Mouchiroud, L.; Koster, J.; Ofman, R.; Baas, F.; Waterham, H.R.; et al. A Homozygous Missense Mutation in ERAL1, Encoding a Mitochondrial RRNA Chaperone, Causes Perrault Syndrome. Hum. Mol. Genet. 2017, 26, 2541–2550. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Du, M.; Li, D.; Wen, S.; Xie, J.; Li, Y.; Chen, A.; Zhang, K.; Xu, P.; Jia, M.; et al. Mutations in FASTKD2 Are Associated with Mitochondrial Disease with Multi-OXPHOS Deficiency. Hum. Mutat. 2020, 41, 961–972. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Oláhová, M.; Kishita, Y.; Garone, C.; Kremer, L.S.; Yagi, M.; Uchiumi, T.; Jourdain, A.A.; Thompson, K.; D’Souza, A.R.; et al. Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2017, 101, 525–538. [Google Scholar] [CrossRef]
- Lessel, D.; Schob, C.; Küry, S.; Reijnders, M.R.F.; Harel, T.; Eldomery, M.K.; Coban-Akdemir, Z.; Denecke, J.; Edvardson, S.; Colin, E.; et al. De Novo Missense Mutations in DHX30 Impair Global Translation and Cause a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 101, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.D.; Pineda-Alvarez, D.E.; Hadley, D.W.; Keaton, A.A.; Agochukwu, N.B.; Raam, M.S.; Carlson-Donohoe, H.E.; Kamat, A.; Chandrasekharappa, S.C. De Novo Deletion of Chromosome 20q13.33 in a Patient with Tracheo-Esophageal Fistula, Cardiac Defects and Genitourinary Anomalies Implicates GTPBP5 as a Candidate Gene. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 862–865. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, M.T.; Sloan, K.E. The Mitochondrial Epitranscriptome: The Roles of RNA Modifications in Mitochondrial Translation and Human Disease. Cell. Mol. Life Sci. 2018, 75, 241–260. [Google Scholar] [CrossRef]
- Laptev, I.; Dontsova, O.; Sergiev, P. Epitranscriptomics of Mammalian Mitochondrial Ribosomal RNA. Cells 2020, 9, 2181. [Google Scholar] [CrossRef]
- Lopez Sanchez, M.I.G.; Cipullo, M.; Gopalakrishna, S.; Khawaja, A.; Rorbach, J. Methylation of Ribosomal RNA: A Mitochondrial Perspective. Front. Genet. 2020, 11, 761. [Google Scholar] [CrossRef]
- Sergiev, P.V.; Aleksashin, N.A.; Chugunova, A.A.; Polikanov, Y.S.; Dontsova, O.A. Structural and Evolutionary Insights into Ribosomal RNA Methylation. Nat. Chem. Biol. 2018, 14, 226–235. [Google Scholar] [CrossRef]
- Bykhovskaya, Y.; Mengesha, E.; Wang, D.; Yang, H.; Estivill, X.; Shohat, M.; Fischel-Ghodsian, N. Human Mitochondrial Transcription Factor B1 as a Modifier Gene for Hearing Loss Associated with the Mitochondrial A1555G Mutation. Mol. Genet. Metab. 2004, 82, 27–32. [Google Scholar] [CrossRef]
- Cotney, J.; McKay, S.E.; Shadel, G.S. Elucidation of Separate, but Collaborative Functions of the RRNA Methyltransferase-Related Human Mitochondrial Transcription Factors B1 and B2 in Mitochondrial Biogenesis Reveals New Insight into Maternally Inherited Deafness. Hum. Mol. Genet. 2009, 18, 2670–2682. [Google Scholar] [CrossRef]
- Koeck, T.; Olsson, A.H.; Nitert, M.D.; Sharoyko, V.V.; Ladenvall, C.; Kotova, O.; Reiling, E.; Rönn, T.; Parikh, H.; Taneera, J.; et al. A Common Variant in TFB1M Is Associated with Reduced Insulin Secretion and Increased Future Risk of Type 2 Diabetes. Cell Metab. 2011, 13, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Garone, C.; D’Souza, A.R.; Dallabona, C.; Lodi, T.; Rebelo-Guiomar, P.; Rorbach, J.; Donati, M.A.; Procopio, E.; Montomoli, M.; Guerrini, R.; et al. Defective Mitochondrial RRNA Methyltransferase MRM2 Causes MELAS-like Clinical Syndrome. Hum. Mol. Genet. 2017, 26, 4257–4266. [Google Scholar] [CrossRef]
- Laptev, I.; Shvetsova, E.; Levitskii, S.; Serebryakova, M.; Rubtsova, M.; Bogdanov, A.; Kamenski, P.; Sergiev, P.; Dontsova, O. Mouse Trmt2B Protein Is a Dual Specific Mitochondrial Metyltransferase Responsible for M5U Formation in Both TRNA and RRNA. RNA Biol. 2020, 17, 441–450. [Google Scholar] [CrossRef]
- Powell, C.A.; Minczuk, M. TRMT2B Is Responsible for Both TRNA and RRNA M5U-Methylation in Human Mitochondria. RNA Biol. 2020, 17, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Metodiev, M.D.; Lesko, N.; Park, C.B.; Cámara, Y.; Shi, Y.; Wibom, R.; Hultenby, K.; Gustafsson, C.M.; Larsson, N.-G. Methylation of 12S RRNA Is Necessary for in Vivo Stability of the Small Subunit of the Mammalian Mitochondrial Ribosome. Cell Metab. 2009, 9, 386–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metodiev, M.D.; Spåhr, H.; Loguercio Polosa, P.; Meharg, C.; Becker, C.; Altmueller, J.; Habermann, B.; Larsson, N.-G.; Ruzzenente, B. NSUN4 Is a Dual Function Mitochondrial Protein Required for Both Methylation of 12S RRNA and Coordination of Mitoribosomal Assembly. PLoS Genet. 2014, 10, e1004110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haute, L.V.; Hendrick, A.G.; D’Souza, A.R.; Powell, C.A.; Rebelo-Guiomar, P.; Harbour, M.E.; Ding, S.; Fearnley, I.M.; Andrews, B.; Minczuk, M. METTL15 Introduces N4-Methylcytidine into Human Mitochondrial 12S RRNA and Is Required for Mitoribosome Biogenesis. Nucleic Acids Res. 2019, 47, 10267–10281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Shi, Z.; Guo, J.; Chang, K.; Chen, Q.; Yao, C.-H.; Haigis, M.C.; Shi, Y. The Human Mitochondrial 12S RRNA m 4C Methyltransferase METTL15 Is Required for Mitochondrial Function. J. Biol. Chem. 2020, 295, 8505–8513. [Google Scholar] [CrossRef]
- Laptev, I.; Shvetsova, E.; Levitskii, S.; Serebryakova, M.; Rubtsova, M.; Zgoda, V.; Bogdanov, A.; Kamenski, P.; Sergiev, P.; Dontsova, O. METTL15 Interacts with the Assembly Intermediate of Murine Mitochondrial Small Ribosomal Subunit to Form M4C840 12S RRNA Residue. Nucleic Acids Res. 2020, 48, 8022–8034. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Romeis, B. Mikroskopische Technik; R. Oldenbourg: München, Germany, 1948. [Google Scholar]
- Scherbakov, D.; Duscha, S.; Juskeviciene, R.; Restelli, L.; Frank, S.; Laczko, E.; Boettger, E.C. Mitochondrial Misreading in Skeletal Muscle Accelerates Metabolic Aging and Confers Lipid Accumulation and Increased Inflammation. RNA 2020, 27, 265–272. [Google Scholar] [CrossRef]
- Rozanska, A.; Richter-Dennerlein, R.; Rorbach, J.; Gao, F.; Lewis, R.J.; Chrzanowska-Lightowlers, Z.M.; Lightowlers, R.N. The Human RNA-Binding Protein RBFA Promotes the Maturation of the Mitochondrial Ribosome. Biochem. J. 2017, 474, 2145–2158. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Ke, H.; Shinde, U.; Inouye, M. The Role of RbfA in 16S RRNA Processing and Cell Growth at Low Temperature in Escherichia Coli. J. Mol. Biol. 2003, 332, 575–584. [Google Scholar] [CrossRef]
- Gao, Y.; Bai, X.; Zhang, D.; Han, C.; Yuan, J.; Liu, W.; Cao, X.; Chen, Z.; Shangguan, F.; Zhu, Z.; et al. Mammalian Elongation Factor 4 Regulates Mitochondrial Translation Essential for Spermatogenesis. Nat. Struct. Mol. Biol. 2016, 23, 441–449. [Google Scholar] [CrossRef]
- Perks, K.L.; Rossetti, G.; Kuznetsova, I.; Hughes, L.A.; Ermer, J.A.; Ferreira, N.; Busch, J.D.; Rudler, D.L.; Spahr, H.; Schöndorf, T.; et al. PTCD1 Is Required for 16S RRNA Maturation Complex Stability and Mitochondrial Ribosome Assembly. Cell Rep. 2018, 23, 127–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Perks, K.L.; Rossetti, G.; Rudler, D.L.; Hughes, L.A.; Ermer, J.A.; Scott, L.H.; Kuznetsova, I.; Richman, T.R.; Narayana, V.K.; et al. Stress Signaling and Cellular Proliferation Reverse the Effects of Mitochondrial Mistranslation. EMBO J. 2019, 38, e102155. [Google Scholar] [CrossRef] [PubMed]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A Neurological Perspective on Mitochondrial Disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- Sörensen, L.; Ekstrand, M.; Silva, José, P.; Lindqvist, E.; Xu, B.; Rustin, P.; Olson, L.; Larsson, N.-G. Late-Onset Corticohippocampal Neurodepletion Attributable to Catastrophic Failure of Oxidative Phosphorylation in MILON Mice. J. Neurosci. 2001, 21, 8082–8090. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Levy, M.; Sampson, M.J.; Anflous, K.; Armstrong, D.L.; Brown, S.E.; Sweatt, J.D.; Craigen, W.J. The Role of Mitochondrial Porins and the Permeability Transition Pore in Learning and Synaptic Plasticity. J. Biol. Chem. 2002, 277, 18891–18897. [Google Scholar] [CrossRef] [Green Version]
- Aloni, E.; Ruggiero, A.; Gross, A.; Segal, M. Learning Deficits in Adult Mitochondria Carrier Homolog 2 Forebrain Knockout Mouse. Neuroscience 2018, 394, 156–163. [Google Scholar] [CrossRef]
- Darvas, M.; Mukherjee, K.; Lee, A.; Ladiges, W. A Novel One-Day Learning Procedure for Mice. Curr. Protoc. Mouse Biol. 2020, 10, e68. [Google Scholar] [CrossRef]
- Shaham, O.; Slate, N.G.; Goldberger, O.; Xu, Q.; Ramanathan, A.; Souza, A.L.; Clish, C.B.; Sims, K.B.; Mootha, V.K. A Plasma Signature of Human Mitochondrial Disease Revealed through Metabolic Profiling of Spent Media from Cultured Muscle Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 1571–1575. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.H.; Waymire, K.G.; Cottrell, B.; Trounce, I.A.; MacGregor, G.R.; Wallace, D.C. A Mouse Model for Mitochondrial Myopathy and Cardiomyopathy Resulting from a Deficiency in the Heart/Muscle Isoform of the Adenine Nucleotide Translocator. Nat. Genet. 1997, 16, 226–234. [Google Scholar] [CrossRef]
- Seidel-Rogol, B.L.; McCulloch, V.; Shadel, G.S. Human Mitochondrial Transcription Factor B1 Methylates Ribosomal RNA at a Conserved Stem-Loop. Nat. Genet. 2003, 33, 23–24. [Google Scholar] [CrossRef]
- Poldermans, B.; Roza, L.; Van Knippenberg, P.H. Studies on the Function of Two Adjacent N6,N6-Dimethyladenosines near the 3’ End of 16 S Ribosomal RNA of Escherichia Coli. III. Purification and Properties of the Methylating Enzyme and Methylase-30 S Interactions. J. Biol. Chem. 1979, 254, 9094–9100. [Google Scholar] [CrossRef]
- Lafontaine, D.; Vandenhaute, J.; Tollervey, D. The 18S RRNA Dimethylase Dim1p Is Required for Pre-Ribosomal RNA Processing in Yeast. Genes Dev. 1995, 9, 2470–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, K.; Rife, J.P.; Culver, G. Mechanistic Insight into the Ribosome Biogenesis Functions of the Ancient Protein KsgA. Mol. Microbiol. 2008, 70, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletnev, P.; Guseva, E.; Zanina, A.; Evfratov, S.; Dzama, M.; Treshin, V.; Pogorel’skaya, A.; Osterman, I.; Golovina, A.; Rubtsova, M.; et al. Comprehensive Functional Analysis of Escherichia Coli Ribosomal RNA Methyltransferases. Front. Genet. 2020, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, N.M.; Douthwaite, S. YebU Is a M5C Methyltransferase Specific for 16 S RRNA Nucleotide 1407. J. Mol. Biol. 2006, 359, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Suzuki, T. Fine-Tuning of the Ribosomal Decoding Center by Conserved Methyl-Modifications in the Escherichia Coli 16S RRNA. Nucleic Acids Res. 2010, 38, 1341–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbergenov, R.; Duscha, S.; Fritz, A.-K.; Juskeviciene, R.; Oishi, N.; Schmitt, K.; Shcherbakov, D.; Teo, Y.; Boukari, H.; Freihofer, P.; et al. Mutant MRPS5 Affects Mitoribosomal Accuracy and Confers Stress-Related Behavioral Alterations. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Richman, T.R.; Ermer, J.A.; Davies, S.M.K.; Perks, K.L.; Viola, H.M.; Shearwood, A.-M.J.; Hool, L.C.; Rackham, O.; Filipovska, A. Mutation in MRPS34 Compromises Protein Synthesis and Causes Mitochondrial Dysfunction. PLoS Genet. 2015, 11, e1005089. [Google Scholar] [CrossRef]
- Lagouge, M.; Mourier, A.; Lee, H.J.; Spåhr, H.; Wai, T.; Kukat, C.; Silva Ramos, E.; Motori, E.; Busch, J.D.; Siira, S.; et al. SLIRP Regulates the Rate of Mitochondrial Protein Synthesis and Protects LRPPRC from Degradation. PLoS Genet. 2015, 11, e1005423. [Google Scholar] [CrossRef]
- Richman, T.R.; Spåhr, H.; Ermer, J.A.; Davies, S.M.K.; Viola, H.M.; Bates, K.A.; Papadimitriou, J.; Hool, L.C.; Rodger, J.; Larsson, N.-G.; et al. Loss of the RNA-Binding Protein TACO1 Causes Late-Onset Mitochondrial Dysfunction in Mice. Nat. Commun. 2016, 7, 11884. [Google Scholar] [CrossRef] [Green Version]
- Rudler, D.L.; Hughes, L.A.; Perks, K.L.; Richman, T.R.; Kuznetsova, I.; Ermer, J.A.; Abudulai, L.N.; Shearwood, A.-M.J.; Viola, H.M.; Hool, L.C.; et al. Fidelity of Translation Initiation Is Required for Coordinated Respiratory Complex Assembly. Sci. Adv. 2019, 5, eaay2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rackham, O.; Busch, J.D.; Matic, S.; Siira, S.J.; Kuznetsova, I.; Atanassov, I.; Ermer, J.A.; Shearwood, A.-M.J.; Richman, T.R.; Stewart, J.B.; et al. Hierarchical RNA Processing Is Required for Mitochondrial Ribosome Assembly. Cell Rep. 2016, 16, 1874–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochel, F.; Slama, A.; Touati, G.; Desguerre, I.; Giurgea, I.; Rabier, D.; Brivet, M.; Rustin, P.; Saudubray, J.-M.; DeLonlay, P. Respiratory Chain Defects May Present Only with Hypoglycemia. J. Clin. Endocrinol. Metab. 2005, 90, 3780–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardeitchik, T.; Mohamed, M.; Ruzzenente, B.; Karall, D.; Guerrero-Castillo, S.; Dalloyaux, D.; van den Brand, M.; van Kraaij, S.; van Asbeck, E.; Assouline, Z.; et al. Bi-Allelic Mutations in the Mitochondrial Ribosomal Protein MRPS2 Cause Sensorineural Hearing Loss, Hypoglycemia, and Multiple OXPHOS Complex Deficiencies. Am. J. Hum. Genet. 2018, 102, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Averina, O.A.; Vysokikh, M.Y.; Permyakov, O.A.; Sergiev, P.V. Simple Recommendations for Improving Efficiency in Generating Genome-Edited Mice. Acta Nat. 2020, 12, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.C.; Peters, J.; Martin, J.E.; Ball, S.; Nicholson, S.J.; Witherden, A.S.; Hafezparast, M.; Latcham, J.; Robinson, T.L.; Quilter, C.A.; et al. SHIRPA, a Protocol for Behavioral Assessment: Validation for Longitudinal Study of Neurological Dysfunction in Mice. Neurosci. Lett. 2001, 306, 89–92. [Google Scholar] [CrossRef]
- Huang, C.-C.; Hsu, M.-C.; Huang, W.-C.; Yang, H.-R.; Hou, C.-C. Triterpenoid-Rich Extract from Antrodia Camphorata Improves Physical Fatigue and Exercise Performance in Mice. Evid.-Based Complement. Altern. Med. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-Y.; Huang, W.-C.; Liu, C.-C.; Wang, M.-F.; Ho, C.-S.; Huang, W.-P.; Hou, C.-C.; Chuang, H.-L.; Huang, C.-C. Pumpkin (Cucurbita Moschata) Fruit Extract Improves Physical Fatigue and Exercise Performance in Mice. Molecules 2012, 17, 11864–11876. [Google Scholar] [CrossRef]
- Chen, K.; Li, N.; Fan, F.; Geng, Z.; Zhao, K.; Wang, J.; Zhang, Y.; Tang, C.; Wang, X.; Meng, X. Tibetan Medicine Duoxuekang Capsule Ameliorates High-Altitude Polycythemia Accompanied by Brain Injury. Front. Pharmacol. 2021, 12, 680636. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Cai, G.; Kong, F.; Wang, X.; Liu, Y.; Yang, C.; Wang, D.; Teng, L. Antifatigue Activity of Liquid Cultured Tricholoma Matsutake Mycelium Partially via Regulation of Antioxidant Pathway in Mouse. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Kaur, R.; Jaggi, A.S.; Bali, A. Beneficial Role of Central Anticholinergic Agent in Preventing the Development of Symptoms in Mouse Model of Post-Traumatic Stress Disorder. J. Basic Clin. Physiol. Pharmacol. 2020, 31, 20190196. [Google Scholar] [CrossRef] [PubMed]
- Hult, E.M.; Bingaman, M.J.; Swoap, S.J. A Robust Diving Response in the Laboratory Mouse. J. Comp. Physiol. B 2019, 189, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.L.; Hovda, D.A.; Giza, C.C. Ontogeny of Rat Recognition Memory Measured by the Novel Object Recognition Task. Dev. Psychobiol. 2009, 51, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaskin, S.; Tardif, M.; Cole, E.; Piterkin, P.; Kayello, L.; Mumby, D.G. Object Familiarization and Novel-Object Preference in Rats. Behav. Process. 2010, 83, 61–71. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The Novel Object Recognition Memory: Neurobiology, Test Procedure, and Its Modifications. Cogn. Process 2012, 13, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Eagle, A.; Wang, H.; Robison, A. Sensitive Assessment of Hippocampal Learning Using Temporally Dissociated Passive Avoidance Task. BIO-PROTOCOL 2016, 6, e1821. [Google Scholar] [CrossRef] [Green Version]
- Xiang, C.; Li, Z.-N.; Huang, T.-Z.; Li, J.-H.; Yang, L.; Wei, J.-K. Threshold for Maximal Electroshock Seizures (MEST) at Three Developmental Stages in Young Mice. Zool Res. 2019, 40, 231–235. [Google Scholar] [CrossRef]
- Ågamo, A.; Ögren, S.O.; Abizaid, A.; Aboitiz, F.; Absalom, A.; Adell, A.; Adelt, I.; Alici, Y.; Allgulander, C.; Almeida, O.; et al. Encyclopedia of Psychopharmacology: A Springer Live Reference; Springer: Berlin/Heidelberg, Germany, 2011; ISBN 978-3-642-27772-6. [Google Scholar]
- Graham, B. Noninvasive, in Vivo Approaches to Evaluating Behavior and Exercise Physiology in Mouse Models of Mitochondrial Disease. Methods 2002, 26, 364–370. [Google Scholar] [CrossRef]
- Serchov, T.; van Calker, D.; Biber, K. Light/Dark Transition Test to Assess Anxiety-like Behavior in Mice. BIO-PROTOCOL 2016, 6, e1957. [Google Scholar] [CrossRef]
- Bourin, M.; Hascoët, M. The Mouse Light/Dark Box Test. Eur. J. Pharmacol. 2003, 463, 55–65. [Google Scholar] [CrossRef]
- Lojda, Z.; Gossrau, R.; Schiebler, T.H. Enzyme Histochemistry: A Laboratory Manual; Springer: Berlin/Heidelberg, Germany, 1979; ISBN 978-3-540-09269-8. [Google Scholar]
- Luna, L. Histopathologic Methods and Color Atlas of Special Stains and Tissue Artifacts; American Histolabs: Gaithersburg, MD, USA, 1993. [Google Scholar]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and in Vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, S.K.; Layton, C.; Bancroft, J.D. (Eds.) Bancroft’s Theory and Practice of Histological Techniques, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 978-0-7020-6864-5. [Google Scholar]
- Aibara, S.; Andréll, J.; Singh, V.; Amunts, A. Rapid Isolation of the Mitoribosome from HEK Cells. J. Vis. Exp. 2018, 140, 57877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayal, A.A.; Medvedeva, N.V.; Nekrasova, T.M.; Duhalin, S.D.; Surin, A.K.; Minin, A.A. Desmin Interacts Directly with Mitochondria. Int. J. Mol. Sci. 2020, 21, 8122. [Google Scholar] [CrossRef] [PubMed]
- Reschke, M.; Clohessy, J.G.; Seitzer, N.; Goldstein, D.P.; Breitkopf, S.B.; Schmolze, D.B.; Ala, U.; Asara, J.M.; Beck, A.H.; Pandolfi, P.P. Characterization and Analysis of the Composition and Dynamics of the Mammalian Riboproteome. Cell Rep. 2013, 4, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Golde, W.T.; Gollobin, P.; Rodriguez, L.L. A Rapid, Simple, and Humane Method for Submandibular Bleeding of Mice Using a Lancet. Lab. Anim. 2005, 34, 39–43. [Google Scholar] [CrossRef]
- Diehl, K.H.; Hull, R.; Morton, D.; Pfister, R.; Rabemampianina, Y.; Smith, D.; Vidal, J.M.; van de Vorstenbosch, C. European Federation of Pharmaceutical Industries Association and European Centre for the Validation of Alternative Methods A Good Practice Guide to the Administration of Substances and Removal of Blood, Including Routes and Volumes. J. Appl. Toxicol. 2001, 21, 15–23. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Averina, O.A.; Laptev, I.G.; Emelianova, M.A.; Permyakov, O.A.; Mariasina, S.S.; Nikiforova, A.I.; Manskikh, V.N.; Grigorieva, O.O.; Bolikhova, A.K.; Kalabin, G.A.; et al. Mitochondrial rRNA Methylation by Mettl15 Contributes to the Exercise and Learning Capability in Mice. Int. J. Mol. Sci. 2022, 23, 6056. https://doi.org/10.3390/ijms23116056
Averina OA, Laptev IG, Emelianova MA, Permyakov OA, Mariasina SS, Nikiforova AI, Manskikh VN, Grigorieva OO, Bolikhova AK, Kalabin GA, et al. Mitochondrial rRNA Methylation by Mettl15 Contributes to the Exercise and Learning Capability in Mice. International Journal of Molecular Sciences. 2022; 23(11):6056. https://doi.org/10.3390/ijms23116056
Chicago/Turabian StyleAverina, Olga A., Ivan G. Laptev, Mariia A. Emelianova, Oleg A. Permyakov, Sofia S. Mariasina, Alyona I. Nikiforova, Vasily N. Manskikh, Olga O. Grigorieva, Anastasia K. Bolikhova, Gennady A. Kalabin, and et al. 2022. "Mitochondrial rRNA Methylation by Mettl15 Contributes to the Exercise and Learning Capability in Mice" International Journal of Molecular Sciences 23, no. 11: 6056. https://doi.org/10.3390/ijms23116056
APA StyleAverina, O. A., Laptev, I. G., Emelianova, M. A., Permyakov, O. A., Mariasina, S. S., Nikiforova, A. I., Manskikh, V. N., Grigorieva, O. O., Bolikhova, A. K., Kalabin, G. A., Dontsova, O. A., & Sergiev, P. V. (2022). Mitochondrial rRNA Methylation by Mettl15 Contributes to the Exercise and Learning Capability in Mice. International Journal of Molecular Sciences, 23(11), 6056. https://doi.org/10.3390/ijms23116056