
Searching for the Molecular Basis of Partial Deafness

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
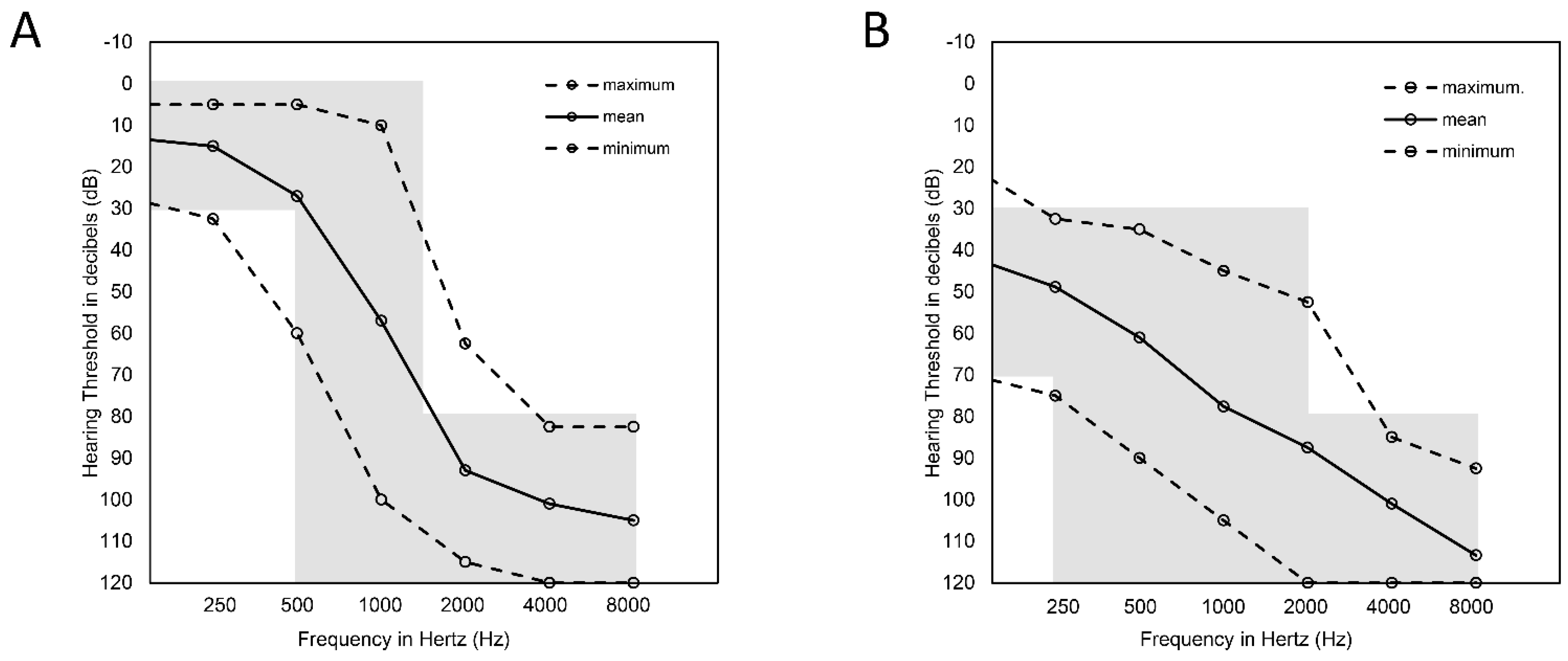
2.1. Audiological Data
2.2. Genetic Results
2.2.1. Genetic Background of HL in the PDT-EC Group
2.2.2. Genetic Background of HL in the PDT-EAS Group
3. Discussion
4. Materials and Methods
4.1. Study Subjects
4.2. Analysis of the Audiological Data
4.3. Genetic Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skarzynski, H.; Lorens, A.; Piotrowska, A.; Anderson, I. Partial deafness cochlear implantation provides benefit to a new population of individuals with hearing loss. Acta Otolaryngol. 2006, 126, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Skarzynski, H.; Lorens, A.; Piotrowska, A.; Skarzynski, P.H. Hearing preservation in partial deafness treatment. Med. Sci. Monit. 2010, 16, CR555–CR562. [Google Scholar] [PubMed]
- Skarzynski, H.; Lorens, A.; D’Haese, P.; Walkowiak, A.; Piotrowska, A.; Sliwa, L.; Anderson, I. Preservation of residual hearing in children and post-lingually deafened adults after cochlear implantation: An initial study. ORL J Otorhinolaryngol. Relat. Spec. 2002, 64, 247–253. [Google Scholar] [CrossRef] [PubMed]
- von Ilberg, C.; Kiefer, J.; Tillein, J.; Pfenningdorff, T.; Hartmann, R.; Sturzebecher, E.; Klinke, R. Electric-acoustic stimulation of the auditory system. New technology for severe hearing loss. ORL J. Otorhinolaryngol. Relat. Spec. 1999, 61, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Lenarz, T. Cochlear implants: Selection criteria and shifting borders. Acta Otorhinolaryngol. Belg. 1998, 52, 183–199. [Google Scholar]
- Skarzynski, H.; Lorens, A.; Piotrowska, A. A new method of partial deafness treatment. Med. Sci. Monit. 2003, 9, CS20–CS24. [Google Scholar]
- Skarzynski, H. Long-term results of partial deafness treatment. Cochlear Implant. Int. 2014, 15, S21–S23. [Google Scholar] [CrossRef]
- Skarzynski, H.; Lorens, A.; Dziendziel, B.; Skarzynski, P.H. Expanding pediatric cochlear implant candidacy: A case study of electro-natural stimulation (ENS) in partial deafness treatment. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1896–1900. [Google Scholar] [CrossRef]
- Marazita, M.L.; Ploughman, L.M.; Rawlings, B.; Remington, E.; Arnos, K.S.; Nance, W.E. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am. J. Med. Genet. 1993, 46, 486–491. [Google Scholar] [CrossRef]
- Korver, A.M.; Smith, R.J.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.; Lustig, L.R.; Usami, S.I.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Primers 2017, 3, 16094. [Google Scholar] [CrossRef] [Green Version]
- Shearer, A.E.; Shen, J.; Amr, S.; Morton, C.C.; Smith, R.J.; Newborn Hearing Screening Working Group of the National Coordinating Center for the Regional Genetics Networks. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet. Med. 2019, 21, 2614–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, A.; Hildebrand, M.; Smith, R. Hereditary Hearing Loss and Deafness Overview. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1434/ (accessed on 1 March 2022).
- Ozieblo, D.; Obrycka, A.; Lorens, A.; Skarzynski, H.; Oldak, M. Cochlear Implantation Outcome in Children with DFNB1 locus Pathogenic Variants. J. Clin. Med. 2020, 9, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Husile, H.; Yang, L.; Cao, Y.; Li, X.; Huo, W.; Bai, H.; Liu, Y.; Wu, Q. A novel pathogenic variant in OSBPL2 linked to hereditary late-onset deafness in a Mongolian family. BMC Med. Genet. 2019, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Bolz, H.; Bolz, S.S.; Schade, G.; Kothe, C.; Mohrmann, G.; Hess, M.; Gal, A. Impaired calmodulin binding of myosin-7A causes autosomal dominant hearing loss (DFNA11). Hum. Mutat. 2004, 24, 274–275. [Google Scholar] [CrossRef]
- Snoeckx, R.L.; Huygen, P.L.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; PLoSki, R.; Murgia, A.; et al. GJB2 mutations and degree of hearing loss: A multicenter study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Oguchi, T.; Ohtsuka, A.; Hashimoto, S.; Oshima, A.; Abe, S.; Kobayashi, Y.; Nagai, K.; Matsunaga, T.; Iwasaki, S.; Nakagawa, T.; et al. Clinical features of patients with GJB2 (connexin 26) mutations: Severity of hearing loss is correlated with genotypes and protein expression patterns. J. Hum. Genet. 2005, 50, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.W.; Kim, D.B.; Choi, J.Y.; Park, H.J.; Lee, J.D.; Hur, D.G.; Bae, S.H.; Jung, D.J.; Lee, S.H.; Kim, U.K.; et al. Molecular and clinical characterization of the variable phenotype in Korean families with hearing loss associated with the mitochondrial A1555G mutation. PLoS ONE 2012, 7, e42463. [Google Scholar] [CrossRef]
- Kim, Y.; Han, J.H.; Yoo, H.S.; Choi, B.Y. Molecular aetiology of ski-slope hearing loss and audiological course of cochlear implantees. Eur. Arch. Otorhinolaryngol. 2022. [Google Scholar] [CrossRef]
- Rim, J.H.; Noh, B.; Koh, Y.I.; Joo, S.Y.; Oh, K.S.; Kim, K.; Kim, J.A.; Kim, D.H.; Kim, H.Y.; Yoo, J.E.; et al. Differential genetic diagnoses of adult post-lingual hearing loss according to the audiogram pattern and novel candidate gene evaluation. Hum. Genet. 2021, 141, 915–927. [Google Scholar] [CrossRef]
- Song, M.H.; Jung, J.; Rim, J.H.; Choi, H.J.; Lee, H.J.; Noh, B.; Lee, J.S.; Gee, H.Y.; Choi, J.Y. Genetic Inheritance of Late-Onset, Down-Sloping Hearing Loss and Its Implications for Auditory Rehabilitation. Ear Hear. 2020, 41, 114–124. [Google Scholar] [CrossRef]
- Yokota, Y.; Moteki, H.; Nishio, S.Y.; Yamaguchi, T.; Wakui, K.; Kobayashi, Y.; Ohyama, K.; Miyazaki, H.; Matsuoka, R.; Abe, S.; et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci. Rep. 2019, 9, 4408. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Moteki, H.; Kitajiri, S.I.; Kitano, T.; Nishio, S.Y.; Yamaguchi, T.; Wakui, K.; Abe, S.; Ozaki, A.; Motegi, R.; et al. Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss. Genes 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, H. Hereditary hearing loss; about the known and the unknown. Hear. Res. 2019, 376, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Weegerink, N.J.; Schraders, M.; Oostrik, J.; Huygen, P.L.; Strom, T.M.; Granneman, S.; Pennings, R.J.; Venselaar, H.; Hoefsloot, L.H.; Elting, M.; et al. Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J. Assoc. Res. Otolaryngol. 2011, 12, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, M.; Nishio, S.Y.; Sakurai, Y.; Hattori, M.; Tsukada, K.; Moteki, H.; Kojima, H.; Usami, S. The patients associated with TMPRSS3 mutations are good candidates for electric acoustic stimulation. Ann. Otol. Rhinol. Laryngol. 2015, 124, 193S–204S. [Google Scholar] [CrossRef]
- Safka Brozkova, D.; Poisson Markova, S.; Meszarosova, A.U.; Jencik, J.; Cejnova, V.; Cada, Z.; Lastuvkova, J.; Raskova, D.; Seeman, P. Spectrum and frequencies of non GJB2 gene mutations in Czech patients with early non-syndromic hearing loss detected by gene panel NGS and whole-exome sequencing. Clin. Genet. 2020, 98, 548–554. [Google Scholar] [CrossRef]
- Garcia-Garcia, G.; Berzal-Serrano, A.; Garcia-Diaz, P.; Villanova-Aparisi, R.; Juarez-Rodriguez, S.; de Paula-Vernetta, C.; Cavalle-Garrido, L.; Jaijo, T.; Armengot-Carceller, M.; Millan, J.M.; et al. Improving the Management of Patients with Hearing Loss by the Implementation of an NGS Panel in Clinical Practice. Genes 2020, 11, 1467. [Google Scholar] [CrossRef]
- Naito, T.; Nishio, S.Y.; Iwasa, Y.; Yano, T.; Kumakawa, K.; Abe, S.; Ishikawa, K.; Kojima, H.; Namba, A.; Oshikawa, C.; et al. Comprehensive genetic screening of KCNQ4 in a large autosomal dominant nonsyndromic hearing loss cohort: Genotype-phenotype correlations and a founder mutation. PLoS ONE 2013, 8, e63231. [Google Scholar] [CrossRef]
- Smits, J.J.; Oostrik, J.; Beynon, A.J.; Kant, S.G.; de Koning Gans, P.A.M.; Rotteveel, L.J.C.; Klein Wassink-Ruiter, J.S.; Free, R.H.; Maas, S.M.; van de Kamp, J.; et al. De novo and inherited loss-of-function variants of ATP2B2 are associated with rapidly progressive hearing impairment. Hum. Genet. 2019, 138, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Usami, S.I.; Nishio, S.Y.; Moteki, H.; Miyagawa, M.; Yoshimura, H. Cochlear Implantation From the Perspective of Genetic Background. Anat. Rec. 2020, 303, 563–593. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Joo, K.; Oh, J.; Han, J.H.; Park, H.R.; Lee, S.; Oh, D.Y.; Woo, S.J.; Choi, B.Y. Severe or Profound Sensorineural Hearing Loss Caused by Novel USH2A Variants in Korea: Potential Genotype-Phenotype Correlation. Clin. Exp. Otorhinolaryngol. 2020, 13, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekin, A.M.; de Ceulaer, G.; Govaerts, P.; Bayazit, Y.; Wuyts, W.; Van de Heyning, P.; Topsakal, V. A New Pathogenic Variant in the TRIOBP Associated with Profound Deafness Is Remediable with Cochlear Implantation. Audiol. Neurootol. 2021, 26, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Huang, S.S.; Qiu, S.W.; Su, Y.; Wang, W.Q.; Xu, H.Y.; Xu, J.C.; Kang, D.Y.; Dai, P.; Yuan, Y.Y. Congenital sensorineural hearing loss as the initial presentation of PTPN11-associated Noonan syndrome with multiple lentigines or Noonan syndrome: Clinical features and underlying mechanisms. J. Med. Genet. 2021, 58, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Thoenes, M.; Zimmermann, U.; Ebermann, I.; Ptok, M.; Lewis, M.A.; Thiele, H.; Morlot, S.; Hess, M.M.; Gal, A.; Eisenberger, T.; et al. OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet. J. Rare Dis. 2015, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Xing, G.; Yao, J.; Wu, B.; Liu, T.; Wei, Q.; Liu, C.; Lu, Y.; Chen, Z.; Zheng, H.; Yang, X.; et al. Identification of OSBPL2 as a novel candidate gene for progressive nonsyndromic hearing loss by whole-exome sequencing. Genet. Med. 2015, 17, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.I.; Oh, K.S.; Kim, J.A.; Noh, B.; Choi, H.J.; Joo, S.Y.; Rim, J.H.; Kim, H.Y.; Kim, D.Y.; Yu, S.; et al. OSBPL2 mutations impair autophagy and lead to hearing loss, potentially remedied by rapamycin. Autophagy 2022, 1–22. [Google Scholar] [CrossRef]
- Horn, H.F.; Brownstein, Z.; Lenz, D.R.; Shivatzki, S.; Dror, A.A.; Dagan-Rosenfeld, O.; Friedman, L.M.; Roux, K.J.; Kozlov, S.; Jeang, K.T.; et al. The LINC complex is essential for hearing. J. Clin. Investig. 2013, 123, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Masterson, J.; Yildirim, B.; Gokkaya, E.; Tokgoz Yilmaz, S.; Tekin, M. A Novel Variant in SYNE4 Confirms its Causative Role in Sensorineural Hearing Loss. Balk. Med. J. 2018, 35, 196–198. [Google Scholar] [CrossRef]
- Ozieblo, D.; Pazik, J.; Stepniak, I.; Skarzynski, H.; Oldak, M. Two Novel Pathogenic Variants Confirm RMND1 Causative Role in Perrault Syndrome with Renal Involvement. Genes 2020, 11, 1060. [Google Scholar] [CrossRef]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| PDT-EC Group | PDT-EAS Group | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family ID | Patient ID | Age at HL Onset (Years) | Age at Hearing Examination (Years) | HL | HAs | CI | Family ID | Patient ID | Age at HL Onset (Years) | Age at Hearing Examination (Years) | HL | HAs | CI | ||||
| Sporadic/Familial | Progression | Symmetry | Sporadic/Familial | Progression | Symmetry | ||||||||||||
| 1 | 943 | 4 | 20 | sporadic | − | + | + | − | 21 | 1371 | 4 | 26 | sporadic | − | + | − | + |
| 2 | 1242 | 3 | 40 | sporadic | − | − | + | − | 22 | 4759 | 15 | 77 | familial | + | + | + | + |
| 3 | 3403 | 24 | 32 | sporadic | − | + | − | + | 23 | 4933 | 25 | 60 | familial | − | + | + | − |
| 4 | 5218 | 3 | 9 | familial | − | + | + | + | 24 | 5047 | 15 | 36 | familial | + | + | − | + |
| 5 | 6949 | 6 | 14 | sporadic | − | + | + | + | 25 | 8507 | congenital | 6 | sporadic | − | + | + | + |
| 6 | 7646 | 28 | 37 | sporadic | + | + | − | + | 26 | 9243 | 15 | 57 | familial | − | + | + | − |
| 7 | 8138 | 4 | 9 | sporadic | − | + | − | + | 27 | 9322 | 18 | 44 | sporadic | − | + | + | − |
| 8 | 8689 | 25 | 48 | familial | N/A | + | + | − | 28 | 9418 | 20 | 48 | sporadic | N/A | + | + | − |
| 9 | 9148 | 14 | 32 | familial | − | + | + | − | 29 | 9425 | 5 | 20 | sporadic | N/A | + | + | − |
| 10 | 9302 | 6 | 43 | sporadic | N/A | − | + | − | 30 | 9508 | 6 | 52 | familial | N/A | + | + | + |
| 11 | 9632 | 30 | 39 | familial | − | + | − | − | 31 | 9772 | 32 | 59 | familial | − | + | − | + |
| 12 | 9661 | 12 | 33 | familial | + | + | + | + | 32 | 10045 | congenital | 5 | familial | N/A | + | + | − |
| 13 | 9754 | 35 | 52 | familial | + | + | − | + | 33 | 10309 | 15 | 17 | sporadic | N/A | + | − | − |
| 14 | 9774 | 20 | 23 | sporadic | N/A | − | + | − | 34 | 10331 | 45 | 51 | sporadic | N/A | + | + | − |
| 15 | 9785 | 4 | 14 | familial | + | + | + | − | 35 | 10332 | 50 | 60 | familial | + | + | + | − |
| 16 | 9994 | 20 | 36 | familial | N/A | + | + | − | 36 | 10385 | 17 | 20 | sporadic | + | + | + | − |
| 17 | 10069 | 6 | 9 | sporadic | − | + | + | − | 37 | 10892 | 39 | 66 | familial | N/A | + | + | − |
| 18 | 11108 | 3 | 31 | familial | − | − | + | − | 38 | 11023 | 3 | 6 | familial | − | + | + | − |
| 19 | 13960 | 7 | 30 | sporadic | − | − | − | − | 39 | 11054 | 20 | 44 | sporadic | − | + | + | + |
| 20 | 14220 | 4 | 15 | sporadic | N/A | + | + | + | 40 | 11162 | 19 | 44 | sporadic | + | + | + | − |
| Family ID | Proband ID | Gene | Variant cDNA Level | Variant Protein Level | Population Frequencies | Pathogenicity Predictions | ACMG Classification | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gnomAD | UK10K | EVS | SIFT | PolyPhen-2 | Mutation Taster | LRT | CADD | SpliceAI | ||||||
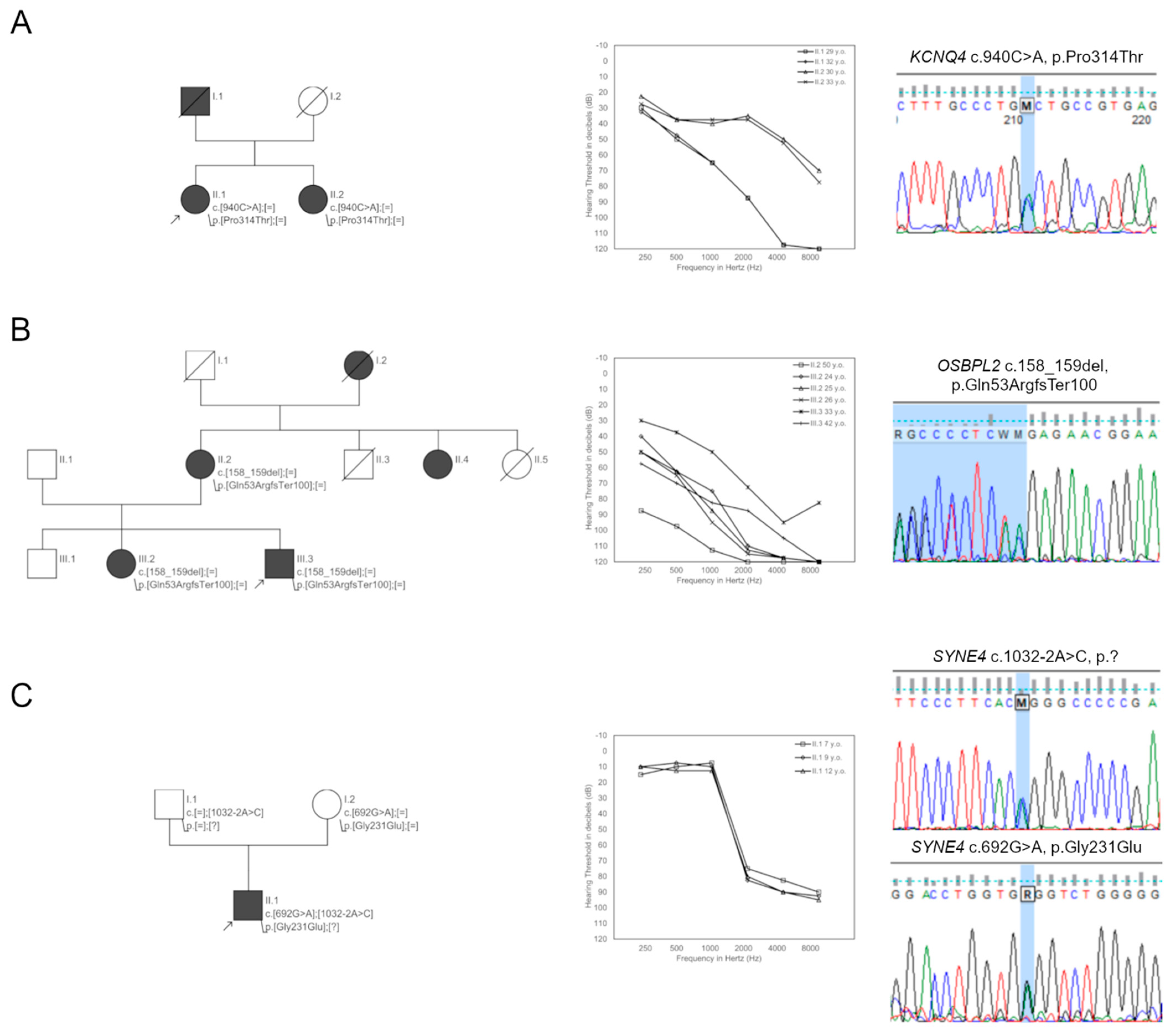
| Family9 | 9148 | KCNQ4 | c.940C>A | p.(Pro314Thr) | 0 | 0 | 0 | D | D | D | D | 29.9 | – | VUS |
| Family12 | 9661 | OSBPL2 | c.158_159del | p.(Gln53Argfs*100) | 0 | 0 | 0 | – | – | – | – | – | – | P |
| Family14 | 9774 | CDH23 | c.2005C>T | p.(Pro669Ser) | 0.000007 | 0 | 0 | D | D | D | D | 32 | – | VUS |
| c.2864G>T | p.(Arg955Leu) | 0.00001 | 0.00001 | 0 | D | D | D | 27 | – | LP | ||||
| Family17 | 10069 | SYNE4 | c.1032-2A>C | p.(?) | 0 | 0 | 0 | – | – | D | – | 19.6 | A | P |
| c.692G>A | p.(Gly231Glu) | 0.0001 | 0 | 0 | D | D | D | N | 24.5 | A | LP | |||
| Family21 | 1371 | TMC1 | c.1919T>C | p.(Leu640Pro) | 0 | 0 | 0 | D | D | D | D | 29.5 | – | VUS |
| c.2030T>C | p.(Ile677Thr) | 0.00001 | 0 | 0 | D | P | D | D | 26.4 | – | VUS | |||
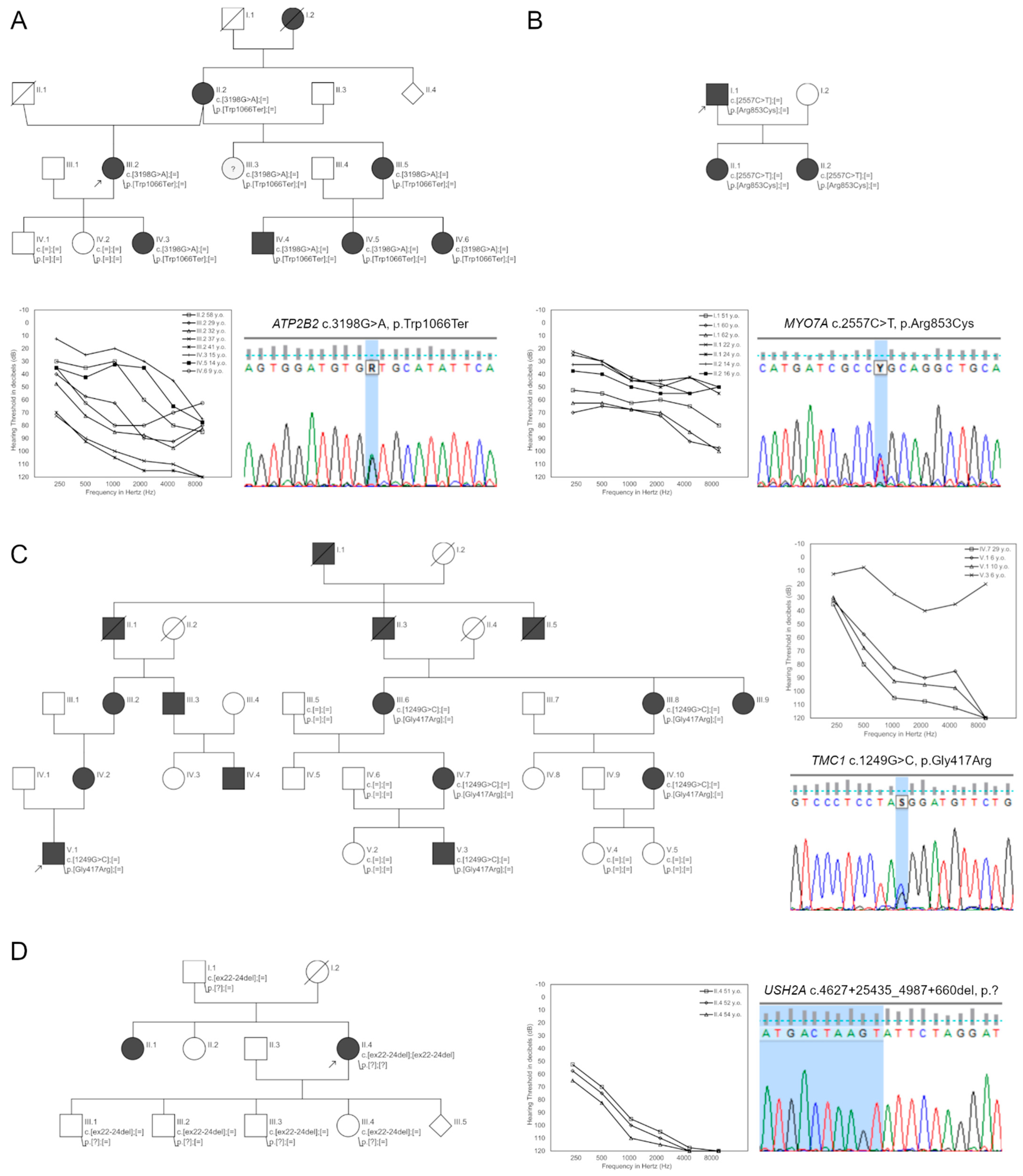
| Family24 | 5074 | ATP2B2 | c.3198G>A | p.(Trp1066Ter) | 0 | 0 | 0 | T | D | D | D | 46 | – | LP |
| Family25 | 8507 | LOXHD1 | c.2575C>T | p.(Arg859Trp) | 0.0004 | 0 | 0.0002 | D | D | D | D | 14.8 | – | VUS |
| c.1228C>T | p.(Gln410Ter) | 0 | 0 | 0 | – | – | D | T | 38 | A | LP | |||
| Family30 | 9508 | USH2A | c.4627+25435_4987+660del | p.(?) | – | – | – | – | – | – | – | – | – | P |
| c.4627+25435_4987+660del | p.(?) | – | – | – | – | – | – | – | – | – | P | |||
| Family33 | 10309 | TMPRSS3 | c.1343T>C | p.(Met448Thr) | 0.00001 | 0 | 0 | D | T | D | D | 24.1 | – | P |
| c.1276G>A | p.(Ala426Thr) | 0.001 | 0.0026 | 0.0014 | D | D | D | D | 29.7 | – | P | |||
| Family34 | 10331 | PTPN11 | c.1226G>C | p.(Gly409Ala) | 0.00002 | 0 | 0.00015 | T | D | D | D | 12.6 | – | VUS |
| Family35 | 10332 | MYO7A | c.2557C>T | p.(Arg853Cys) | 0 | 0 | 0 | T | D | D | D | 34 | – | P |
| Family36 | 10385 | TRIOBP | c.3004del | p.(Ala1002Leufs*3) | 0 | 0 | 0 | – | – | – | – | – | – | LP |
| c.5014G>T | p.(Gly1672Ter) | 0.0004 | 0.0002 | 0.0003 | – | – | D | – | 37 | – | P | |||
| Family37 | 10892 | MYO6 | c.1417A>G | p.(Ile473Val) | 0 | 0 | 0 | D | D | D | D | 23.7 | – | VUS |
| Family38 | 11023 | TMC1 | c.1249G>C | p.(Gly417Arg) | 0 | 0 | 0 | D | D | D | D | 23.7 | – | LP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oziębło, D.; Bałdyga, N.; Leja, M.L.; Skarżyński, H.; Ołdak, M. Searching for the Molecular Basis of Partial Deafness. Int. J. Mol. Sci. 2022, 23, 6029. https://doi.org/10.3390/ijms23116029
Oziębło D, Bałdyga N, Leja ML, Skarżyński H, Ołdak M. Searching for the Molecular Basis of Partial Deafness. International Journal of Molecular Sciences. 2022; 23(11):6029. https://doi.org/10.3390/ijms23116029
Chicago/Turabian StyleOziębło, Dominika, Natalia Bałdyga, Marcin L. Leja, Henryk Skarżyński, and Monika Ołdak. 2022. "Searching for the Molecular Basis of Partial Deafness" International Journal of Molecular Sciences 23, no. 11: 6029. https://doi.org/10.3390/ijms23116029
APA StyleOziębło, D., Bałdyga, N., Leja, M. L., Skarżyński, H., & Ołdak, M. (2022). Searching for the Molecular Basis of Partial Deafness. International Journal of Molecular Sciences, 23(11), 6029. https://doi.org/10.3390/ijms23116029